

Abstract
The omega (ome) gene product is a modifier of larval cuticle protein 5 and its alleles (and duplicates) in the third instar of Drosophila melanogaster. Using deletion mapping the locus mapped to 70F-71A on the left arm of chromosome 3. A homozygote null mutant (ome 1) shows a pleiotropic phenotype that affected the size, developmental time of the flies, and the fertility (or perhaps the behavior) of homozygous mutant males. The omega gene was verified as producing a dipeptidyl peptidase IV (DPPIV) by genetic analysis, substrate specificity and pH optimum. The identity of the gene was confirmed as CG32145 (cytology 70F4) in the Celera Database (Berkeley Drosophila Genome Project), which is consistent with its deletion map position. The genomic structure of the gene is described and the decrease in DPPIV activity in the mutant ome1 is shown to be due to the gene CG32145 (omega). The D. melanogaster omega DPPIV enzyme was partially purified and characterized. The exons of the ome1 mutant were sequenced and a base substitution mutation in exon 4 was identified that would yield a truncated protein caused by a stop codon. A preliminary study of the compartmentalization of the omega DPPIV enzyme in several organs is also reported.
| Abbreviations: | |
|---|---|
| DPPIV | dipeptidyl peptidase IV |
| LCP5 & LCP6 | third instar larval cuticle proteins 5 & 6 |
| ome & ome1 | omega locus name (CG32145) and mutant allele in D. melanogaster |
| pNA | paranotroanilide |
Keywords: Drosophila melanogaster, omega, gene identification, enzyme compartmentalization; DPPIV, CD26, dipeptidyl peptidase IV
Introduction
Dipeptidyl peptidase IV (DPPIV, EC3.4.14.5) is a multifunctional enzyme that is important in the processing of numerous oligopeptides in many organisms from bacteria to mammals. It belongs to the prolyl oligopeptidase/S9 enzyme family characterized by a Ser-Asp-His catalytic triad in the C- terminal region. In humans and most mammals, the prolyl oligopeptidase gene family consists of at least: prolyl endopeptidase (PEP), acylaminoacyl peptidase (ACPH), dipeptidyl peptidase IV (DPPIV) and three proteins that have close sequence homology to DPPIV (fibroblast activation protein (FAP), DPPX and DPP10) and two peptidases called DPP8 and DPP9 that have lower sequence homology with DPPIV. The enzymes in this family share highly conserved structural features and moderately conserved amino acid sequence homology (Chen et al., 2003). The DPPIV enzyme and its close relatives are more highly conserved in the C-terminal (an (α/β - hydrolase domain) than in the N-terminal region where the proteins form a seven-bladed propeller region (Abbott et al., 2000 and included references). The N-terminal region has one important 7 amino acid conserved sequence in most of the family members (DW(V/L)YEEE) and the first two glutamic acid residues in this sequence have been shown to be necessary for enzyme activity (Abbott et al., 1999).
DPPIV is a highly specific dipeptidyl aminopeptidase that is characterized by the release of an N-terminal dipeptide, Xaa-Yaa-|-Zaa-, from a polypeptide, preferentially when Yaa is proline, provided Zaa is neither proline nor hydroxyproline (Prosite; BRENDA). The reactivity of the enzyme is related to the amino acid in the penultimate position that can be, after proline and alanine, the following amino acids, with reactivity in this order: pro>ala>>>ser>gly>val (and sometimes thr and leu). In addition, the structure of the substrate's length and/or amino acid sequence immediately surrounding the scissile bond and in the C terminal region can also affect reactivity and/or specificity (Boonacker, et al., 2003; Kühn–wache et al., 2003; Mentlein, 1999; Ludwig et al., 2002; Gault et al., 2003, De Meester et al., 2003).
In many organisms DPPIV is involved not only in the proteolytic cleavage of dipeptides from the NH2-terminus of a variety of biologically active peptides, but also in activation of signal transduction and cell matrix adherence processes. In humans DPPIV (also known as CD26) functions to process (primarily to inactivate) several peptides, many of which are implicated in regulation of immune, inflammatory, nervous and endocrine functions (Abbott et al., 1999; Thoma et al., 2003). No obvious phenotype is associated with this enzyme in humans other than the apparent involvement in glucose regulation and changes in its expression in association with various disease conditions (Boonacker, et al., 2003; OMIM). Fischer rats lacking a functional DPPIV gene have no visible phenotype, show no change in growth rate and only minor physiological effects relating to glucose tolerance (Boonacker, et al., 2003). The enzyme seems to be a ubiquitous housekeeping enzyme, but one whose synthesis is regulated in some tissues (Boonacker, et al., 2003)
In insects, the enzyme activity was recognized in D. melanogaster when transgenic flies with the flounder antifreeze protein gene were found to process the antifreeze protein by removal of an XP dipeptide in the hemolymph. (Peters et al., 1993) The enzyme has been reported in the blowfly, Calliphora vicina, where a DPPIV activity was isolated and shown to cleave the ecdysiostatic peptide trypsin-modulating oostatic factor of Neobellieria (Sarcophaga) bullata and in the brain and intestine of the cockroach, where it is thought to be involved in the inactivation of several tachykinin related peptides (Martensen et al., 1998; Nässel et al., 2000). In the cockroach high activity was obtained from the membrane fraction of the intestine and some 10 fold less was found in brain membranes. Both tissues also showed a smaller amount of soluble activity (Nässel et al., 2000). Suggested substrates for insect DPPIV include the antibacterial cecropins, which are shown to be activated by an aminopeptidase activity from Hyalophora cecropia hemolymph (Boman et al., 1989).
Five genes in FlyBase are expected to code for DPPIV-like proteins in Drosophila melanogaster, CG11034, CG11319, CG32145 (omega), CG9059 and CG3744 (Flybase). The mutant designated omega1 (ome1) was recovered in a screen for cuticle protein variants and identified as a recessive modifier of LCP5 (the larval cuticle protein 5) and its alleles, shifting their migration pattern in native gel PAGE (Fig. 1) (Chihara et al., 1986).
Figure 1.
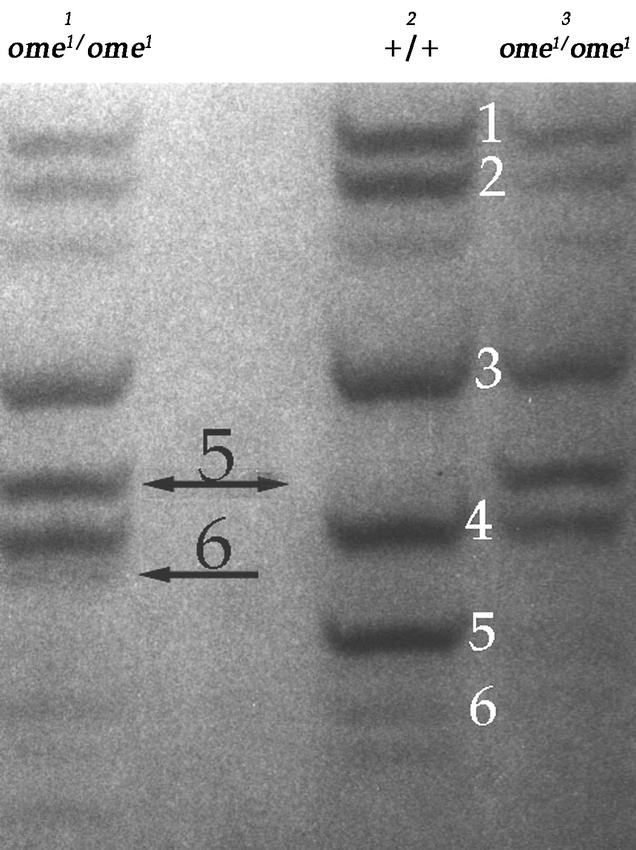
The third instar larval cuticle proteins on a 15% acrylamide nondenaturing gel stained with Coomaasie Blue. Lane 1: The cuticle proteins of third instar larvae from the wildtype stock (oreR) crossed into an ome1 background, Lane 2: wildtype, Lane 3: ome1/ome1 homozygote stock. The wildtype bands are numbered in white. The arrows indicate the modified protein bands 5 and 6. Protein 6 is variant in the homozygote mutant (see Chihara and Kimbrell, 1986).
In this paper we confirm that the wildtype gene omega is coded for by CG32145 and is a DPPIV with specificity similar to, but not identical with, human DPPIV. We clarify the action of the omega DPPIV enzyme on the third instar cuticle proteins LCP5 and LCP6, a protein related to LCP5 and most probably a variant of a duplicated LCP5 (Charles et al., 1998). We present data showing that omega DPPIV has specificity that distinguishes it from other DPPIV enzymes in the fly. Data on the distribution of the enzyme in several organs, and partial characterization of a partially purified epithelial membrane fraction preparation of the enzyme are also provided. This work further confirms the nature of the ome1 mutant as deficient in a DPPIV enzyme.
Materials and Methods
Fly stocks
ome1 and Lcp10rho are ethylmethane sulfonate induced mutants previously described by Chihara and Kimbrell (1986). The wildtype strain is an Oregon R stock.
Fertility analysis
Five virgin females were placed in a vial with a single male for five days at which point the females were separated into individual vials and the male discarded. The offspring in all six vials (the original vial and the five individual vials) were scored for eleven days following the emergence of the first adult fly. Ten replicates of each cross were performed as listed. 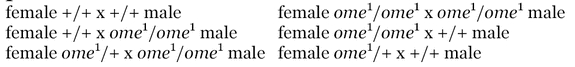
Modification of Oregon R “wildtype” LCP6 in the ome1 background
Virgin ome1 female flies were crossed to wildtype males. The offspring males were backcrossed to virgin ome1 female flies. Integument (cuticle) was dissected from the back-cross third instar larvae and run on non-denaturing gels as described in Chihara and Kimbrell (1986). The larval proteins were examined for heterozygous cuticle protein variants in the homozygous ome1 background similar to those seen in Fig. 1.
Deficiency mapping
ome1 virgin females were mated to deficiency stocks to determine the cytological location of omega. A minimum of 8 F1 larval cuticles were hand dissected to reduce the probability of missing the homozygote ome1 in any cross to less than 0.4%. Cuticle proteins were extracted and run on gels as described by Chihara and Kimbrell (1986).
Protein micro-sequencing
Third instar larval cuticles were prepared in batches of 50 cuticles from wildtype, ome1 and Lcp10rho and the proteins run on gels for analysis as described by Charles et al. (1998).
Collections for developmental studies
To obtain eggs, stocks of young flies (2–3 days old) were transferred to empty bottles that were then inverted onto apple juice agar plates (Ashburner, 1989) that had been coated with yeast paste at 25° C. A 2–hour pre–lay was followed by two hours of egg laying for collection. Eggs were transferred to vials of standard food in groups of 50. Triplicates of 400 eggs were counted in each experiment. For the pupal stage, white prepupae were transferred to new vials and the time recorded to within one hour of pupariation. Flies were counted as they emerged in 2–hour increments.
Preparation of larval enzyme extracts
Late third instar larvae were placed on a glass plate (covered with aluminum foil and in snug contact with an ice platform) and rolled with a pipet (used like a rolling pin), or a solid brass metal cylinder (∼2 in. in diameter, weighing about 1 lb, and wrapped in aluminum foil), depending on the number of larvae, to extrude their insides. The carcasses were then washed with cold Ringer's solution and homogenized in Buffer 1, [0.5mM Phenylthiourea 0.38M Sucrose 0.1M Tris–HCl pH7.5] in the proportion of 10 ml buffer/250 larval carcasses. The homogenate was centrifuged and washed in Buffer 1. The wash was added to the first extract and called the “cytosol” fraction. The pellet was re–extracted with the Triton–X containing Buffer 2 [same as Buffer 1 with 1% Triton–X] using 500 µl/250 larvae in the same way as above, and this extract was labeled the membrane fraction.
Enzyme assays
Chromogenic substrates and inhibitors were purchased from Bachem (www.bachem.com). The ingredients for buffers were purchased from Sigma (www.sigmaaldrich.com). Human DPPIV was a generous gift from Dr. Hans–Ulrich Demuth (of ProbioDrug), or purchased from Sigma. The standard end point assay was modified from Mentlein (1989). Stock solutions of Gly-Pro-4-para nitroaniline and Gly-Pro-β nalphthylamide, or other chromogenic peptidase substrates, were made in dimethylsulfoxide at a concentration of 100 mM or 200 mM. For nitroanilide substrates, 80 µl membrane fraction (or 500µl cytosol) was incubated at pH 7.5 or 8 (0.1M Tris–HCl buffer) at 37° C for 15min. The final substrate concentration was 0.5 mM. The reaction was terminated by the addition of 10 µl 1M ZnCl2, and the samples centrifuged for 2 min, at 14,000 × g in a micro centrifuge. For –naphthylamide substrates the method of Mentlein and Struckhof (1989) was used with the following modifications: 50 µl of 5 mM substrate solution (diluted in 0.1M Tris–Hcl, pH 7.5 or 8.0, from the stock solution) was mixed with 400 µl or 200 µl 0.1M Tris–HCl, and then incubated with 50 µl membrane fraction enzyme extract, or 250 µl “cytosol”, for 30 min at 37° C. The kinetics assay for DPPIV hydrolysis was monitored by a Hewlett–Packard (www.hp.com) diode array spectrophotometer using Hewlett–Packard Chemstation software in the kinetics mode. The reaction was measured for 3 min at room temperature. An initial rate was obtained with units of AU/min/µg protein.
pH optimum
The pH optimum was determined using the standard end–point assay (Martensen et al., 1998) with a buffer mixture of 75 mM each, Bicine, bis-Tris, and HEPES, the mixture was adjusted to the desired pH (pH range 5.5 to 10). The substrate was Gly–Pro–βNA.
Inhibitors
For phenylmethanesulfonyl fluoride and ZnCl2 inhibition of the crude extract the enzyme preparation was incubated with inhibitor for 2 hours before being added to substrate solution. The enzyme reaction was then measured by the kinetics assay as described above for 3 min. For inhibition using the partially purified membrane fraction, the enzyme–inhibitor samples were assayed with the standard end–point assay. PMSF and bacitracin were dissolved in DMSO, ZnCl2 in 1mM Tris buffer, bestatin and diprotin A in methanol. Inhibitors were added to enzyme extracts (at a final inhibitor concentration of 0.1mM or 1mM) and pre-incubated for 15 minutes at 37° C. All reactions were controlled against Tris buffer or added DMSO.
Partial purification of DPPIV using DEAE columns
Each crude enzyme preparation (7ml for membrane fraction – equivalent to approximately 3500 larvae – and 120 ml for the cytosol fraction – about 6000 larval equivalents) was applied to a column (3cm × 23.5cm) of DEAE–Sepharose fast flow (Amersham, www5.amershambiosciences.com) equilibrated with 0.1M NaCl/20 mM triethanolamine buffer, pH 8.5. The column was eluted with a linear gradient of 0.1–0.5 M NaCl/20 mM triethanolamine buffer (500 ml total elution volume), and fractions were collected (8–9 ml/fraction). For the purification of the membrane fraction, 2% Triton X–100 was also present in the column eluent. Every third tube was assayed for enzyme activity. Active fractions were combined and concentrated by ultrafiltration using Jumbosept centrifugal concentrators (30,000 kD Molecular weight cutoff, Pall Gelman, www.pall.com) for 1 hour at 4° C at 3,000 g. Final concentrate volume was 4–5 ml. The column was washed with 200 ml of 1.0 M NaCl/20 mM triethanolamine buffer, then 500 ml 0.1M NaCl/20 mM triethanolamine buffer between elutions. For further purification and molecular mass determination either crude extract or the concentrate from the DEAE column was applied onto a Superdex S–200 gel filtration column (2 cm × 69 cm). 1.0 ml of extract was loaded and the column was eluted with 200 ml of 0.1M NaCl/20 mM triethanolamine buffer (pH 8.5). 2.0 ml fractions were collected and every third tube was tested for dipeptidyl peptidase activity. The active fractions were pooled and used for characterization. The column was washed with 500 ml of the same buffer between elutions.
HPLC test for specificity of enzymes
High pressure liquid chromatograpy (HPLC) was performed using an Alltech (www.alltechweb.com) Alltima C18 column (150 mm, 4.6mm, 5mm pore size), with an elution gradient of 10–50% acetonitrile/0.1% trifluoroacetic acid over 15 minutes, then 50–10% over 5 min. Eluent was detected at 216 nm. The solvents used were 99.8% acetonitrile/0.1% trifluoroacetic acid and 0.1% trifluoroacetic acid in MiliQ water (the acetonitrile and trifluoroacetic acid were from J.T. Baker, www.jtbaker.com). 20mM triethanolamine buffer was filtered through a 0.2 micron tuffrin filter (Gelman Acrodisc) and used for all solutions and dilutions of enzymes and substrates. Human DPPIV was used as a control and for qualitative comparison for all enzyme and substrate reactions. The human DPPIV was stored and used as a 1–10,000 dilution (0.005µg/ml, from ProbioDrug [www.probiodrug.de] or Sigma) in a phosphate buffer. Extracts for processing of the trypsin modulating oostatic factor (Asn–Pro–Thr–Asn– Leu–His – MW696), substance P (Arg–Pro–Lys– Pro–Gln–Gln–Phe–Phe–Gly–Leu–Met – MW1349), and bradykinin (Arg–Pro–Pro–Gly– Phe–Ser–Pro–Phe–Arg – MW1060) were tested. The peptides were stored as stock solutions at 1 mM concentration (in 20 mM triethanolamine buffer). Final reaction concentrations were 100 µM substrate with 2% (by volume) partially purified membrane fraction in a reaction volume of 100 µl for the trypsin modulating oostatic factor and bradykinin, and 100 µM substance P with 10% (by volume) membrane fraction in a reaction volume of 100 µl. Substrates were incubated with enzyme in 20 mM triethanolamine buffer for 90 minutes at 37° C. The reaction was then stopped by heat denaturing the mixture at 90° C for 15 minutes. Human DPPIV trials used 10 µU or 50 µU human enzyme with 100 µM substrate in 100 µl reaction volume. Controls were heat denatured at 90° C for 15 min prior to incubation. 20 µl of product was loaded for HPLC. Prior to loading samples were filtered through a 0.2 micron cellulose acetate syringe filter that had been pre-rinsed in MiliQ water and 20mM triethanolamine buffer.
Organ preparations
All organs were dissected in insect Ringer's solution, transferred and stored frozen in Buffer 1 at −70° C until pooled and processed. Organs were homogenized either by blending in a pre–cooled Micro Waring blender for 90 seconds on high or by a pellet pestle that fit in a 1.5 ml Eppendorf tube depending on the size of the preparation. An aliquot was reserved for protein assay and the remaining homogenate was transferred to Oakridge tubes and centrifuged for 3 hrs at 4° C, 20,000 g or for 1 hour at 49,000 g at 4° C. The supernatant was collected (labeled cytosol fraction) and stored at −70 °C. The pellet was washed with Buffer 1 and centrifuged at 20,000 g at 4° C for 5 minutes. The supernatant was discarded and the pellet was extracted with Buffer 2 by homogenizing in an ice cold blender or tube and pestle for 2 minutes. The homogenate was centrifuged at 49,000 g at 4° C for 30 minutes. The supernatant was collected (labeled membrane fraction) and stored at −70° C, while the pellet was discarded. Crude homogenate, cytosol fraction, and membrane fraction were then assayed for protein content and DPPIV activity.
2–3 day old males were dissected for testes and accessory glands combined. Testes were quick frozen in liquid nitrogen. For assay they were defrosted and pooled to give a concentration of 1 testes/µl Buffer 1. Late third instar brains (lobes and ventral ganglion) were dissected as above and homogenized at a concentration of 1 brain/µl (wildtype) and 1.6 brains/µl (ome1).
The adult brains were prepared from combined males and females. Adults were placed on ice to immobilize them and heads were removed in 1X Ringer solution under the light microscope. The heads were immediately placed in a 1.5ml Eppendorf tube filled with liquid nitrogen with the tube immersed in liquid nitrogen. At the end of each dissection session the Eppendorf tube was taken out of the pool of liquid nitrogen, allowing the liquid nitrogen inside the tube to evaporate. Immediately after all the liquid nitrogen had dispersed, 200 µl ice cold Buffer 1 was introduced into the tube and the heads were stored at −70° C. Brains were homogenized at a concentration of 1 brain/µl Buffer 1. For homogenization, heads were suspended in liquid nitrogen in a cold mortar and pestle and ground into a powder that was then transferred into a cold 1.5 ml Eppendorf tube. Using a pestle for the Eppendorf tube, the samples were further homogenized, brought to a volume of 800µl/100 heads with Buffer 1 and spun in an ultracentrifuge at 20,000 × g for 4 hrs to recover the cytosol fraction. The remaining pellet was treated as above.
Ovaries were collected from aged virgin or inseminated females. Ovaries (both lobed structures including the oviduct and uterus recognized as one unit) were isolated as above and pooled, at a concentration of 1 ovary/5 µl Buffer 1, and 3 ovaries/µl of Buffer 2 for the membrane fraction. Each sample was processed in the same manner as for testes, except the lipid layer was removed and assayed separately.
Cloning of genomic wildtype DNA
Exons 3–10 were cloned from a P1 clone (DS00646 – obtained from the Berkeley Drosophila Genome Project) digested with HindIII. Using a DIG labeled probe of the degenerate probe 1 (Table 1) and EST LD21715, positive colonies were isolated and the DNA recovered and cloned using standard procedures into pBluescript® II KS + (Stratagene, www.probiodrug.de). Standard procedures using the various probes as indicated were used to identify and confirm the genomic DNA in all cloning work (Table 1). Clones of exon 1 and 2 were made after the Berkeley Drosophila Genome Project published the D. melanogaster genome sequence that made it possible to identify the gene sequence in the database. Thus we used the published sequence to generate primers for isolation of the DNA by PCR from BAC 16F22 (obtained from Berkeley Drosophila Genome Project). Primers were: Exon 1: Forward – ATGTCTCGGGAAATGCCACC, Reverse – CCATCCAAACTGACTTTAGG. Exon 2: Forward – GCCGAACTGGAAACTGAATG Reverse – GGGGAAACAATCTTTTTTTTGGGG. A standard PCR procedure was used with an annealing temperature of 53.2° C. The PCR products were verified with DIG labeled probes for exons 1 & 2 respectively (Table 1) and then ligated into Bluescript SK+ as above. The extent of the cloned inserts was determined by sequencing (Seqwright).
Table 1.
Hybridization probes and their corresponding melting temperature.

Probes
The oligonucleotide probe for DWV(I)YEEE (degenerate) was purchased from Genemed Synthesis (www.genemedsyn.com). Other oligonucleotide probes were obtained from Operon technologies, Inc (www.operon.com). Probe sequences were determined by reverse translation of consensus protein sequences or afterwards directly chosen from the Celera candidate genomic DNA sequence (see Table 1). The oligo probes were 3′ end labeled with the Digoxigenin Oligonucleotide Tailing Kit (Boehringer Mannheim, www.roche.com), following the manufacturer's instructions. The full length EST LD 21715 was obtained from the Berkeley Drosophila Genome Project.
Genomic DNA preparation
Genomic DNA was extracted from the wildtype strain and the ome1 mutant of D. melanogaster according to protocol #48 in Ashburner (1989), then digested by BamH I. Control mouse genomic DNA was kindly provided by Xiaodong Shen (UCSF, USA).
Sequencing of omega1 genomic DNA
ome1 DNA was prepared from whole flies and sent to Laragen Inc. for PCR sequencing. Primers were designed using the Gadfly sequence. Sequencing was done twice on both strands and the sequence compared with the wildtype sequence in Gadfly. Differences from wildtype that occurred only in both sequenced strands were considered valid.
Results and Discussion
The mutant phenotype
None of the co-dominant cuticle protein variants identified from the original mutant screen (Chihara et al., 1986) had any detrimental or visible phenotype, whereas the recessive ome1 mutant stock that also had a modified larval cuticle protein (Fig. 1), seemed somewhat small and developmentally “slow”. To confirm this observation, the size and development of the ome1 stock was measured. ome1 showed no difference in the wet weight of eggs, third instar larvae, or pupae, however the ome1 adults (females and males) were smaller by about 15–20% (Table 2). The homozygous ome1 flies showed a delay in developmental time of approximately one day over the life cycle (Fig. 2), and no decrease in developmental time during the pupal period. There also was no difference in the percent of flies that emerged when timed from the white prepupae (Fig. 3). It was concluded that the developmental delay was in the larval period. This amounted to about a 20% delay (1 day out of 5) across the larval stages.
Table 2.
Percent wet weight - ome1/wild type. 400 animals at each stage were weighed in groups of 100. The eggs, pupae and larvae were washed and blotted dry.
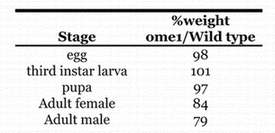
Figure 2.
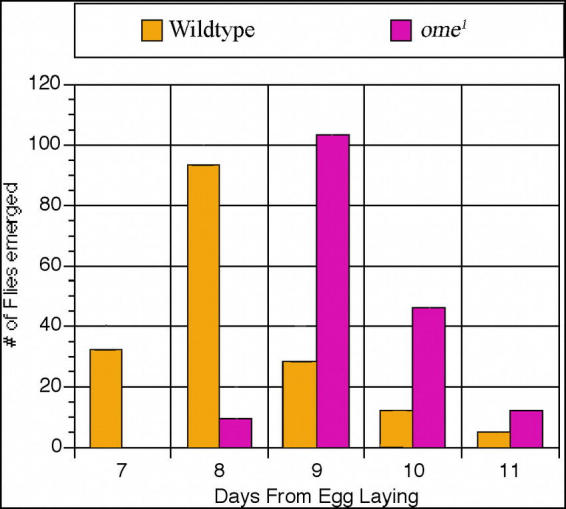
Number of flies emerged per day. ome1 is the homozygote mutant.
Figure 3.
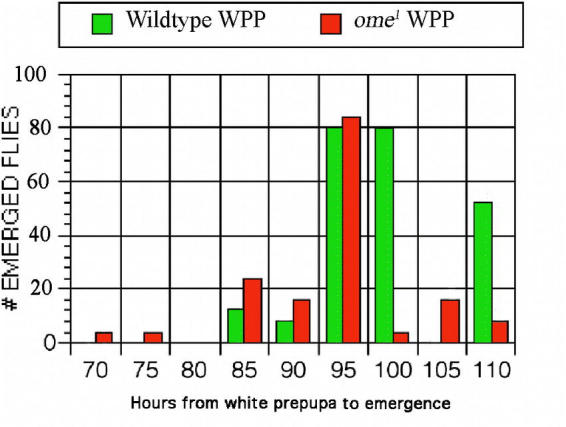
The time of pupation measured from the formation of the white prepupa.
The ome1 homozygote males show a decrease of about 40% both in the ability to fertilize females (Fig. 4) and in the percentage of progeny flies per female among successfully fertilized females (Fig. 5). Two tailed Student's t–tests were significant and gave P value levels between 10−3 and 10−6.
Figure 4.
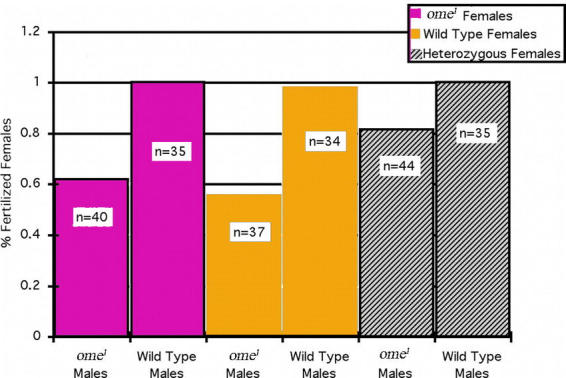
Percent fertilized females. For each cross, single males were introduced to a vial containing five females on day one. On day five the male was discarded and the females were separated into individual vials. Those females that produced offspring post day five were considered fertilized. In all cases the ome1 males were less successful at fertilizing females. ‘n’ is the number of females in the sample. ‘n < 50’ indicates that some females died/escaped prior to determining whether or not they had been successfully fertilized.
Figure 5.
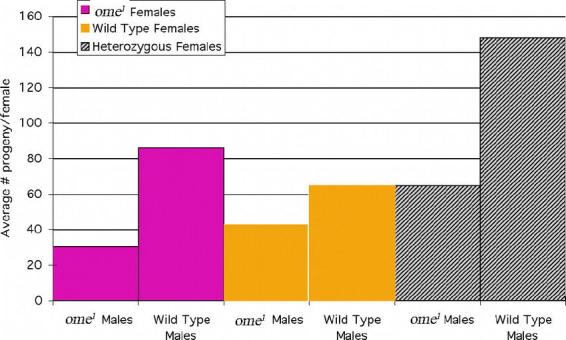
Average number of progeny per female. Of the females successfully fertilized, it is clear that the ome1 mutant males produced fewer living progeny/female. Note the apparent “hybrid vigor” illustrated in the heterozygote female crosses. In this case as well, however, the ome1 males produced fewer progeny per female than the wildtype males. With ome1 homozygote females a two tailed student's t test gave: P = 7.7(×10−5, with wildtype females: P = 2.3 × 10−6, heterozygote females: P = 9.9 × 10−3, where P is the probability that the samples are taken from the same population and therefore have equal means. Number of females is the same as in Fig. 4 (../ref/figure4.html).
Cause of the shift in the mobility of LCP 5 and 6 in the ome1 mutant
In order to elucidate the mechanism of modification of protein LCP 5 in the third instar larval cuticle protein the ome1 mutant was backcrossed the into a wildtype stock (mms) to check the wildtype proteins directly in an ome1 background (Chihara et al., 1986) (Fig. 1). Recombinants showed that both protein 5 and 6 were affected by the mutation. The N–terminal ends of the mature proteins in the wildtype and ome1 homozygote recombinants were sequenced (Fig. 6). This sequence makes it clear that the shift seen in the ome1 larval cuticle protein 5, and its alleles and related duplicates (Charles et al., 1998), can be explained by the lack of a DPPIV– like processing of the N– terminal dipeptide arginine-proline after the signal peptide is cleaved.
Figure 6.

Micro sequencing data of the N terminal residues of mature third instar Drosophila melanogaster cuticle proteins. Note the extra N-terminal residues included in ome1 and Lcp10rho mutant proteins. Wild-type stocks are: mms-multiple marked stock, and CS - Canton S (Chihara et al., 1986 (../#chiharaEA86) ). The sequenced N-terminal ends of Lcp5 and Lcp6 are a 100% match with virtual sequence of cp65Agb1/2 from the 65A5-6 locus (Charles et al., 1997 (../#charlesEA97) ). Lower case amino acids indicate the putative signal peptide cleavage site from the genomic DNA virtual translation.
The Lcp10rho protein is produced by a co–dominant ethyl methanesulfonate–induced mutation and is not shifted in the ome1 background, i.e. is not cleaved by the enzyme. It is clearly related to LCP5 by N–terminal sequencing (Fig. 6), but it carries a serine residue in the penultimate position that must make it refractive to ome1 DPPIV processing. Although a few DPPIV enzymes are known to cleave after a penultimate serine residue, most do not or do so with much lower rates (Mentlein, 1999; Lambeir et al., 2001). In any event, the omega wildtype enzyme does not process this protein before it is exported to the cuticle.
The omega gene maps to 70F–71A on chromosome 3L by deletion
Prior to the publication of the Drosophila genomic sequence a deletion map of the ome1 mutant was constructed. A series of crosses with deficiency chromosomes placed this DPPIV gene in 70F–71A (Fig. 7). This is consistent with the hybridization map position determined by the Berkeley Drosophila Genome Project cytology for the EST LD 21715 and for the gene CG32145 (originally designated CG9370), thus confirming the identity of the omega enzyme activity with CG32145, the omega locus.
Figure 7.
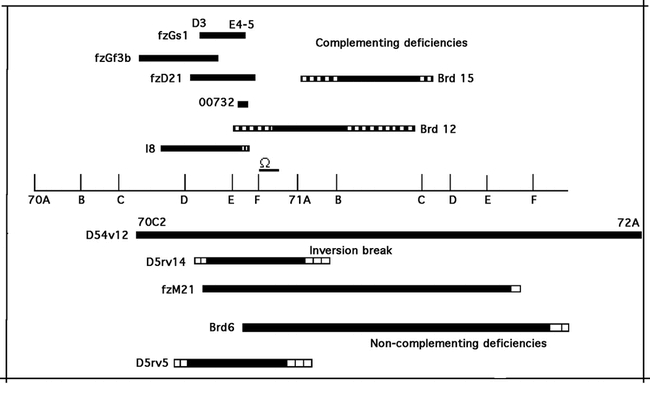
A deficiency map of the omega gene. Hatched bars represent uncertain ends. Deficiencies were obtained from the Bloomington Stock Center.
Gene structure and protein comparisons
A comparison of genomic sequence, our sequence and the full length EST LD21715 (Berkeley Drosophila Genome Project) was used to derive the intron-exon structure of the gene. Analysis of the sequence both from our studies and from the Berkeley Drosophila Genome Project database yields several interesting comparisons between this Drosophila DPPIV (Fig. 8) and human DPPIV gene structure.
Figure 8.
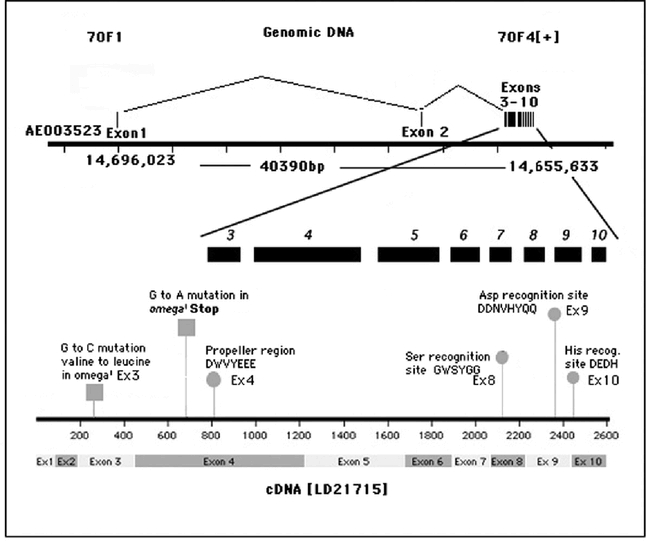
Exon structure of omega. Circles on ome+ cDNA indicate active site positions as labeled. The squares pinpoint the positions of differences in the ome1 cDNA as ascertained from genomic sequencing.
The Drosophila gene omega includes a very large intron in the untranslated 5′ end of the RNA (all of exon 1) whereas the first very large intron is between exons 2 and 3 of the human gene (Abbott et al., 1994). The original isolation of the omega DNA was accomplished by comparing alignments of over 12 eukaryotic DPPIV enzymes and recognizing the consensus sequence DWVYEEE. This highly conserved sequence, found in most, if not all, of the DPPIV family of proteases, is the conserved 7 amino acid sequence of the propeller region (DW(V/L)YEEE), (Abbott et al., 1999) that is common to S9b proteases (Fig. 9b). It is split between exons 8 and 9 in the human but is found in the single exon 4 in omega. Two glutamine residues (glu205 & glu206 human and glu227 & glu228 in omega) are extremely highly conserved (Abbott et al., 1999; and this report Fig. 9a) and are required for enzyme activity in human DPPIV (Abbott et al., 1999). Each of the amino acid residues of the Ser–Asp–His catalytic active site reside in a separate exon in both species. In omega the position of the conserved serine recognition sequence GWSYGG is in exon 8 in contrast to the human and mouse DNA where it is split between two exons (21 and 22) (Abbott et al., 1994; 1995). It has been suggested that the ancestral gene for the DPPIV family in humans is either DPP8 or DPP9 in that they have the smallest gene size and the fewest exons (Abbott et al., 2000; Chen et al., 2003). The serine recognition site in these two genes (DPP8 & DPP9) is contained within a single exon, as it is in omega and the other Drosophila putative DPPIV proteins (Flybase). A phylogenetic tree (Fig. 9c) is consistent with the omega protein being related to the ancestral DPPIVs, with gene CG3744 being more closely related to DPP8 & DPP9, and genes CG11319 and CG9059 more distantly related and appearing as outgroups.
Figure 9b.
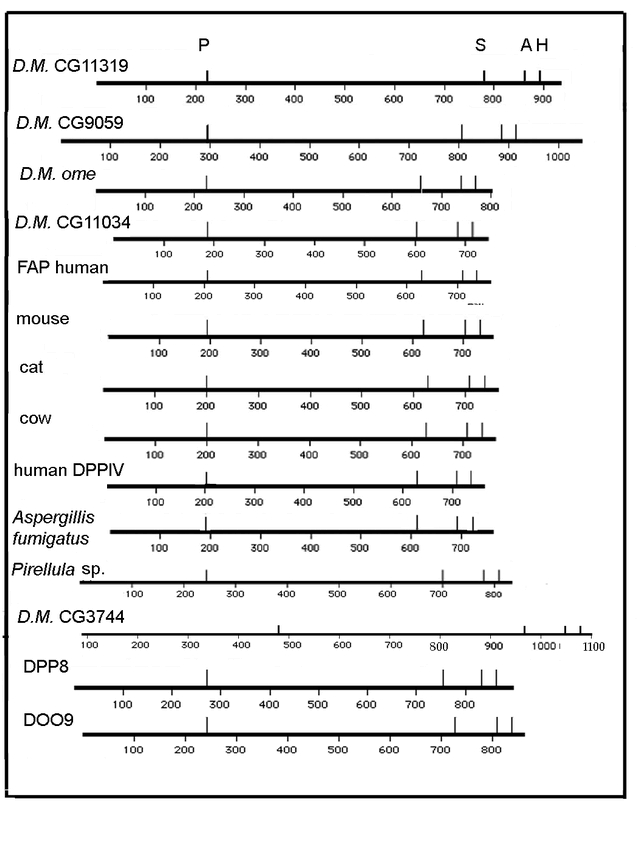
Broad structural comparison of several DPPIV proteins. DPPIV protein maps have been aligned to the conserved sequence found within the N- terminal region (DW(V/L)YEEE) that contains the double glutamic acid required for DPPIV activity (Abbott et al. 1999) This sequence is perfectly conserved except as indicated in Figure 9a. The spacing between the omega conserved sites of the N- terminal conserved sequence and catalytic triad most resembles the mammals, Aspergillus, and CG11034. The spacing of active sites for the other putative D. melanogaster DPPIV genes does not match any of the others as well. The spacing for CG3744 most closely resembles DPP9.
Figure 9a.
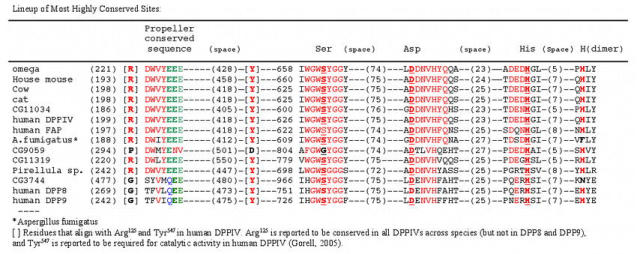
A pile up of the main highly conserved sequences of the DPPIVs and related proteins (MacVector Clustal analysis). The numbers in parentheses indicate distance between the end of one and the beginning of the next sequence. The highly conserved residues are in red. The catalytic triad is shown in underlined red bold, the conserved glutamic acids are in green except for the QEE of the DPP8 related proteins (Q in blue). Note the substitution of Gly for Ser in CG9059 (black G). The His residue reportedly necessary for dimer fomation is indicated under (dimer) in red bold (Chen et al. 2004) and the residues that align with the required Tyr (Tyr547 in human DPPIV(Bjelke et al. 2004; Gorrell, 2005)) and the highly conserved Arg125 (Gorrell, 2005) are shown in square brackets.
Figure 9c:
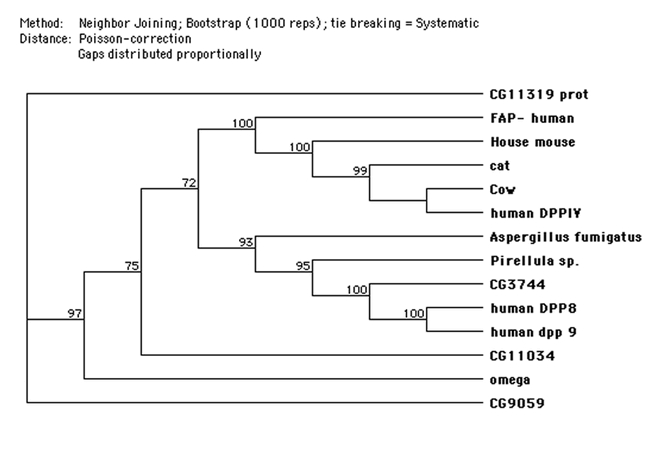
A phylogenetic tree generated by the MacVector (for MAC) program. Sequences for non- Drosophila proteins from Uniprot (www.expasy.org/prot).
Thus, it would seem that the human gene represents a splitting of most of the ancestral exons into the 26 we see now, with exons varying in length from 15 to 65 residues. We have compared the 300 5′ flanking bases of omega to those of the human DPPIV gene and find 41% identity in sequence. Like the human upstream region there is no TATA box, and there is only one complete AP1 site. In D. melanogaster the AP1 is on the minus strand.
The virtual protein sequences of the four additional putative DPPIV genes in D. melanogaster were compared to each other and to several other DPPIV–like proteins, including human DPPIV and the related proteins FAP, DPP8 and DPP9. The pileup of conserved sequences (Fig. 9a) of several known DPPIVs and DPPIV–like proteins, including the D. melanogaster genes, shows the active site consensus sequences and the relation between these DPPIV–like proteins and the DPPIVs. A simplified alignment of the proteins to display the spacing between the main conserved sites, the N–terminal S9b conserved sequence, and the Ser–Asp–His catalytic triad, shows considerable agreement between the mammalian and D. melanogaster DPPIVs. (Fig. 9b) The distance between the 7 base conserved sequence (DWVYEEE) and the beginning of the catalytic triad is very similar for the mammalian proteins and for D. melanogaster omega and CG11034 (Fig. 9a & Fig. 9b). The internal spacing for the Ser–Asp–His triad is fairly well conserved across the list, but the distance from the 7 base consensus sequence, DWVYEEE, to the triad differs notably for the D. melanogaster genes CG9059, CG11319 and CG3744 and for human DPP8 & 9. In human DPPIV Tyr547 was shown to be required for activity of the enzyme (Gorrell, 2005), and this residue is perfectly conserved across these proteins except for CG9059, which is also missing the highly conserved Arg125 residue. Thoma et al. (2003) report that the distance between the double Glu residues and the catalytic serine is important in limiting cleavage to dipeptides and the size and shape of the region surrounding the P1 proline restricts the specificity of the enzyme to proline (and a few other amino acids in this position with lower activity). Note that of the four other D. melanogaster proteins, only the configuration for the CG11034 is overall similar in this respect to omega and the other eukaryotic proteins. The other putative D. melanogaster DPPIV proteins CG9059, CG3744 and CG11319 (Flybase, Abbott et al., 1999) are quite different, either in the distance of the N terminal end to the DWVYEEE consensus sequence or in the distance from it to the serine recognition site. The alignment for these proteins shows a similar consistency in the sequences of the four active sites as well as the spacing (Fig. 9b). Note that only the CG3744 gene of the D. melanogaster putative DPPIV proteins has the N– terminal (propeller) motif QEE seen in the more closely related human proteins DPP8 (Uniprot Accession # Q6 V1x1) and Dpp9 (Uniprot Accession # Q6UAL0) (Fig. 9a). Note that for the proteins shown in Fig. 9a, all display the conserved histidine (human His750 and omega His779) residue reported by Chen et al, (2004) to be required for human DPPIV dimer formation with the exception of the Aspergillus enzyme and CG3744. Presumably the Aspergillus enzyme functions as a monomer. However, the DPP8 and DPP9 proteins are also reported to function as monomers (human DPPIV does not) indicating this conserved histidine is not an adequate indicator of dimerization for these proteins (Abbott et al., 2000).
Pair–wise comparisons among the mammalian proteins show amino acid similarity (including identities and similarities) ranging from 80% to 93%, while the D. melanogaster omega – mammalian DPPIV similarities ranged from 30 to 46%. Pair-wise comparisons of omega protein with CG 11319 and omega with CG11034 show them to be as similar to each other as they are to the mammalian proteins (43% and 46% respectively), and the similarity to CG9059 and CG3744 is much less (19% & 17% respectively). CG3744 compared to DPPIV 8 and 9 give similarities of 31 and 32% respectively. It is certainly possible that these differences relate to different substrate specificities. CG9059 lacks the serine residue within the consensus sequence of the serine recognition site, the highly conserved tyrosine within the hydrolase domain, and one of the conserved glutamic acids in the propeller sequence, and so presumably is not active as a DPPIV. At least one of the 3 additional D. melanogaster gene products (CG3744, CG11319 and CG11034) must have DPPIV activity as we see it as residual activity in the ome1 mutant. This will be further discussed below.
Cloning of wildtype omega gene
The genomic exons of ome+ were cloned into three separate vectors for further analysis. The exon 1 clone included about 1000 bases of upstream sequence and the exon 2 clone about 180 bases of intron 1 and about 85 bases of intron 2. Exons 3–10 were incorporated into one clone of about 5kb. All were cloned in Bluescript SK+. These clones will be provided to any researcher who might want them.
The ome1 mutation is a result of a nonsense mutation in exon 4
All of the exons and 140 bases upstream of the ome1 mutant allele were sequenced on both strands. The entire DNA of the exons and upstream region was wildtype with two clear exceptions. Two base substitutions were found, one in exon 3 that changed a valine to a leucine (GTT to CTT) and one in exon 4 that converted a tryptophan codon to a stop codon (TGG to TAG). (Fig. 8). We presume this stop codon is the cause of loss of enzyme activity. The ome1 exon sequences have been placed in GenBank: Genbank accession numbers: AY245164, AY245165, AY245166, AY245167, AY245168, AY245169, AY245170, and AY245171
The omega enzyme is a fly DPPIV (EC 3.4.14.5)
DPPIV is a serine protease with specificity for cleaving on the carboxy side of a penultimate proline residue at the N terminal end of a protein (or, with less activity, an alanine and, even slower, when a ser, gly or val). The enzyme is most frequently membrane bound, although there are soluble forms reported (Perner et al., 1999; Durinx et al., 2000). To test if the missing enzyme in ome1 homozygotes was DPPIV we made crude homogenates of both wildtype and ome1 larval “epithelia” (see Methods for carcass preparation), since we assumed that this tissue would contain the enzyme in order to process the secreted cuticle protein. The wildtype extract had prolyl specific dipeptidase activity on artificial substrates (see below) to a much higher level than did the ome1 mutant extract.
DPPIV activity of both the membrane and soluble fractions from D. melanogaster wildtype and ome1 third instar larval “epithelia” homogenates were assayed with the chromogenic substrates Gly–Pro–4–nitroanilide (Gly–Pro–pNA) or Gly–Pro–β–naphthylamide (Gly–Pro–βNA). Gly–Phe–pNA was used as a control to estimate the non–specific peptidase activity from the crude extract homogenate. Using a kinetics assay, DPPIV activity (per µg protein) in the wildtype crude membrane fraction at pH 7.5 upon Gly–Pro–pNA is 3 times that of the ome1 membrane fraction; for membrane fractions from both wildtype and ome1 homogenate there was no enzyme activity upon Gly–Phe–pNA. This was the first indication that the ome1 mutant contained a residual DPPIV enzymatic activity (Fig. 10a). Cytosol fractions for both the wildtype and ome1 from the homogenate showed some activity upon both Gly–Pro–pNA and Gly–Phe–pNA. The level of the enzyme activity differed between the two substrates, but there was no major difference between wildtype and ome1 for both substrates (Fig. 10a).
Figure 10a.
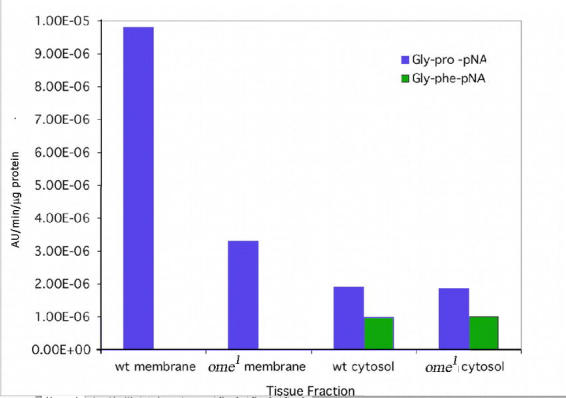
wt = wildtype, Gly-pro-pNA (kinetics assay, average of four experiments).
On the natural substrate (Lcp5) we saw preferential degradation of the ome1 Lcp5 protein, but concurrent degradation of contaminating proteins by proteases in the .png preparation made quantification impossible. However, we did find that the wildtype extract degraded the unprocessed ome1 LCP5 more rapidly than the ome1 extract and human DPPIV did not process it at all (data not shown). This is suggestive of differing specificities for the D. melanogaster and human homologs of DPPIV as well as for the major and residual activities of the D. melanogaster enzymes.
The molecular weight of LCP5 is as large as the largest peptides reported for most human DPPIV substrates; the molecular weight after the signal peptide is removed is about 9.6kD for 86 amino acids. Most substrates we found reported for human DPPIV ranged between 66 and 77 amino acids RANTES (P13501)–68 amino acids, 76 & 77 amino acids for the chemokines CCL2 (P13500) and IP–10 (P02778) respectively, after the signal peptides are cleaved (UniprotKB/Swissprot entries). The only substrate we found larger than this was CXCL9 (MIG) that has 103 amino acids and is cleaved considerably slower than most of the other substrates (De Meester et al., 2003).
Using the more sensitive end point assay, Ala–Phe–Pro–pNA was tested with the wildtype and ome1 membrane fractions and there was a large difference between them. The wildtype membrane fraction cleaved Ala–Phe–Pro–pNA, while the ome1 membrane fraction had no activity; the wildtype cytosol fraction showed ∼30% higher activity on Ala–Phe–Pro–pNA than ome1 (see Fig. 10b). The only identified D. melanogaster tripeptidyl peptidase in the genomic database is TPPII and this enzyme does not cleave substrates with the Ala–Phe–Pro N–terminal sequence (Renn, 1998, Brenda). To test if this result could be explained by the cleavage of Alanine from the tripeptide followed by DPPIV activity, Ala–pNA was tested. The Ala–pNA was cleaved by membrane and cytosol fractions of both the wildtype and ome1 without significant difference between them (See Fig. 10c). We interpret these data to mean that both membrane fractions cleaved the Ala N terminal amino acid from the Ala–Phe–Pro pNA, equally well, and that the wildtype can, but the ome1 mutant can not, continue the degradation of the remaining Phe–Pro–pNA substrate.
Figure 10b.
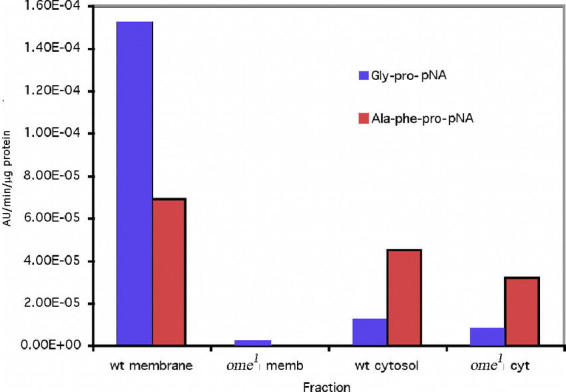
wt = wildtype, Effect of crude homogenate of wildtype and ome1 on the artificial substrate Ala-phe-pro-pNA compared to Gly-pro-pNA.
Figure 10c.
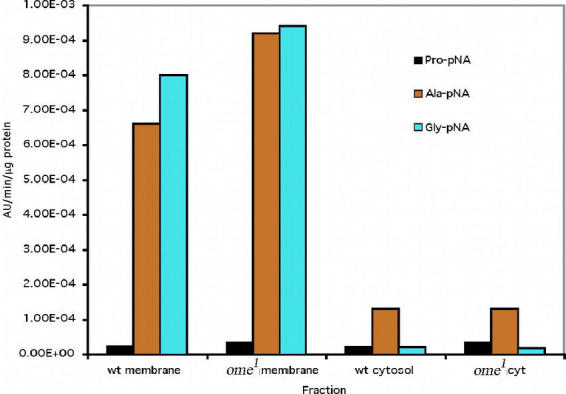
wt = wildtype, Ala-pNA, Pro-pNA and Gly-pNA, (average of three experiments) - using the end point assay.
Furthermore, Gly–pNA and Pro–pNA were tested with the wildtype and ome1 crude extract to clarify that it was DPPIV in wildtype that cleaved the Gly–Pro–pNA rather than that Gly was first cleaved by one enzyme other than DPPIV, then Pro was cleaved. Both membrane and cytosol fractions from larval crude extract cleaved Gly–pNA, and there was no difference between the wildtype and ome1. However, cytosol fractions cleaved Gly–pNA to a much lower degree, compared to the membrane fractions (Fig. 10c). Both membrane and cytosol fractions from wildtype and ome1 have little activity upon Pro–pNA (Fig. 10c). The cytosol fractions of both wildtype and ome1 showed some non–specific activity for the Gly–Phe substrate as well as equal and very low levels of activity on the Gly–Pro substrate. This suggests that there is another DPPIV like enzyme in the cytosol unrelated to the omega gene (see later discussion). The crude extract of the membrane fraction from epithelial tissue of ome1 larvae showed a decrease of at least 66 to 80% in activity for either substrate Gly–Pro–pNA or Gly–Pro–βNA (data not shown) over the same membrane fraction prepared from ome+ homozygotes. Given the lack of difference in the activity of the cytosol fraction between wildtype and mutant, we conclude that the missing omega enzyme is primarily a membrane bound protein in this tissue, which is consistent with its function of processing the larval cuticle proteins, LCP5 & 6(lcp65A).
Partial purification of membrane fraction activity
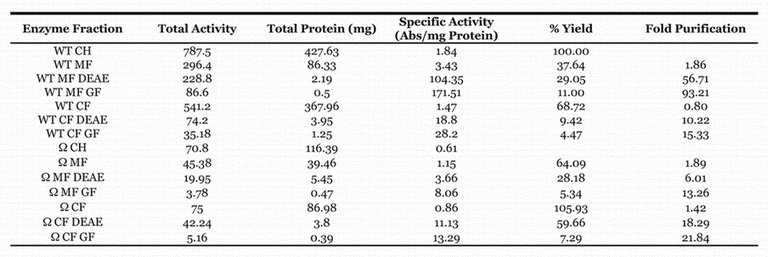
Larval carcass enzyme preparations from the cytosol fractions were purified about 20 fold by the DEAE column, but the overall purification decreased by approximately half after the gel filtration elution. The wild–type membrane fraction gave better partial purification, but the ome1 membrane activity was only purified about 13–fold due to the low level of starting activity and decrease in enzyme activity. The membrane fraction activity obtained from the two columns (MFGF >90 fold purification) was used for further characterization (Table 3).
Table 3.
Summary of purification of larval carcass DPP IV activity. Table Legend: Abbreviations: MF:Membrane Fraction, CF: Cytosol Fraction, CH: Crude Homogenate, GF: Sample run through the DEAE column then the GelFiltration Column, DEAE: Extracts run though DEAE column only, WT: Wild-type. Percent Yield was calculated by the total activity at a given step/total activity of the crude extract. Fold purification was calculated as specific activity of a purified sample/specific activity of the crude extract.
Molecular weight determination
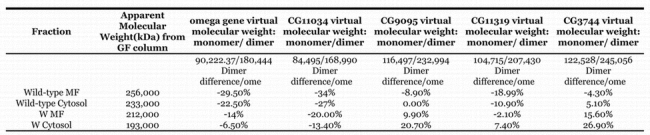
The partially purified enzyme activity of each of the two membrane fractions (wildtype and ome1) is shown in Figure 11a and the cytosol fractions are shown in Fig. 11b. The four fractions eluted at different molecular weights as shown in Fig. 11a & Fig. 11b and Table 4. When compared to the predicted MW of the omega wildtype gene product (90,222 D) it appears that the enzyme functions as a dimer, and most probably has been post–translationally modified as well. These data are consistent with those seen in mammalian DPPIVs where the enzyme is highly glycosylated and commonly functions as a dimer. Virtual translations for the gene transcripts of CG9059 and CG3744 give dimer molecular weights too large for the mutant ome1 residual activity. CG11319 is too large for the cytosolic residual activity. Since the known DPPIVs that are similar to omega DPPIV seem to be glycosylated dimers (Polgar, 2002), these data are consistent with omega DPPIV and CG11034 product being glycosylated proteins. The data also support the idea that the residual activity in the ome1 mutant is coded for by CG11034.
Figure 11a.
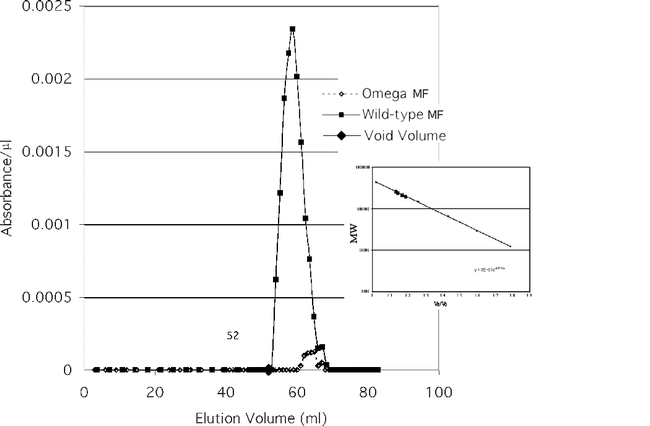
Enzyme activity of partially purified Wildtype and ome1 membrane fractions eluted from the Gel Filtration column. The insert shows the molecular mass of standards and fractions.
Figure 11b.
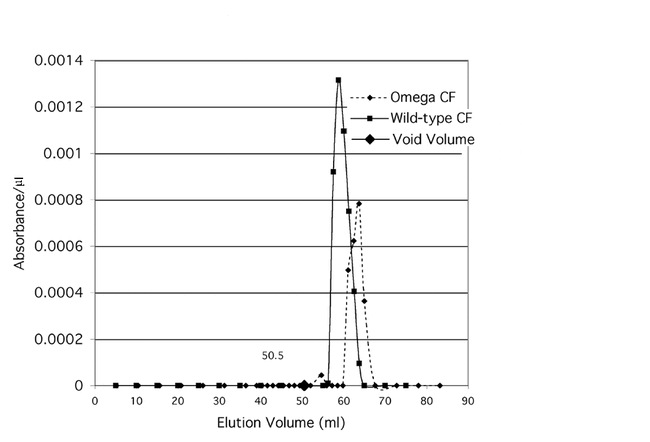
Enzyme activity of partially purified wildtype and ome1 cytosol fractions eluted from the Gel Filtration column.
Table 4.
Elution ratios and estimated molecular weights of the DPPIV preparations. Each value is an average of 3 trials +/– (SE%). For the purposes of Molecular Weight determination, the elution volume used to determine the Elution to Void Volume ratio (VE/VO) is the volume of the highest absolute enzyme activity.

Since most DPPIVs in vivo are glycosylated (Polgar, 2002), glycosylation is a likely explanation for the differences between calculated and observed molecular weights. In addition, when the D. melanogaster enzyme was exposed to endoglycosidases 2 and 3, the amount of DPPIV activity present in both wildtype and omega membrane fractions decreased by 85%, while when exposed to another endoglycosidase (endoglycosidase 1) in a similar assay, the activity was unaffected (data not shown). A possible explanation for this is that the enzymes are glycosylated and the removal of certain sugar groups (by endoglycosidase 2 and 3) disrupted the DPPIV activity, although a recent study indicates that glycosylation is not critical for activity of the human enzyme (Aertgeerts et al., 2004). The amino acid sequence of the human protein has 8 possible glycosylation sites when analyzed by NetNGlyc 1.0 for human proteins. (Analysis from http://www.cbs.dtu.dk/services/NetNGlyc)
Thus the apparent molecular weight of human DPPIV is 110–120 kD for the monomer, and 264 kD for the functional dimer (De Meester et al., 1992; Polgar, 2002) while the calculated molecular weight from this sequence is 88,278 D (∼177 kD for a homodimer). (http://www.expasy.org/cgi-bin/niceprot.pl?P27487). And (http://mpr.nci.nih.gov/prow/guide/61856995_g.htm)
When plugged into the NetNGlyc 1.0 human analysis program the omega DPPIV amino acid sequence possessed 4 possible N–linked glycosylation sites. If D. melanogaster proteins are legitimately analyzed in this way, this could explain the apparent molecular weight of 256 kD for a gene product predicted to have a molecular weight of 180.4 kD as a dimer (Table 5). Mammalian DPPIVs can have as much as 22.7% of their weight as sugar moieties. (Brenda). In comparison to human DPPIV, the increase of molecular weight of the omega DPPIV from the predicted molecular weight (from the amino acid sequence) to the estimated in vivo molecular weight is comparable. (Table 5) The data are consistent with the CG11034 gene coding for the residual DPPIV and also functioning as a dimer, with a dimeric weight of 168,990D (Table 5).
Table 5.
Comparison of molecular weights to predicted molecular weights for the five putative D. melanogaster DPPIVs. %difference of virtual wt - estimated wt/estimated weight. Only omega and CG11034 fit the expected MW value allowing for glycosylation.
The pH optimum
The pH optimum for the crude enzyme showed a broad peak around pH 8.0 (data not shown). The partially purified wildtype fraction shows two distinct pH optima in the membrane fraction (Fig. 12a) and a broad peak in the cytosol (Fig. 12b) a pattern consistent with more than one enzyme. Both graphs indicate that we have at least two different enzymes capable of cleaving the artificial substrate used for assay. The ome1 mutant is clearly missing the enzyme with the higher pH optimum in both compartments. We thus assume that the pH optimum for the wildtype omega gene product (omega DPPIV) is approximately 8.5 in both the membrane bound and soluble form.
Figure 12a.
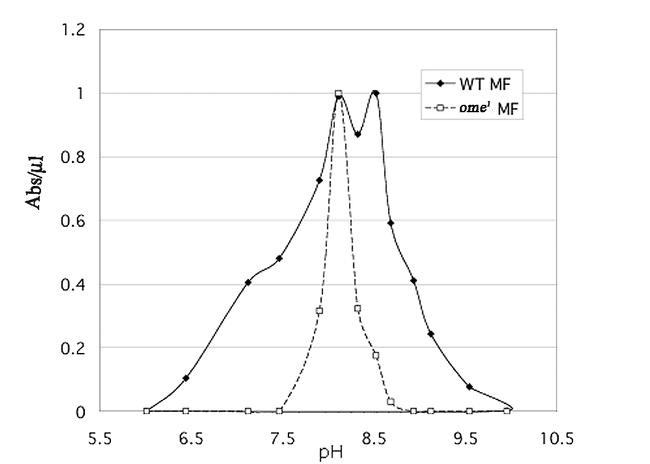
(a) pH curve for the partially purified membrane fraction of wildtype (WT MF) and ome1 (ome1 MF) DPPIV.
Figure 12b.
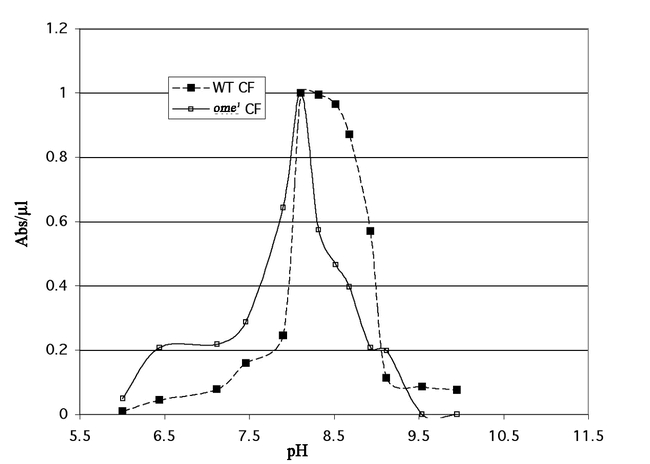
(b) pH curve for the partially purified cytosol fraction of wildtype (WT CF) and ome1 (ome1 CF) DPPIV.
Inhibitors
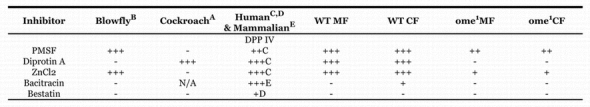
Two DPPIV inhibitors, PMSF, a general inhibitor of serine proteases and ZnCl2, a known inhibitor of mammalian DPPIV, were tested on D. melanogaster third instar larval crude carcass extract. Diprotin A, bacitracin and bestatin were also tested on the partially purified extracts. 2mM ZnCl2 completely eliminated the enzyme activity. 2.5 mM PMSF decreased the DPPIV activity by 78% for membrane fraction. As with the pH there are differences between the wildtype and mutant sensitivities, again indicating that different enzymes are functioning in the two strains (Table 6). The wildtype enzyme extract behaved more like human enzyme in its response to the inhibitors then either the ome1 mutant residual enzyme activity or the blowfly and cockroach enzymes, however it must be kept in mind that the wildtype extract is a mixture of at least two DPPIVs.
Table 6.
Inhibition of enzyme activity. +: sensitivity between 50–75%,++:sensitivity between 25–50%,+++: sensitivity 25% or less A: From Nassel et. al 2000 B: From Martensen et al 1998 C: From De Meester et al, 1992 and Mentlein et al, 1986 D: Buckley et al, 2004 E: From O'Connor and O'Cuinn, 1986
Characterization of activity on known substrates for DPPIV
In order to determine if the nature of the activity of the partially purified enzyme preparation was comparable to human DPPIV we tested the partially purified MF on several substrates and compared the degradation products with the activity of Human DPPIV. Bradykinin is a nonapeptide (Arg–Pro–Pro–Gly–Phe–Ser–Pro– Phe–Arg) with a proline following the penultimate proline. As such it should not be, and was not, degraded either by human or Membrane Fraction DPPIV (data not shown).
The trypsin modulating oostatic factor (Asn–Pro–Thr–Asn–Leu–His) is a hexapeptide found in the blowfly, Calliphora vicina, and is believed to be a natural substrate for DPPIV in that fly (Martensen et al., 1998). Both human DPPIV and the partially purified membrane fraction were able to cleave the Asn–Pro dipeptide from the N– terminal end. (Fig. 13A–E). The trypsin modulating oostatic factor control has a peak at 4.8 – 5 minutes. Once incubated in the presence of either enzyme the peak shifts to 3.75 minutes, consistent with a tetrapeptide. The additional dipeptide is expected to run in the solvent front (dipeptide controls not shown).
Figure 13a–e.
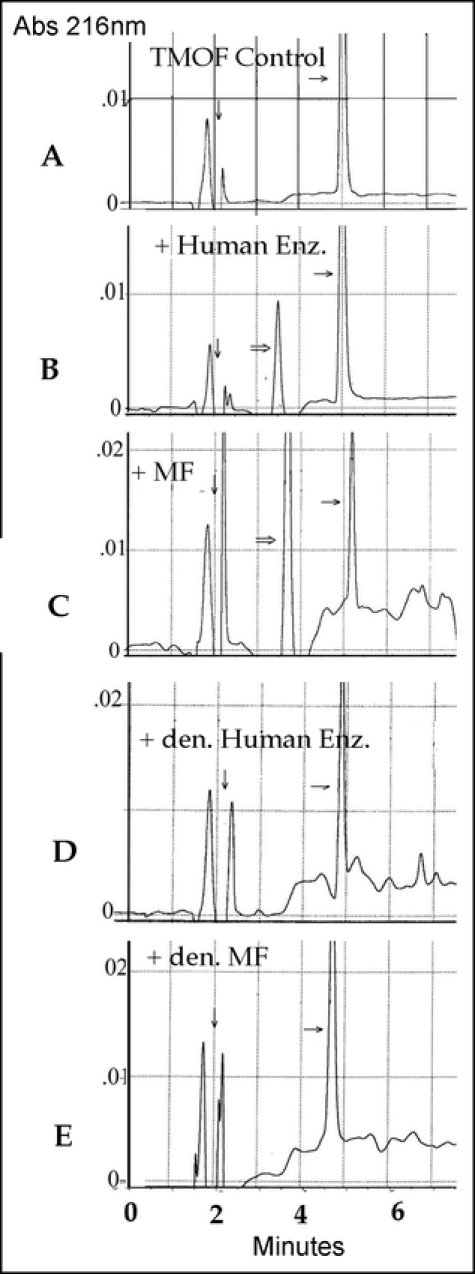
The HPLC tracings of TMOF degraded by human DPPIV and partially purified membrane fraction from carcass adhering cells. den. = denatured by boiling for 10 min. The dipeptide products were not resolved (control dipeptides ran with the solvent front). ? Control substrate; ? Cleaved product; ? between solvent front peaks. Both membrane fraction and the human enzyme gave unexplained noise around the smaller peptide peaks.
Substance P (Arg–Pro–Lys–Pro–Gln–Gln–Phe– Phe–Gly–Leu–Met) was then tested to verify that the membrane fraction enzyme was removing the dipeptide as a unit. We compared the cleavage of human and membrane fraction enzyme and the same degradation products were observed from both enzymes (Fig. 13F–H). The substance P control exhibits a peak at 12 minutes. Digests show three peaks: the substance P peak at 12 minutes, and two new peaks at 5.8 and 7.2 minutes. The appearance of two product peaks is consistent with DPPIV activity. Both Substance P and the first cleavage product, a 9 amino acid molecule (Lys–Pro–Gln–Gln–Phe–Phe–Gly–Leu–Met), have the N– terminal structure recognized by a DPPIV type enzyme. If the activity present in the larval extract were, for example, a DPPII, then one would expect 4 unique substrate peaks for the enzyme extract (Fig. 13F–H). Thus the D. melanogaster enzyme activity is consistent with a classic DPPIV dimeric enzyme with a slightly alkaline pH optimum, and the ability to remove an N– terminal X–proline dipeptide.
Figure 13f–h.
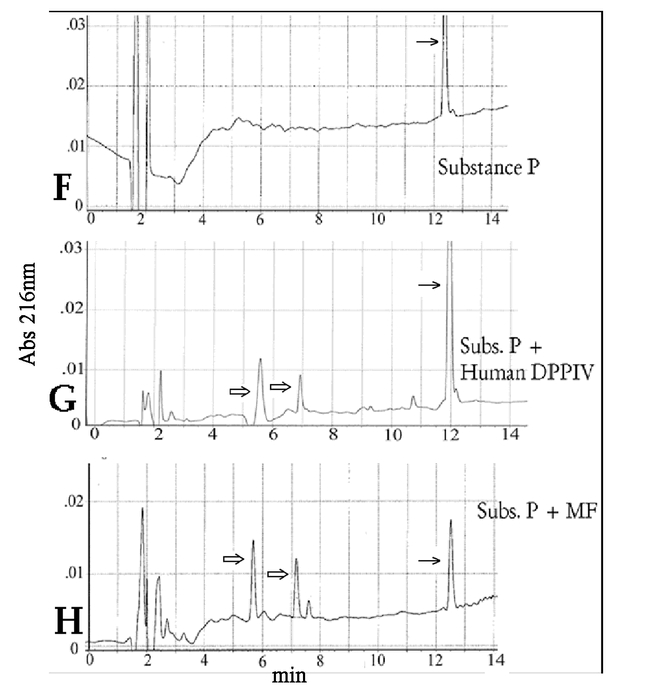
The HPLC tracings of substance P degraded by human DPPIV and partially purified membrane fraction from carcass adhering cells. den. = denatured by boiling for 10 min. The dipeptide products were not resolved (control dipeptides ran with the solvent front). ? Control substrate; ? Cleaved product; ? between solvent front peaks. Both membrane fraction and the human enzyme gave unexplained noise around the smaller peptide peaks.
The substrate and inhibitor specificities, membrane association, and pH optimum of the omega gene protein are consistent with the characteristics of DPPIV in other organisms. The conserved sequences known to be characteristic of the DPPIV enzymes are found in both omega and CG11034 protein sequences. The molecular weights and evidence of dimerization in the omega enzyme activity lead us to conclude that the omega gene codes for a major D. melanogaster DPPIV and that in the carcass of the larva the genes omega and CG11034 are coding for the main enzymes. The data indicate that there are other DPPIV–like enzymes in the various D. melanogaster tissues. This is consistent with the information in GadFly indicating that there are four other possible DPPIV–like enzymes. Given that cuticle protein 5 is not modified at all by the ome1 mutant or its extracts we suggest that the omega gene is the primary enzyme with this activity and that the substrate specificities of the other enzyme(s) are different.
Differential compartmentalization of the DPPIV activity
Using whole animals, two microarray expression studies have shown that the mRNA for omega is expressed during embryogenesis at about 12 hours of embryonic development (Flybase: Gene Expression Data) and in larvae and pupae from 18 hours prior to pupariation through 12 hours post pupariation. The peak of this activity was at pupariation (White et al., 1999). We have looked at the activity in membrane and cytosol from several isolated organs of the late third instar larva and the adult.
The distribution of the enzyme activity differs between organs in both stages. In the larva the brain (and ventral ganglion) and the carcass were examined. The compartmentalization of the brain activity is opposite that of the epithelium (carcass). The main activity in the carcass is omega and is primarily in the membrane fraction, whereas the main activity of the brain is in the cytosol and is split between the omega gene product and the residual activity (Fig. 10A, Fig. 14 A–B and Table 7). Adult brain, testes and ovary were tested in the same way. Adult brains had relatively low overall activity, with the major activity level in the cytosol and the omega enzyme more important in the membrane fraction. In the adult cockroach similar compartmentalization was found between membrane and soluble forms with major activity in the membrane fraction. The cockroach also had similarly low levels of activity in the adult brain (Nässel et al., 2000). The adult testes had quite high activity (per µg protein) and most of it was in the cytosol. The membrane fraction activity was split almost equally between the omega enzyme and the residual activity. The adult ovaries, however, partition in exactly the opposite way, with high activity in the membrane fraction and relatively little in the cytosol. In the ovaries the main enzyme seems to be the omega product (Table 7).
Figure 14a–d.
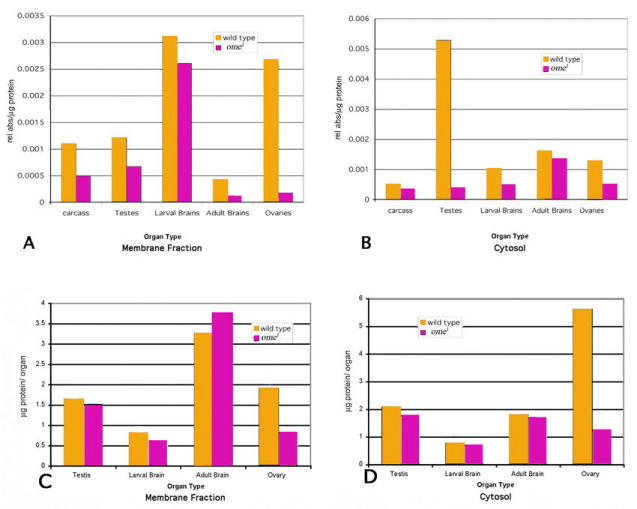
Each organ prep (except for carcass ñ see above) was 100 organs. Each bar represents an average of at least four preparations. a: Membrane fraction comparison of total DPPIV activity/µg protein. b: Cytosol fraction comparison of DPPIV activity/µg protein. c: µg protein/organ for membrane fraction. d: µg protein/organ for cytosol fraction. Ovaries data are for aged virgins. Inseminated and virgin ovaries were assayed from 5–10 day old females. The data presented here are for virgin ovaries, as these data were less variable than for ovaries from inseminated females. In inseminated females the average activity for membrane fraction of ome1 was ∼53% of wildtype and for the cytosol ∼23% of wildtype.
Table 7.
Comparison of compartmentalization of DPPIV activities in various organs. * averaged over 10 experiments. ** average of at least four experiments.

Three organs had high activity levels (larval brains, adult ovaries and testes) for combined DPPIV activity. In ovaries and testes the major portion of the activity was omega DPPIV, while the larval brain activity seems to be fairly equally split between omega and the residual enzyme activity. All the ome1 organs were only marginally smaller than their wildtype counterparts with the possible exception of the adult brains (heads) and ovaries (Fig. 14 C–D). Although the larvae did not show a difference in weight, the adults were slightly smaller (Table 2).
The omega gene DPPIV is more important in the cytosol of the adult brain, but the combined enzyme activity here is relatively low. In addition, the larval brain membrane fraction has a high enzyme activity, but it does not seem to be the omega gene product. It will be interesting to discover which of the alternate DPPIV genes accounts for the “residual” DPPIV activity in each of these organs, as well as what connection loss of the enzyme has to the observed phenotypes.
The D. melanogaster omega DPPIV seems to be a ubiquitous enzyme, which is characteristic of mammalian DPPIV, but does not seem to be a “simple housekeeping enzyme”. There is clearly differential compartmentalization of the omega DPPIV activity into membrane bound and soluble, as well as differential expression across the life cycle and organs of the fly. The difference in activity per unit protein between the wildtype and the mutant in the testes cytosol and in the membrane fraction of the ovary indicates that the enzyme has an important role in these two tissue compartments. Why the loss of omega DPPIV seems to be associated with a decrease of male fertility, but not with female fertility remains to be seen.
The search for other natural substrates for the D. melanogaster DPPIVs must include the immunity proteins and proteins that could effect the fertility of the males. Among the immunity proteins the diptericin precursor has a signal sequence followed by two amino acid pairs Y P M P thought to be removed by a DPPIV activity. Defensin, drosocin, and metchnikowin also have a signal sequence followed by X – P that is also believed to be removed by Dipeptidase activity during activation (Data from PIR. A47103, M55432, Q24396). It remains to be seen which DPPIV activity in the fly processes these important proteins.
Acknowledgments
This work was supported by Grants to Carol J. Chihara — Grant # 9987556 from The National Science Foundation and from the University of San Francisco Faculty Development Fund.
References
- Abbott CA, Baker E, Sutherland GR, McCaughan GW. Genomic organization, exact localization, and tissue expression of the human CD26 (dipeptidyl peptidase IV) gene. Immunogenetics. 1994;42:331–338. doi: 10.1007/BF01246674. [DOI] [PubMed] [Google Scholar]
- Abbott CA, Baker E, Sutherland GR, and McCaughan GW. 1995 Genomic organization, exact localization, and tissue expression of the human CD26 (dipeptidyl peptidase IV) gene. Immunogenetics. 42:76. (correction). [DOI] [PubMed] [Google Scholar]
- Abbott CA, McCaughan GW, Gorrell MD. Two highly conserved glutamic acid residues in the predicted propeller domain of dipeptidyl peptidase IV are required for its enzyme activity. FEBS Letters. 1999;458:278–284. doi: 10.1016/s0014-5793(99)01166-7. [DOI] [PubMed] [Google Scholar]
- Abbott CA, Yu DMT, Woollatt E, Sutherland GR, McCaughan GW, Gorrell M. Cloning, expression and chromosomal localization of a novel human dipeptidyl peptidase (DPP) IV homolog, DPPP8. European Journal of Biochemistry. 2000;267:6140–6150. doi: 10.1046/j.1432-1327.2000.01617.x. [DOI] [PubMed] [Google Scholar]
- Aertgeerts K, Ye S, Shi L, Prasad SG, Witmer D, Chi E, Sang BC, Wijnands RA, Webb DR, Swanson RV. N-linked glycosylation of dipeptidyl peptidase IV (CD26): Effects on enzyme activity, homodimer formation, and adenosine deaminase binding. Protein Science. 2004;13:145–54. doi: 10.1110/ps.03352504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M. 1989 Drosophila A Laboratory Manual. Cold Spring Harbor Laboratory Press. [Google Scholar]
- Bjelke JR, Christensen J, Branner S, Wagtmann N, Olsen C, Kanstrup AB, Rasmussen HB. Tyrosine 547 constitutes an essential part of the catalytic mechanism of dipeptidyl peptidase IV. The Journal of Biological chemistry. 2004;279:34691–34697. doi: 10.1074/jbc.M405400200. [DOI] [PubMed] [Google Scholar]
- Boman HG, Boman AI, Andreu D, Li Z, Merrifield RB, Schlenstedt G, Zimmermann R. Chemical synthesis and enzymic processing of precursor forms of cecropins A and B. The Journal of Biological Chemistry. 1989;264:5852–5860. [PubMed] [Google Scholar]
- Boonacker E, Van Noorden CJF. The multifunctional or moonlighting protein CD26/DPPIV. European Journal of Cell Biology. 2003;82:53–73. doi: 10.1078/0171-9335-00302. [DOI] [PubMed] [Google Scholar]
- Brenda. The Comprehensive Enzyme Information System. http://www.brenda.uni-koeln.de/php/result_flat.php3?ecno=3.4.14.5 1/25/2004. [Google Scholar]
- Buckley SJ, Collins PJ, O'Conner BF. The purification and characterization of novel dipeptidyl peptidase IV-like activity from bovine serum. International Journal of Biochemistry and Cell Biology. 2004;36:1281–96. doi: 10.1016/j.biocel.2003.02.001. [DOI] [PubMed] [Google Scholar]
- Charles J–P, Chihara CJ, Nejad S, Riddiford LM. A cluster of cuticle protein genes of Drosophila melanogaster at 65A: sequence, structure and evolution. Genetics. 1997;147:1213–1224. doi: 10.1093/genetics/147.3.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles J–P, Chihara CJ, Nejad S, Riddiford LM. Identification of proteins and developmental expression of RNAs encoded by the 65A cuticle protein gene cluster in Drosophila melanogaster. Insect Biochemistry and Molecular Biology. 1998;28:131–138. doi: 10.1016/s0965-1748(97)00107-0. [DOI] [PubMed] [Google Scholar]
- Chien CH, Huang LH, Chou CY, Chen YS, Han YS, Chang GG, Liang PH, Chen X. One site mutation disrupts dimer formation in human DPP–IV proteins. The Journal of Biological Chemistry. 2004;270:52338–52345. doi: 10.1074/jbc.M406185200. [DOI] [PubMed] [Google Scholar]
- Chen T, Ajami K, McCaughan G, Gorrell M, and Abbott CA. 2003 Dipeptidyl Peptidase IV Gene Family: The DPIV family. In Hildebrandt M, Klapp, B, Hoffmann T, Demuth HU, editors. Dipeptidyl Aminopeptidase in Health and Disease. Kluwer Academic/Plenum Publishers, New York. [DOI] [PubMed] [Google Scholar]
- Chihara CJ, Kimbrell DA. The cuticle proteins of Drosophila melanogaster: genetic localization of a second cluster of third instar genes. Genetics. 1986;114:393–404. doi: 10.1093/genetics/114.2.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Meester I, Vanhoof G, Hendriks D, Demuth HU, Yaron A, Scharpé S. Characterization of dipeptidyl peptidase IV (CD26) from human lymphocytes. Clinica Chimica Acta. 1992;210:23–34. doi: 10.1016/0009-8981(92)90042-o. [DOI] [PubMed] [Google Scholar]
- De Meester I, Lambeir AM, Proost P, and Scharpé S. 2003 Dipeptidyl peptidase IV substrates, an update on in vitro peptide hydrolysis by human DPPIV. in Hildebrandt M, Klapp B, Hoffmann T, Demuth HU, editors. Dipeptidyl Aminopeptidase in Health and Disease. Kluwer Academic/Plenum Publishers, New York. [DOI] [PubMed] [Google Scholar]
- Durinx C, Lambeir AM, Bosmans E, Falmagne J–B, Berghmans R, Haemers A, Scharpé S, De Meester I. Molecular characterization of dipeptidyl peptidase activity in serum: Soluble CD26/dipeptidyl peptidase IV is responsible for the release of X–Pro dipeptides. European Journal of Biochemistry. 2000;267:5608–5613. doi: 10.1046/j.1432-1327.2000.01634.x. [DOI] [PubMed] [Google Scholar]
- Flybase. http://flybase.bio.indiana.edu. [Google Scholar]
- Gault VA, Flatt PR, Harriott P, Mooney MH, Bailey CJ, O'Harte FPM. Improved biological activity of Gly2– and Ser2– substituted analogues of glucose–dependent insulinotrophic polypeptide. Journal of Endocrinology. 2003;176:133–141. doi: 10.1677/joe.0.1760133. [DOI] [PubMed] [Google Scholar]
- Gorrell M. Dipeptidyl peptidase IV and related enzymes in cell biology and liver disorders. Clinical Science. 2005;108:277–292. doi: 10.1042/CS20040302. [DOI] [PubMed] [Google Scholar]
- Kühn–wache K, Hoffmann T, Manhart S, Brandt W, and Demuth H. 2003 The specificity of DP IV for natural substrates is peptide structure determined. in Hildebrandt M, Klapp B, Hoffmann T, Demuth HU, editors. Dipeptidyl Aminopeptidase in Health and Disease. Kluwer Academic/Plenum Publishers, New York. [DOI] [PubMed] [Google Scholar]
- Lambeir A–M, Curinx C, Proost P, Damme JV, Scharpé S, De Meester I. Kinetic study of the processing by dipeptidyl–peptidase IV/CD26 of nueropeptides involved in pancreatic insulin secretion. FEBS letters. 2001;507:327–330. doi: 10.1016/s0014-5793(01)02982-9. [DOI] [PubMed] [Google Scholar]
- Ludwig A, Schiemann F, Mentlein R, Lindner G, Brandt E. Dipeptidyl peptidase IV(CD26) on T cells cleaves the CXC chemokine CSCL11 (I–TAC) and abolishes the stimulating but not the desensitizing potential of the chemokine. Journal of Leukocyte Biology. 2002;72:183–191. [PubMed] [Google Scholar]
- Martensen I, Koolman J, Mentlein R. Proline–specific dipeptidyl peptidase from the blue blowfly Calliphora vicina hydrolyzes in vitro the ecdysiostatic peptide trypsin–modulating oostatic factor (Neb–TMOF) Archives of Insect Biochemistry and Physiology. 1998;37:146–157. doi: 10.1002/(SICI)1520-6327(1998)37:2<146::AID-ARCH3>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Mentlein R. Dipeptidyl–peptidase IV (CD26) – role in the inactivation of regulatory peptides. Regulatory Peptides. 1999;85:9–24. doi: 10.1016/s0167-0115(99)00089-0. [DOI] [PubMed] [Google Scholar]
- Mentlein R, Rix H, Feller AC, Heymann E. Characterization of dipeptidyl peptidase IV from lymphocytes of chronic lymphocytic leukemia of T–type. Biochimica et Biophysica acta. 1986;45:567–74. [PubMed] [Google Scholar]
- Mentlein R, Struckhoff G. Purification of two dipeptidyl aminopeptidases II from rat brain and their action on proline–containing neuropeptides. Journal of Neurochemistry. 1989;52:1284–1293. doi: 10.1111/j.1471-4159.1989.tb01877.x. [DOI] [PubMed] [Google Scholar]
- Nagakura T, Yasuda N, Yamazaki K, Ikuta H, Yoshikawa S, Asano O, Tanaka I. Improved glucose tolerance via enhanced glucose-dependent insulin secretion in dipeptidyl peptidase IV– deficient Fischer rats. Biochemical and Biophysical Research Communications. 2001;284:501–506. doi: 10.1006/bbrc.2001.4999. [DOI] [PubMed] [Google Scholar]
- Nässel DR, Mentlein R, Bollner T, Karlsson A. Proline–specific dipeptidyl peptidase activity in the cockroach brain and intestine: partial characterization, distribution, and inactivation of tachykinin–related peptides. Journal of Comparative Neurology. 2000;418:81–92. doi: 10.1002/(sici)1096-9861(20000228)418:1<81::aid-cne6>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- O'Connor BF, 'Cuinn G. Post proline dipeptidyl–aminopeptidase from synaptosomal membranes of guinea pig brain. European Journal of Biochemistry. 1986;154:329–35. doi: 10.1111/j.1432-1033.1986.tb09401.x. [DOI] [PubMed] [Google Scholar]
- OMIM. Online Mendelian Inheritance in Man. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM. [Google Scholar]
- Perner F, Gyuris T, Rakoczy G, Sarvary E, Gorog D, Szalay F, Kunos I, Szonyi L, Peterfy M, Takacs L. Dipeptidyl peptidase activity of CD26 in serum and urine as a marker of cholestasis: Experimental and clinical evidence. Journal of Laboratory and Clinical Medicine. 1999;134:56–67. doi: 10.1016/s0022-2143(99)90054-9. [DOI] [PubMed] [Google Scholar]
- Peters ID, Rancourt DE, Davies PL, Walker VK. Isolation and characterization of an antifreeze protein precursor from ransgenic Drosophila : evidence for partial processing. Biochimica et Biophysica Acta. 1993;1171:247–254. doi: 10.1016/0167-4781(93)90062-i. [DOI] [PubMed] [Google Scholar]
- Polgar L. The prolyl oligopeptidase family. Cellular and Molecular Life Sciences. 2002;59:349–62. doi: 10.1007/s00018-002-8427-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renn SCP, Tomkinson B, Taghert PH. Characterization and cloning of tripeptidyl peptidase II from the fruit fly, Drosophila melanogaster. Journal of Biological Chemistry. 1998;273:19173–19182. doi: 10.1074/jbc.273.30.19173. [DOI] [PubMed] [Google Scholar]
- Thoma R, Löffler B, Stihle M, Huber W, Ruf A, Hennig M. Structural basis of proline–specific exopeptidase acitivity as observed in human dipeptidyl peptidase IV. Structure. 2003;11:947–959. doi: 10.1016/s0969-2126(03)00160-6. [DOI] [PubMed] [Google Scholar]
- White KP, Rifkin SA, Hurban P, Hogness DS. Microarray analysis of Drosophila development during metamorphosis. Science. 1999;286:2179–2184. doi: 10.1126/science.286.5447.2179. [DOI] [PubMed] [Google Scholar]