

Abstract
Three different methods for analyzing natural microbial community diversity were combined to maximize an estimate of the richness of bacterioplankton catabolizing riverine dissolved organic matter (RDOM). We also evaluated the ability of culture-dependent quantitative DNA-DNA hybridization, a 16S rRNA gene clone library, and denaturing gradient gel electrophoresis (DGGE) to detect bacterial taxa in the same sample. Forty-two different cultivatable strains were isolated from rich and poor solid media. In addition, 50 unique clones were obtained by cloning of the bacterial 16S rDNA gene amplified by PCR from the community DNA into an Escherichia coli vector. Twenty-three unique bands were sequenced from 12 DGGE profiles, excluding a composite fuzzy band of the Cytophaga-Flavobacterium group. The different methods gave similar distributions of taxa at the genus level and higher. However, the match at the species level among the methods was poor, and only one species was identified by all three methods. Consequently, all three methods identified unique subsets of bacterial species, amounting to a total richness of 97 operational taxonomic units in the experimental system. The confidence in the results was, however, dependent on the current precision of the phylogenetic determination and definition of the species. Bacterial consumers of RDOM in the studied estuary were primarily both cultivatable and uncultivable taxa of the Cytophaga-Flavobacterium group, a concordant result among the methods applied. Culture-independent methods also suggested several not-yet-cultivated β-proteobacteria to be RDOM consumers.
Estuaries are generally considered to be net heterotrophic as a consequence of the discharge of riverine dissolved organic matter (RDOM) (6, 10, 20, 28). The bacterial species consuming RDOM produce particulate carbon and energy to fuel the marine ecosystem through an external source, in contrary to bacterioplankton production, which is based on the consumption of indigenous DOM. They thereby play an important role in the estuarine biogeochemistry similar to that of primary producers. It was recently shown that a limited set of cultivatable bacteria, primarily in the Flavobacterium-Cytophaga group, were consumers of RDOM in the northern Baltic Sea (11). However, the number of not-yet-cultured bacterial taxa consuming RDOM and therefore the total richness of these bacteria are not known. Culture-independent methods have been proposed to provide a less biased picture of the richness of bacterial communities than culture-dependent methods (2, 3, 24) because of the selective pressure imposed by the requirement of the latter for growth on a solid substrate, leading to the isolation of a plate-growth-adapted subpopulation from the communities. However, a different view has been forwarded by other researchers stating that cultivatable bacteria may constitute the majority of the total bacterial numbers in samples (15, 19) on the basis of DNA-DNA hybridization between bacterial isolates and community DNA from natural samples. The two views are not necessarily mutually exclusive. If there is a tendency for actively growing cells to form colonies on commonly applied rich solid media, these bacteria may be selectively isolated. Quantification of the abundance in natural samples by DNA-DNA hybridization may therefore provide a good estimate of the actively growing and dominant species, as this method is not related to the abundance of the species in question on the plate surface. Dominant species on the plate surface may be minorities in natural samples and vice versa. At the same time, it is likely that many species fail to grow at all on the plate surface of a given medium or show so few CFU that they are overlooked in the isolation step. The cultivation-dependent strategy may therefore underestimate the richness in samples, which is better estimated by culture-independent methods.
Culture-independent methods may detect species that are missed by plating, provided that the amplification efficiency is high enough. However, they are typically dependent on PCR and other molecular biological techniques. Several potential biases have been shown or are conceivable for the required extraction of community DNA, the PCR step, and other enzymatic reactions (see, for example, reference 30). Also, cloning into vectors or separation of 16S ribosomal DNA (rDNA) by denaturing or temperature gradient electrophoresis has its own potential shortcomings regarding accurate separation of taxa (14).
Investigations in which several phylogenetic methods are compared by use of the same sample are almost completely lacking (however, see reference 9). Even less is known about their absolute accuracy, as “standards” for natural bacterial assemblages are not available. Comparisons of the abilities of several different methods to analyze the phylogeny of microbial communities in the same sample are therefore needed.
In this study, we estimated the richness of the RDOM consumers among not-yet-cultivated bacteria in an experimental system designed to promote the growth of RDOM-catabolizing estuarine bacteria (11). A culture-dependent method and two culture-independent methods were used to estimate the richness of given samples and then were compared with respect to their abilities to estimate the richness and diversity of bacteria.
Our first hypothesis was that the culture-dependent method would identify a significantly smaller subset of the species identified by the PCR-based methods because of the potential selection of bacteria competent to grow on solid media in the former strategy. Our second hypothesis was that the dominant taxa determined by PCR-based methods would be mainly affiliated with the Cytophaga-Flavobacterium group, as previously determined by use of cultured bacteria (11). The performance of the methods should also ideally be independent of season or treatment investigated.
MATERIALS AND METHODS
Dilution culture experiments.
Estuarine dilution cultures from the northern Baltic Sea were prepared by the addition of DOM from the Öre River. The overall aim with this experimental setup was to examine which estuarine bacterial species consume RDOM (for more details, see reference 11). The dilution culture experiments with two treatments were conducted during March, May, and November 1998. One treatment was amendment with RDOM (+RDOM); the other treatment did not have this amendment but was adjusted to the same volume with a brackish saltwater medium (−RDOM). Both treatments were inoculated with bacterioplankton. The control treatment without an inoculum (11) was not analyzed in this study due to the low bacterial numbers obtained. Two replicates of all experimental bottles were incubated with gentle shaking (50 rpm) in the dark at an in situ temperature for approximately 2 weeks (377 to 408 h), until the bacterial communities reached the stationary phase. Subsamples for further studies were taken from the stationary phase.
DNA extraction, 16S rDNA amplification, and sequencing.
Picoplankton organisms from 1-liter samples were collected by filtration through 0.2-μm-pore-size polysulfone filters (Supor-200; Gelman Inc.). Total cellular DNA was extracted by placing the filters in Tris-EDTA buffer (pH 8.0), followed by sonication (Turner Sonic & Materials VCX; 120 W, 5-s pulse and 5-s pause for 2 min) and lysis of cells with lysozyme (final concentration, 1 mg ml−1; 1 h at 37°C) and proteinase K (final concentration, 100 μg ml−1; 1 h at 37°C). DNA was further purified as described below. The obtained mixed-community DNA was used for amplification of the 16S rRNA gene followed by denaturing gradient gel electrophoresis (DGGE) and clone library analysis.
Genotypic characterization of isolated strains was performed with single purified colonies. Cells were lysed with proteinase K and cetyltrimethylammonium bromide (CTAB)-NaCl treatment, followed by chloroform-phenol extraction to extract chromosomal DNA (29). The whole 16S rDNA gene from isolated bacteria was amplified by using Taq polymerase (Boehringer Mannheim). Bacterial universal primers 27F (3′-AGAGTTTGATCATGGCTCAG-5′) and 1492R (3′-TACGGYTACCTTGTTACGACTT-5′) were used for amplification (12). The PCR product was purified with PCR Kleen spin columns (Bio-Rad Inc.), and the nucleotide sequences were determined from the purified ∼800-bp partial fragment of the 16S rDNA gene.
Screening and abundance of isolated species.
Reverse-hybridization experiments were conducted to screen for dominant species in the cultures. The community DNAs from all seasons and treatments were radioactively labeled with a random priming kit (High Prime DNA labeling kit; Boehringer) and [α-32P]dATP (3,000 to 6,000 Ci mmol−1; Amersham Pharmacia Biotech). The labeled community DNA from each treatment and season was then hybridized to cells of all 312 isolated strains. The hybridization conditions and detection method were as specified below. On the basis of the reverse hybridization, isolates showing a strong dot blot hybridization signal were quantified by the total DNA-DNA hybridization protocol originally described by Pinhassi et al. (15). Chromosomal DNA from an isolate was labeled by using a random priming kit (described above). A standard curve for bacterial abundance versus signal intensity was used to convert the hybridization signal to bacterial numbers. The standards were hybridized together with unknown samples from the cultures. Bacterial counts were obtained by direct microscopic enumeration as described by Kisand et al. (11). A dilution series were then made by suspending bacteria in autoclaved, 0.2-μm-filtered brackish water. Duplicate samples containing from 1 × 104 to 5 × 106 cells per dot were applied to hybridization membranes (Hybond-N+; Amersham) by using a blotting apparatus (Gibco BRL). Cells were lysed as described below. Bacterial cells from experimental water (volume of 10 or 20 ml) were filtered with the same membranes as bacterial standards. The samples were lysed in the blotting apparatus by covering the slots with 100 μl of 0.5 M NaOH and incubating the samples twice for 5 min each. The solution was filtered dry, and the slots were covered with 100 μl of 1 M Tris-HCl (pH 7.4) for 5 min. Finally, the dots were covered with 100 μl of 1.5 M NaCl- 0.5 M Tris-HCl (pH 7.4), and the solution was filtered dry. The DNA was linked to the membranes by optimal cross-linking (1,200 mJ cm−2; 14 s).
Membranes were prehybridized in a solution consisting of 10% dextran sulfate, 1% sodium dodecyl sulfate, and 100 μg of salmon sperm DNA/ml for at least 2 h at 69°C in a hybridization incubator (Robbins Scientific model 400). Denatured probe was added to the hybridization tube (Robbins). Membranes with community DNA and a standard for each isolate were hybridized in the same hybridization tube overnight at 69°C. Membranes were washed with 5× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate)-0.1% sodium dodecyl sulfate solution at 74°C for 45 min and in 2× SSC at room temperature for 45 min. Washed membranes were exposed on a PhosphorImager (Molecular Dynamics) for detection of the hybridization signal. The relationship between hybridization signal and number of bacteria was obtained from the slope of the standard curve for each isolate.
DGGE.
An estimate of the richness and diversity of community bacterioplankton DNA was also obtained by using DGGE. The specific 16S rDNA region at positions ∼341 to ∼928 was PCR amplified as described by Teske et al. (25). The resulting amplicon was run on a polyacrylamide gel at 200 V for a maximum of 12 h at 60°C in a 20 to 70% denaturing gradient. The gel was stained with SybrGreen I or SybrGold (Molecular Probes) and analyzed with STORM (Molecular Dynamics) and a Fluor-S MultiImager (Bio-Rad).
In order to confirm the identities of DGGE bands with sequences from isolates and clones, 22 unique and visually distinguishable bands were carefully cut from 12 different lanes on the polyacrylamide gel by using a sterile scalpel. DNA was extracted from the polyacrylamide gel by using a Qiaex II DNA purification kit (Qiagen Inc.), and purified DNA fragments were reamplified and reanalyzed by DGGE as described above. In most cases, reamplified DNA fragments were cloned into the pGEM Easy-T vector system (Promega Inc.) prior to sequencing. Finally, 16S rDNA fragments obtained from DGGE were sequenced as described above and compared to longer sequence fragments obtained from isolates and the clone library. DGGE images for band detection and integrated band area intensities were analyzed by using Quantity One computer software (version 4.2.3; Bio-Rad).
16S rRNA gene clone library.
The amplified 16S rDNA was inserted into the pGEM Easy-T vector system. Competent Escherichia coli DH5α was used for electrophoretic transformation with a Bio-Rad Gene Pulser II apparatus (25 μF, 400 Ω, 2.5 kV). Transformants were plated on Luria-Bertani agar plates (with 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside [X-Gal] and isopropyl-β-d-thiogalactopyranoside [IPTG]) and grown at 37°C overnight. Positive (white colony morphotype) transformants were streaked for isolation, cells were lysed in Milli-Q water, and M13 and M13R primers from the lacZ region of the vector were used to amplify the inserts. The proper lengths of the inserts were evaluated on agarose gels (fragment comparison with a 1-kb DNA ladder; Gibco BRL), and the inserts were subjected to sequencing as described above.
Phylogenetic comparison.
The 16S rDNA sequences obtained from the isolates were affiliated with taxa by using current empirical criteria with some exceptions, as the degree of rDNA similarities to species is not strictly defined (7, 8, 12). If 16S rDNA showed >97% similarity to any sequence in the databases searched, then the species name of the homologous sequence was used. If 16S rDNA similarity of >97% occurred among several novel sequences from this study, then the taxa were treated as a single species only if additional tests corroborated the match. The additional tests were as follows. Isolates showing>97% similarity were further evaluated by total genomic DNA-DNA hybridization. If two isolates showed <70% DNA-DNA similarity, then they were classified as different taxa (26). 16S rDNA sequences from the clone library and sequences of excised DGGE bands were compared by using DGGE profiles. Isolates with sequences showing >97% similarity but different DNA melting temperatures and therefore distinct bands were treated as different taxa.
Statistical and phylogenetic analyses.
The significance of the dispersion was tested by using the multivariate randomization test with the grouping factors RDOM levels, seasons, and duplicates (P < 0.05) (13). To estimate the representation of the phylotypes, the clone coverage was calculated with the following equation:
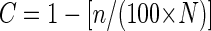 |
(1) |
where C is the coverage, n is the number of single clones, and N is the total number of clones in the clone library. The sequences were aligned by using the secondary structure of the small-subunit rRNA. A phylogenetic tree was constructed by using comparisons between distance (the Kimura two-parameter model [10a]). The permutation test was used to evaluate the significance of the tree, and the jackknife method was used to estimate the significance of branches.
Nucleotide sequence accession numbers.
The sequences reported here have been submitted to GenBank under accession numbers AF388881 to AF388908, AF493059 to AF493066, and AF494469 to AF494507.
RESULTS
Comparison of richness.
Sequence data from the 16S rRNA gene were the basis for the phylogenetic comparison of the methods. When comparing sequences, we used the common criterion in which 16S rDNA sequences that are >97% similar are defined as the same species. In addition, however, we used the requirement that genomic DNA-DNA hybridization should show a similarity of >70% to classify two sequences as the same species, when this was possible (cultivated bacteria only). Also, partial 16S rDNA sequences of the same species should have similar melting temperatures shown by similar migrations in DGGE.
The overall number of different taxa (i.e., richness) showed good correspondence among the methods, with about a 50% lower value for the PCR-DGGE method (Table 1). The total number of different species identified by all three methods was 96. This number was clearly higher than that obtained with any of the methods alone (Table 1). The distribution of taxa among major groups also revealed differences among the methods. On a higher phylogenetic level, the most pronounced difference was observed for the γ- and β-proteobacteria. The cultivation method resulted in more γ-proteobacteria, while the culture-independent PCR-based analysis resulted in more β-proteobacteria.
TABLE 1.
Bacterial taxa identified by different methods in the same experimental systema
| Organism | Number of OTUs found by:
|
Match of OTUs in the following methods:
|
Total no. of unique OTUs | ||||
|---|---|---|---|---|---|---|---|
| Isolationb | Cloningc | PCR-DGGEd | Isolation and cloninge | Isolation and DGGEf | Cloning and DGGEg | ||
| Cytophaga-Flavobacterium group | 12 | 13 | NR | 3 | NR | NR | 22 |
| α-Proteobacteria | 4 | 5 | 5 | 2 | 1 | 2 | 10 |
| β-Proteobacteria | 4 | 18 | 10 | 1 | 0 | 5 | 24 |
| γ-Proteobacteria | 20 | 11 | 7 | 1 | 1 | 1 | 35 |
| ɛ-Proteobacteria | — | 1 | — | 0 | 0 | 0 | 1 |
| Gram positive (G+C rich) | 2 | 2 | 1 | 0 | 0 | 0 | 5 |
| Total | 42 | 50 | 23 | 97 | |||
Phylogenetic affiliation was based on 16S rDNA sequences for all 97 samples analyzed. NR, species not resolved, phylotypes were impossible to resolve due to problems with fuzzy DGGE bands in the Cytophaga-Flavobacterium group. —, species not found.
16S rDNA was obtained by initial cultivation of single-cell colonies on solid agar.
Cloning of 16S rDNA amplicons from mixed-community DNA.
Partial sequencing of amplicons following PCR-DGGE analysis.
Number of taxa found by isolation and cloning.
Number of taxa found by isolation and PCR-DGGE.
Number of taxa found by cloning and PCR-DGGE.
On the species level, a poor match was found among the different methods. Only one species (Methylobacterium sp. strain GOBB3-216) was detected by all three methods, given the three criteria outlined above. This meant that the 16S rDNA gene sequences obtained for this bacterium by the different methods were similar at the >97% level and that the genomic DNA lacked significant similarity (i.e., a DNA-DNA hybridization similarity of <70%) to that of other bacteria. Also, the mobilities of the ∼580-bp-long 16S rDNA fragment matched in a DGGE analysis, regardless of whether it originated from a pure isolate, the clone library,or an excised and sequenced DGGE band.
Cultured isolates and clone library sequences had another seven matches on the species level, distributed among α-, β-, and γ-proteobacteria and three members of the Cytophaga-Flavobacterium group (Table 1). A comparison of the clone library sequences and the sequences obtained from PCR-DGGE bands resulted in seven matches, distributed among α-, β-, and γ-proteobacteria (Fig. 1 and Table 1).
FIG. 1.

Phylogenetic tree (Kimura two-parameter distance optimized criteria) for all of the identified and sequenced taxa: •, isolates; ▪, clones in the 16S rRNA gene library; ♦, PCR-DGGE bands. Taxa were assigned as the same species if sequences showed a similarity of >97% or if different DGGE bands showed a sequence similarity of 98 to 99.9%. Analysis was based on 410- to 430-bp-long 16S rDNA fragments from positions ∼310 to ∼740 bp (according to E. coli numbering). The significance of the branches was estimated by the jackknife resampling method (values of >50% are shown). CFB, Cytophaga-Flavobacterium group; Gm+, gram positive.
Diversity of cultivatable bacteria.
Usually, quite a few colony morphology types (two to seven) dominated on plates with solid growth medium (either rich or poor). Rare colony morphology types were occasionally found on just one of the three replicate plates. A higher richness was found by reverse quantitative DNA-DNA hybridization (QDH) with 312 cultured isolates applied to hybridization membranes and probed with radiolabeled DNA. At least 18 species were detectable by this method (11). Several abundant colonies on the plates, such as Flavobacterium sp. strains GOBB3-317-2, GOBB3-209, and GOBB3-309, were also found to be dominant in the community DNA, as measured by QDH. In addition, several isolated bacteria which were abundant in the community DNA were found to be rare on the plates, for example, Flavobacterium sp. strains GOBB3-C101 and GOBB3-C103-3. A clear response of the cultivatable population to the +RDOM treatment was demonstrated by QDH (Fig. 2a), where five or six species achieved high numbers in the enriched treatment independent of season.
FIG. 2.

Diversity of brackish water bacteria in +RDOM- and −RDOM-treated samples during three different seasons, as analyzed by isolation-dependent DNA-DNA hybridization and isolation-independent cloning of 16S rDNA. (a) Relative abundance of isolated bacteria in the total community DNA (QDH). (b) Relative abundance of different clones in the clone library.
Diversity estimated by clone frequency.
The most numerous taxa among the cloned phylotypes were β-proteobacteria and members of the Cytophaga-Flavobacterium group (Table 1). A rare clone identified as ɛ-protebacteria was also detected in the clone library. Individual treatments in the experimental system were applied to 12 to 18 different clones. In total, the number of clones identified was 50.
An estimate of the diversity of phylotypes by the cloning method was obtained from the frequencies of specific clones in the library (Fig. 2b). This value was probably a crude estimate of the bacterial abundance in the samples, as amplification and cloning efficiencies may affect clone frequencies, apart from the abundance of species-specific DNA in the original sample (17, 21). Most of the frequently seen clones were β-proteobacteria and members of the Cytophaga-Flavobacterium group.
The average coverage (equation 1) from different samples was 77% ± 3% (mean and standard error; n = 12). This value indicated that about 20 different clones in replicate treatments were not enough to achieve a sufficiently confident estimate of the actual richness in the samples. Also, low coverage meant that the frequencies of the clones did not represent the abundance of the phylotypes well from a statistical point of view. This conclusion was indirectly supported by a comparison of the replicates, which were statistically different (multivariate randomization test; P < 0.05), in contrast to all other parameters measured in the replicates, which were not statistically different (P > 0.05). Rare phylotypes (i.e., only one clone from the entire clone library) were represented by an unidentified member of the ɛ-proteobacteria, one member of the Cytophaga-Flavobacterium group (probably not the genus Flavobacterium), three Flavobacterium spp., and members of the γ-, β-, and α-proteobacteria (six, three, and two, respectively).
Diversity estimated by PCR-DGGE band density.
The total richness estimated by the number of DGGE bands was lower than that calculated from isolates or clone libraries (Table 1). In total, 23 sharp bands were identified and sequenced. Individual lanes (independent samples) contained 8 to 15 sharp bands (Fig. 3). Four bands of β-proteobacteria (Methylophilus sp. strain GOBB3-CL123, Sphingomonas sp. strain GOBB3-CL309, and strains GOBB3-B07-4-3 and GOBB3-CL117 of the Comamonadaceae) were present in both +RDOM and control samples. In addition to the sharp bands, an intense, fuzzy band of Cytophaga-Flavobacterium bacteria located in the upper part of the gradient was present in all of the samples. The intensities of the DGGE bands, as measured by densitometry, were assumed to provide a relative measure of the amounts of genomic DNAs. Usually, the same few dominant bands appeared in the +RDOM and control samples. A few dominant bands observed among less dense bands were found to be present independent of season or treatment (Fig. 3). Four members of the β-proteobacteria dominated in all treatments by showing the highest band density, amounting to 14 to 28% of the total density.
FIG. 3.

Bacterial diversity in samples analyzed by DGGE. The most dense sequenced bands belonged to Methylophilus sp. (1, GOBB3-B10-1-4; 2, GOBB3-CL123; 3, GOBB3-CL130), Sphingomonas sp. (4, GOBB3-CL309), unidentified γ-proteobacteria (5, GOBB3-B04-10-2; 6, GOBB3-B04-10-7), β-proteobacteria of the family Comamonadaceae (7, GOBB3-CL296; 8, GOBB3-B07-4-3; 9, GOBB3-CL117; 10, GOBB3-CL120; 11, GOBB3-CL121), Stenotrophomonas sp. (12, GOBB3-CL244; 13, GOBB3-CL245), unidentified Firmicutes (14, GOBB3-B02-5-14), Methylophilus sp. (15, GOBB3-216); and Pantoea agglomerans (16, GOBB3-C104). The whole fuzzy band of the Cytophaga-Flavobacterium group typically showed 15 to 50% of the total intensity, while the dominant bands showed intensities of 4 to 30%. Some minor sequenced DGGE bands were excluded from the standard plot. The average intensities of +RDOM- and −RDOM-treated samples were equal, as similar amounts of DNA were loaded, but the actual amounts of chromosomal DNA in the cultures were different due to differences in the total numbers of bacteria.
Response of diversity to RDOM enrichment.
By QDH, a clear response to +RDOM was demonstrated for cultivatable bacteria at all seasons (Fig. 2a). Cultivatable bacteria showing a growth response to +RDOM were mainly affiliated with the Flavobacterium genus in the Cytophaga-Flavobacterium group and with one member of the γ-proteobacteria (Fig. 2a and 4a).
FIG. 4.

Scores averaged for bacterial taxa by the grouping factor (+RDOM/−RDOM) in a discriminant analysis (13). Species with high scores were more typical of the +RDOM treatment, and species with low scores were more typical of the −RDOM treatment. Data are given for isolates (a), clones from the clone libraries (b), and DGGE profiles (c). Note that bacteria in the Cytophaga-Flavobacterium group were likely to be excluded from the DGGE analysis due to the inability to resolve the fuzzy band typical of this group.
The frequencies of clones in the 16S rRNA gene library were more scattered between phylotypes than was the case for the abundance of isolates (Fig. 2b). There was no significant difference in diversity among the seasons for either treatment (multivariate randomization test; P = 0.10). The difference between the +RDOM and −RDOM treatments was also statistically weak (P = 0.08). However, bacteria in the clone library responding to +RDOM were mostly cultured and uncultured flavobacteria and, to a lesser extent, uncultured β- and γ-proteobacteria and uncultured gram-positive bacteria (Fig. 4b). However, no α-proteobacteria seemed to utilize RDOM.
The dominance of flavobacteria was in agreement with the results obtained by QDH for cultivatable bacteria. Flavobacteria and gram-positive bacteria were also represented by a higher number of species in the +RDOM samples than in the control samples. Several species of β- and γ-proteobacteria were equally abundant in both treatments, encompassing common species in the cultures independent of season.
Through quantification of the phylotypes obtained by the PCR-DGGE method as band densities, the species diversity in the +RDOM treatment was significantly different from that in the −RDOM treatment (multivariate randomization test; P = 0.015). The difference in diversity among seasons was also significant (P = 0.005), compared to the results obtained with the culture-dependent method. Due to the PCR step in the PCR-DGGE method, the average raw intensities of +RDOM and −RDOM samples tended to become equal. However, because of the differences in the total numbers of bacteria in the cultures, the amounts of chromosomal DNAs (not measured directly) in the samples were different. Therefore, the same level of intensity of DGGE bands in different samples was interpreted as different amounts of bacteria in terms of abundance in the cultures.
Due to the potential influence of the efficiency of amplification of the target sequence in the original sample, DGGE probably provided only a crude estimate of the most dominant phylotypes. Also, PCR-DGGE is not sensitive enough to detect small amounts of DNAs in a highly diverse system. This factor may have obscured true dominance relationships.
The intensity of the fuzzy band for flavobacteria was further measured and interpreted as the total abundance of these bacteria. This was done because it was not possible to resolve the flavobacteria by DGGE, due to multiple melting domains in the amplified region resulting in indistinguishable fuzzy bands (11a, 27, 31). PCR-DGGE therefore underestimated the true species richness.
Like the cultivation-dependent and cloning methods, the PCR-DGGE method suggested that the Cytophaga-Flavobacterium group was more abundant in the +RDOM treatment than in the control, as measured as the intensity of the slowly migrating fuzzy band (Fig. 4c). Several β- and γ-proteobacteria, as well as some gram-positive bacteria, also were more abundant in the +RDOM treatment than in the control. The PCR-DGGE method did not detect α-proteobacteria as being abundant in the +RDOM treatment, like the cloning method. However, due to methodological problems, it was impossible to analyze which species in the Cytophaga-Flavobacterium group specifically responded to +RDOM on the basis of the PCR-DGGE analysis.
DISCUSSION
Richness of cultivatable and not-yet-cultured bacteria.
The main aim of this study was to identify bacterioplankton catabolizing RDOM in the estuarine environment. To foster a coverage of both cultivatable and not-yet-cultured bacteria, both a culture-dependent method and two culture-independent methods were applied to the study of the richness and diversity in our experimental bottles.
The total richness estimated by the sum of all methods was clearly higher than the richness obtained from each single method (Table 1). The total number of operational taxonomic units (OTUs) found in our study was also clearly higher than that reported from other studies using either culture-dependent or culture-independent approaches used alone (4, 15, 18). The number of OTUs from most studies in which a single method was used was in the range of 40 to 50, also in agreement with the results of the individual methods in our study. Based on our results, the agreement between different studies could be an artifact due to a limitation in detecting the species actually present in the sample. However, it could also be due to the artificial introduction of base variations, resulting in higher variabilities in the determined sequences, as discussed below.
The communities of species identified by the different methods from the same experimental system constituted different sets, with an overlap of about 7% for the isolation and cloning methods, 3% for isolation and DGGE, 9% for cloning and DGGE, and an overlap of 1% for all three. Therefore, the populations of bacterial taxa obtained by the culture-dependent, cloning, and DGGE approaches in principle constituted separate sets with a small overlap at the species level. A better agreement might have been expected since the isolates were abundant members of the enrichment communities, according to DNA-DNA hybridization between species-specific probes and community DNA from the experimental bottles.
Consequently, isolates did not constitute a subset of the PCR-based sets of species, but rather formed a separate set of species. Our original hypothesis was that PCR-based methods both would find the same species as the isolation based method, but also an appreciable number of additional species that escaped the culture conditions applied. However, this was not the case, suggesting that the two PCR-based methods also showed an inherent bias in detecting the species present in the original sample. An important observation was that all three methods appeared to underestimate the total richness in our samples.
The richness of species determined by DGGE was lower than that in the clone library and that of cultivatable bacteria, partly due to the inability to resolve the Cytophaga-Flavobacterium cluster. Characteristic of the DGGE analysis was that several abundant taxa belonged to closely related “clone families” having clearly distinguished bands with low nucleotide sequence differences (>97% similarity). Four such clone families were affiliated with the β- and γ-proteobacteria (Fig. 3). Usually not all of the bands of the particular family were present together in the same sample. This result provided evidence that the bands in a clone family could originate from different organisms, despite their high similarity. However, a fair analysis is not possible without sequencing data from pure cultures. Thus, we cannot exclude the possibility that the artificial introduction of variations during PCR amplification or the natural heterogeneity of 16S rRNA gene copies was responsible for the closely related clone families.
Response to +RDOM.
All methods showed support for a positive response of flavobacteria to +RDOM, but a positive response was also evident for some β- and γ-proteobacteria and gram-positive bacteria (Fig. 2, 3, and 4). Culture-dependent QDH showed the clearest evidence for flavobacteria being important RDOM consumers. With this method, the same group of cultivatable species (mostly the genus Flavobacterium) dominated in the +RDOM dilution cultures independent of the season investigated.
Generally, DGGE and clone frequencies supported the dominance of flavobacteria in the +RDOM treatment (Fig. 2 and 4). We found six phylotypes of flavobacteria in the clone library that were not detected by QDH (Fig. 1). Flavobacteria were impossible to resolve further by DGGE due to the occurrence of multiple melting domains in this taxon and therefore of specific melting temperatures characteristic of the amplified 16S rRNA gene fragments. However, given that flavobacterium 16S rDNA comigrated during DGGE, the dense fuzzy band in the +RDOM treatment lent support for the presence and abundance of flavobacteria as important RDOM consumers.
Thus, the cloning method suggested that at least an additional six not-yet-cultured species of flavobacteria were consumers of RDOM. The DGGE method merely supported the presence of flavobacteria and could not add specific information on the species level. Flavobacteria were therefore likely important for RDOM consumption in this estuarine environment.
In the comparison between QDH and clone frequency, at least one species (Flavobacterium sp. strain GOBB3-209) became specifically important in the +RDOM treatment. The congruence between the methods might have been higher if flavobacteria also had been resolved in the DGGE analysis. Consequently, only QDH could quantitatively and significantly support the finding that a limited set of species, primarily flavobacteria, became dominant in the +RDOM treatment. The frequency of clones lent some support for a response by the species in the same genus (P = 0.08), while PCR-DGGE provided only semiquantitative support for flavobacteria being important RDOM consumers.
Neither of the culture-independent methods suggested that the species exclusively appearing in the +RDOM treatment was dominant in relation to the total bacterial numbers in the cultures (mostly Flavobacterium spp., such as strains GOBB3-CL98, GOBB3-103-1, and GOBB3-209). Other species detected by these methods, however, were relatively abundant in both treatments (e.g., β-proteobacterium strain GOBB3-CL117), suggesting a successful feeding strategy at both substrate conditions offered. These generalists, however, also showed in several instances a relatively higher frequency of occurrence in the +RDOM treatment than in the −RDOM treatment (e.g., β-proteobacterium Methylophilus sp. strain GOBB3-CL123 and uncultured γ-proteobacterium strain GOBB3-B04-10-02). In this comparison, the frequency of a clone was compared with the total number of clones, and the intensity of one DGGE band was compared with the intensities of all bands. Due to the uncertainty in the quantitative accuracy of the culture-independent methods, however, some of these differences may not be real.
Clone frequency was likely not an accurate quantitative measure of the abundance of clones, although dominant clones would be expected to occur at a higher frequency. However, a higher number of clones (several thousands; at least 1 order of magnitude) should be analyzed to obtain statistically reliable quantification of the dominant species and to detect rare species. Development in sequencing techniques (e.g., capillary sequencers on microplates) may allow routine sequencing of the amounts of clones in the near future. However, present isolation-independent methods used in microbial ecology generally lack the possibility of sufficient confident quantification to detect treatment effects at the species level. The reason is either the low numbers of samples analyzed (e.g., clone libraries) or the lack of quantitative control in many steps of PCR-dependent fingerprinting methods (e.g., DGGE and restriction fragment length polymorphism analysis). Also, in most cases, in situ fluorescent hybridization can be applied with appropriate phylogenetic confidence only at a taxonomic resolution above the species level. A more complete identification of the not-yet-cultured RDOM-catabolizing species therefore has to await further method development.
Differences in the abilities to identify taxa.
The largest discrepancy between the QDH method and the PCR-based methods was found for the β-proteobacteria, as few β-proteobacteria were found among the cultured bacteria. Several β-proteobacteria not cultivated in our study could therefore be important RDOM consumers in addition to flavobacteria (Fig. 4b and c).
The results from the culture-independent methods were also less congruent among seasons compared to the cultivatable community. The difference here is difficult to interpret because of methodological uncertainties. We cannot see any reason for a better efficiency of detection of the dominant species by the QDH method (i.e., positive selection). At any rate, it is difficult to question the fact that the species identified as RDOM consumers showed a clear numerical response to +RDOM relative to the controls, where those species would show the same detection efficiency (11). Growing species must therefore have been competent to utilize some of the substrates in the RDOM pool for growth. It was therefore more likely that the quantification of both clones and DGGE bands was too inaccurate to provide reliable quantitative values. Consequently, we considered QDH to provide the most reliable quantification of bacterial species in natural samples among the methods studied, but with the limitation that the species in question must have been cultivated.
Cultivatable versus noncultivatable bacteria.
This study adds weight to the set of studies advocating that a larger fraction of bacterial are cultivatable than are assessed from the number of CFUs and microscopic direct total counts alone (19, 15). Assuming that the phylotypes identified by the PCR-based methods are true species of importance in our samples, the cultivatable fraction would amount to 50% of the studied bacterial community. This value is remarkably high compared to the commonly cited view that less than 3% of aquatic bacteria are cultivatable (1). Part of this discrepancy, however, is due to the fact that the value of 3% is calculated as the fraction of total bacterial cells plating on solid media, rather than a true estimate of the fraction of cultivatable species. It is therefore plausible that a substantial share of the bacterial species in natural surface waters are cultivatable on rich solid media. Most species, however, show a low plating efficiency.
Our results also showed that a significant fraction of the taxa might be overlooked by the culture-dependent method. The cultivatable community constituted a majority of the total counts in the March experiment but only 32 and 47% in the May and November experiments, respectively (11). Even if we assume 20% of the bacteria to be nonliving (and DNA deficient) in these samples, the not-yet-cultured part of the community was probably important, corresponding to <10 to 50%, depending on season.
Cause of the mismatch observed among methods.
Our results prompt the question of why the correspondence among the methods was so low on the species level. Shortcomings of PCR-based methods encompass selective DNA extraction and preferential amplification of certain templates (30). Another poorly recognized phenomenon are the mutational “hot spots,” where random base anomalies are introduced into the amplified product (22). Speksnijder et al. (22) showed that sequence artifacts will be introduced when PCR is used for a diverse set of templates. The result of this phenomenon is a less accurate sequence with high nucleotide variability at hot-spot regions. In our study, this problem may have been especially relevant for the PCR-DGGE method due to the additional cloning step and thus the additional PCR amplification of the mixed DNA templates that were required to sequence the DGGE bands. Chimera formation is the only artifact that is currently possible to identify by generally available tools (e.g., Check Chimera at the Ribosome Database Project data bank).
The presence of multiple copies of 16S rRNA genes in randomly selected environmental isolates is yet another source of sequence variation (5). Such heterogeneity would influence both sequencing results for pure cultures of isolates and the results of DGGE fingerprinting methods, as several DGGE bands may originate from a single bacterium.
Because of this multitude of potential biases, Speksnijder et al. (22) proposed the use of a lower similarity of 16S rDNA when assigning sequences to the same species based on direct amplification of mixed DNA templates in environmental samples. Widening the window defining two OTUs as the same species to 95 to 96% similarity would result in a significantly higher number of similar bacterial species among the methods used in this study (data not shown). The limited resolution of present phylogenetic methods would therefore explain to a large extent the mismatch among the methods in our study. An indication that this was a plausible explanation for the low match among the methods was that we did not find biases among the methods to be as clear on a higher phylogenetic level. If this were the case, then the sum of all OTUs found in this study would have been an overestimate of the true species richness in the experimental system, primarily due to inaccuracies in the PCR-based methods.
A problem with widening the allowance for similar species, however, is that the operational criterion of >97% similarity for similar species is still not sufficiently stringent to appropriately differentiate between taxa with different ecological functions. Even in the range of 98 to 99.9% 16S rDNA similarity, important genetic and phenotypic differences can be profound (23, 16). Thus, methods providing a phylogenetic resolution at or below the species level are required to allow a better understanding of the relationships between bacterial species and their functional roles in the natural environment. A well-founded consensus regarding the species concept for prokaryotes would further aid in the advancement of bacterial ecology.
Acknowledgments
We are grateful to Rocio Cuadros for collecting the DNA samples for DGGE and other analyses. We are also indebted to Simon Tuck for the linguistic corrections.
Funding for this project was kindly provided by the Foundation for Strategic Environmental Research (MISTRA, no. 97238) and The Swedish National Research Council (B-AA/BU 08583-319) to J. Wikner. Umeå Marine Sciences Centre and Hanse Wissenschatskolleg kindly contributed a fellowship to V. Kisand and a valuable supply of field equipment.
REFERENCES
- 1.Amann, R. I. 1995. In situ identification of micro-organisms by whole cell hybridization with rRNA-targeted nucleic acid probes. Microb. Ecol. 33:1-15.
- 2.Amann, R. I., W. Ludwig, and K.-H. Schleifer. 1995. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 59:143-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brock, T. 1987. The study of microorganisms in situ: progress and problems. Symp. Soc. Gen. Microbiol. 41:1-17. [Google Scholar]
- 4.Crump, B. C., E. V. Armbrust, and J. A. Baross. 1999. Phylogenetic analysis of particle-attached and free-living bacterial communities in the Columbia River, its estuary, and the adjacent coastal ocean. Appl. Environ. Microbiol. 65:3192-3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dahllöf, I., H. Baillie, and S. Kjelleberg. 2000. rpoB-based microbial community analysis avoids limitations inherent in 16S rRNA gene intraspecies heterogeneity. Appl. Environ. Microbiol. 66:3376-3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.del Giorgio, P. A., J. J. Cole, and A. Cimbleris. 1997. Respiration rates in bacteria exceed phytoplankton production in unproductive aquatic systems. Nature 385:148-151. [Google Scholar]
- 7.Devereux, R., S. H. He, C. L. Doyle, S. Orkland, D. A. Stahl, J. Legall, and W. B. Whitman. 1990. Diversity and origin of Desulfovibrio species—phylogenetic definition of a family. J. Bacteriol. 172:3609-3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dobson, S. J., T. A. McMeekin, and P. D. Franzmann. 1993. Phylogenetic relationships between some members of the genera Deleya, Halomonas, and Halovibrio. Int. J. Syst. Bacteriol. 43:665-673. [DOI] [PubMed] [Google Scholar]
- 9.Eilers, H., J. Pernthaler, F. O. Glockner, and R. Amann. 2000. Culturability and in situ abundance of pelagic bacteria from the North Sea. Appl. Environ. Microbiol. 66:3044-3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Findlay, S., R. L. Sinsabaugh, D. T. Fischer, and P. Franchini. 1998. Sources of dissolved organic carbon supporting planktonic bacterial production in the tidal freshwater Hudson River. Ecosystems 1:227-239. [Google Scholar]
- 10a.Kimura, M. 1980. A simple method for estimating evolutionary rate of base substitution through comparative studies of nucleotide sequence. J. Mol. Evol. 16:111-120. [DOI] [PubMed] [Google Scholar]
- 11.Kisand, V., R. Cuadros, and J. Wikner. 2002. Phylogeny of culturable estuarine bacteria catabolizing riverine organic matter in the northern Baltic Sea. Appl. Environ. Microbiol. 68:379-388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11a.Kisand, V., and J. Wikner. Limited resolution of 16S rDNA DGGE caused by melting properties and closely related DNA sequences. J. Microbiol. Methods, in press. [DOI] [PubMed]
- 12.Lane, D. 1991. 16S/23S rRNA sequencing, p. 115-175. In M. Goodfellow (ed.), Nucleic acid techniques in bacterial systematics. John Wiley & Sons, Chichester, England.
- 13.Manly, B. 1997. Multivariate data, p. 260-283. In B. F. J. Manly (ed.), Randomization, bootstrap and Monte Carlo methods in biology. Chapman & Hall, Ltd., London, England.
- 14.Muyzer, G., and K. Smalla. 1998. Application of denaturing gradient gel electrophoresis (DGGE) and temperature gradient gel electrophoresis (TGGE) in microbial ecology. Antonie Leeuwenhoek 73:127-141. [DOI] [PubMed] [Google Scholar]
- 15.Pinhassi, J., U. L. Zweifel, and A. Hagström. 1997. Dominant marine bacterioplankton species found among colony-forming bacteria. Appl. Environ. Microbiol. 63:3359-3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pukall, R., O. Pauker, D. Buntefuss, G. Ulrichs, P. Lebaron, L. Bernard, T. Guindulain, J. Vives-Rego, and E. Stackebrandt. 1999. High sequence diversity of Alteromonas macleodii-related cloned and cellular 16S rDNAs from a Mediterranean seawater mesocosm experiment. FEMS Microbiol. Ecol. 28:344. [Google Scholar]
- 17.Rainey, F. A., N. Ward, L. I. Sly, and E. Stackebrandt. 1994. Dependence on the taxon composition of clone libraries for PCR amplified, naturally occurring 16S rDNA on the primer pair and the cloning system used. Experientia 50:796-797. [Google Scholar]
- 18.Rappe, M. S., K. Vergin, and S. J. Giovannoni. 2000. Phylogenetic comparisons of a coastal bacterioplankton community with its counterparts in open ocean and freshwater systems. FEMS Microbiol. Ecol. 33:219-232. [DOI] [PubMed] [Google Scholar]
- 19.Rehnstam, A.-S., S. Bäckman, D. Smith, F. Azam, and A. Hagström. 1993. Blooms of sequence-specific culturable bacteria in the sea. FEMS Microbiol. Ecol. 102:161-166. [Google Scholar]
- 20.Rolff, C., and R. Elmgren. 2000. Use of riverine organic matter in plankton food webs of the Baltic Sea. Mar. Ecol. Prog. Ser. 197:81-101. [Google Scholar]
- 21.Scharf, S. J., G. T. Horn, and H. A. Erlich. 1986. Direct cloning and sequence analysis of enzymatically amplified genomic sequences. Science 233:1076-1078. [DOI] [PubMed] [Google Scholar]
- 22.Speksnijder, A., G. A. Kowalchuk, S. De Jong, E. Kline, J. R. Stephen, and H. J. Laanbroek. 2001. Microvariation artifacts introduced by PCR and cloning of closely related 16S rRNA gene sequences. Appl. Environ. Microbiol. 67:469-472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stackebrandt, E., and B. M. Goebel. 1994. A place for DNA-DNA reassociation and 16S ribosomal RNA sequence analysis in the present species definition in bacteriology. Int. J. Syst. Bacteriol. 44:846-849. [Google Scholar]
- 24.Staley, J., and A. Konopka. 1985. Measurements of in situ activities of nonphotosynthstic microorganisms in aquatic and terrestrial habitats. Annu. Rev. Microbiol. 39:321-346. [DOI] [PubMed] [Google Scholar]
- 25.Teske, A., C. Wawer, G. Muyzer, and N. B. Ramsing. 1996. Distribution of sulfate-reducing bacteria in a stratified fjord (Mariager Fjord, Denmark) as evaluated by most-probable-number counts and denaturing gradient gel electrophoresis of PCR-amplified ribosomal DNA fragments. Appl. Environ. Microbiol. 62:1405-1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wayne, L. G., D. J. Brenner, R. R. Colwell, P. A. D. Grimont, O. Kandler, M. I. Krichevsky, L. H. Moore, W. E. C. Moore, R. G. E. Murray, E. Stackebrandt, M. P. Starr, and H. G. Truper. 1987. Report of the Ad Hoc Committee on Reconciliation of Approaches to Bacterial Systematics. Int. J. Syst. Bacteriol. 37:463-464. [Google Scholar]
- 27.Wieland, G., R. Neumann, and H. Backhaus. 2001. Variation of microbial communities in soil, rhizosphere, and rhizoplane in response to crop species, soil type, and crop development. Appl. Environ. Microbiol. 67:5849-5854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wikner, J., R. Cuadros, and M. Jansson. 1999. Differences in consumption of allochthonous DOC between a lake and an estuary in a temperate watershed. Aquat. Microb. Ecol. 17:289-299. [Google Scholar]
- 29.Wilson, F. 1994. Preparation of genomic DNA from bacteria, p. 2.4.1-2.4.2. In K. Janssen (ed.), Current protocols in molecular biology. Greene Publishing Associates, New York, N.Y. [DOI] [PubMed]
- 30.Wintzingerode, F., U. B. Göbel, and E. Stackebrandt. 1997. Determination of microbial diversity in environmental samples: pitfalls of PCR-based rRNA analysis. FEMS Microbiol. Rev. 21:213-229. [DOI] [PubMed] [Google Scholar]
- 31.Wu, Y., V. M. Hayes, J. Osinga, I. M. Mulder, M. W. Looman, C. H. Buys, and R. M. Hofstra. 1998. Improvement of fragment and primer selection for mutation detection by denaturing gradient gel electrophoresis. Nucleic Acids Res. 26:5432-5440. [DOI] [PMC free article] [PubMed] [Google Scholar]