

Abstract
The lack of an appropriate animal model with multiorgan pathology characteristic of the human form of cystic fibrosis has hampered our understanding of the pathobiology of the disease. We evaluated multiple organs of congenic C57BL/6J cystic fibrosis transmembrane regulator (Cftr)−/− and Cftr+/+ mice maintained from weaning on a liquid diet then sacrificed between 1 and 24 months of age. The lungs of the Cftr−/− animals showed patchy alveolar overdistention, interstitial thickening, and fibrosis, with progression up to 6 months of age. The proximal and distal airway surface was encased with mucus-like material but lacked overt evidence of chronic bacterial infections or inflammation. All Cftr−/− animals showed progressive liver disease, with hepatosteatosis, focal cholangitis, inspissated secretions, and bile duct proliferation; after 1 year of age there was progression to focal biliary cirrhosis. The intercalated, intralobular and interlobular ducts and acinar lumina of the exocrine pancreas, the parotid and submaxillary glands of the Cftr−/− animals were dilated and filled with inspissated material, as well as mild inflammation and acinar cell drop out. Quantitative measurements of the pancreas showed significant acinar atrophy and increased acinar volume in comparison with age-matched Cftr+/+ littermates. The ileal lumen and crypts were filled with adherent fibrillar material. After 3 months of age the vas deferens of the Cftr−/− animals could not be identified. None of the aforementioned pathological changes were observed in the Cftr+/+ littermates fed the same liquid diet. We show, for the first time, that long-lived C578L/6J Cftr−/− mice develop manifestations of cystic fibrosis-like disease in all pathologically affected organs in the human form of cystic fibrosis.
Cystic fibrosis (CF) is an autosomal recessive condition that is caused by mutations in the cystic fibrosis transmembrane regulator gene (CFTR).1 Although other genetic and environmental factors almost certainly contribute to disease pathobiology, CF disease arises from impaired ion conductance on epithelial cell surfaces, which increases the concentration and alters the viscosity of intraluminal contents.2 Multiple organs are affected but the phenotype is extremely heterogeneous.3,4 CF may present in the neonate with bowel obstruction because of meconium ileus, whereas older patients may be plagued by intermittent chronic recurrent ileo-cecal obstruction from adherent mucofeculent material. In the airways, mucus clearance is impaired by the inability of the surface liquid to serve as a medium for ciliary boating of the mucigel. Obstruction of the airways results in chronic bacterial infection and rampant inflammation leads to end-stage lung disease and death. In its severest form, exocrine pancreatic disease begins in utero.5,6 Intraluminal obstruction by inspissated material within the acinar lumina and small ducts leads to progressive acinar atrophy7 and replacement by fat and fibrous tissue.3 Similar alterations occur in the small ducts of the parotid gland, but the pathological effects are less severe.8,9 The biliary canaliculi and small bile ducts are obstructed with inspissated material.3,10–13 Cholangitis develops and the biliary ductules proliferate; with progression most patients develop focal biliary cirrhosis and in some cases extensive multilobular cirrhosis ensues. Some patients develop focal or diffuse hepatic steatosis.13 Approximately 20 percent of patients present with bowel obstruction because of meconium ileus.14 Almost all affected men are infertile because of obstructive azoospermia. The sweat duct, which lacks intraluminal macromolecules, is the only CFTR-expressing ductular system that is not affected by obstructive pathological changes.
The first murine model of CF was established by gene targeting of embryonic stem cells to disrupt the murine cftr gene.15 Other murine models have been developed to model human mutations, including the most common CFTR gene mutation ΔF508 (confers loss of phenylalanine) and models carrying common missense mutations (G551D, R117H).16–20 Most of these animals develop a range of intestinal pathology that may cause fatal complications after bowel obstruction at birth or when weaned to solid chow. However, pathological changes in other CF-affected organs, including the lungs, pancreas, and liver, were reported to be either mild or absent. Rozmahel and colleagues,21 used an exon-1 knockout CF mouse model (CftrM1HSC/CftrM1HSC) to demonstrate that the severity of intestinal disease was influenced by the genetic background, and that modifier genes, could, in part, compensate for the lack of functional intestinal CFTR. Subsequently, Kent and colleagues22 demonstrated that Cftr−/−mice bred into a congenic C57BL/6J consistently developed spontaneous lung disease. Features of the lung disease included defective mucociliary transport, alveolar distention, and interstitial thickening with fibrosis and inflammation.
In this report, we demonstrate that, with further aging, a CF-knockout murine model bred into a congenic C57BL/6J background consistently develops progressive pathology in multiple organs that are strikingly similar to the human manifestations of CF disease.
Materials and Methods
Breeding and Maintenance of Animals
All experimental protocols were conducted after review and approval by the institutional Animal Care Committee. Congenic C57BL/6J heterozygous breeding pairs were maintained on regular mouse chow and continuously bred. To maintain congenic status and prevent genetic drift, each new generation of mice was bred to wild-type C57BL/6J mice, obtained from the Jackson Laboratories (Bar Harbor, ME). This created the production stock from which experimental animals were obtained. Male and female wild-type animals were used in alternate breedings. Offspring were genotyped at 14 days of age using polymerase chain reaction analysis of tail clip DNA. To minimize bowel obstruction and optimize long-term viability, 20- to 23-day-old Cftr−/− mice and their Cftr+/+ littermates were weaned to a liquid diet (Liquidet F3107; Bioserve, Frenchtown, NJ) using glass liquid mouse feeders, prepared in sterile water according to the manufacturer’s instructions.23 Fresh diet and feeders, sterilized by autoclave, were replaced daily. Mice and their offspring were housed in a nonsterile conventional housing unit in microisolators cages, with corncob bedding changed daily, and provided with sterile water in addition to the liquid diet. The colony was maintained at a pathogen-free status by serological screening at a commercial laboratory. Mice were kept in a 12-hour light-dark cycle.
Tissue Collection
Groups of Cftr−/− animals and their wild-type littermates were sacrificed at varying ages [1 to 2 months (Cftr−/−, n = 6; Cftr+/+, n = 6), 3 to 5 months (Cftr−/−, n = 13; Cftr+/+, n = 8), 9 to 12 months (Cftr−/−, n = 8; Cftr+/+, n = 3), and 15 to 24 months (Cftr−/−, n = 5; Cftr+/+, n = 5)]. Mice were anesthetized by intraperitoneal injection of sodium pentobarbital (30 mg/kg) and the abdominal cavity was opened. The inferior vena cava was cut and 1% glutaraldehyde and 4% paraformaldehyde in 0.1 mol/L phosphate buffer (pH 7.4) was perfused through the left ventricle. The airways were perfused through the larynx with the same fixative and the tissues were maintained at a constant pressure. The trachea, lungs, liver, pancreas, ileum parotid, and submaxillary and sublingual glands were harvested and immersed in the same fixative. Tissue from the reproductive tract of males older than 3 months of age, including the intact testis, epididymis, and vas deferens, was collected and fixed in formalin.
Tissue Preparation and Analysis
Transmission Electron Microscopy (TEM)
Portions of the harvested organs were minced into 1-mm3 pieces, fixed for an additional 4 hours in the same fixative and then postfixed for 1 hour in phosphate-buffered 2% osmium tetroxide. Samples were then dehydrated before infiltration, in ascending concentrations of ethanol, embedded in Epon Araldite (JEOL, Peabody, MA), and polymerized at 60°C overnight. Areas of interest were selected by light microscopic examination of 0.5-μm-thick sections, cut by ultra-microtome and stained with toluidine blue. Ultrathin sections exhibiting a pale gold interference color from selected blocks were cut, mounted on grids, and stained with ethanol uranyl acetate and lead citrate before TEM examination.
Scanning Electron Microscopy (SEM)
Portions of the trachea, bronchi, bronchioles, and lungs were prepared for SEM. Median longitudinal sections of lung as well as segments of the airways were postfixed with osmium tetroxide, dehydrated in ethanol, critical point-dried, mounted on stubs, and rendered conductive with a thin layer of sputter-coated gold palladium and then examined under SEM.
Light Microscopy
All remaining tissues were fixed in formalin and prepared for routine histology. Slides were stained with hematoxylin and eosin (H&E), and either Masson’s trichrome, diastase-digested periodic acid-Schiff reagent (PASD), or reticulin stain.
Care was taken to orient the entire male reproductive tract, before embedding and cutting, to ensure correct alignment of all section. As a result, midline sections of the testes and cross sections of epididymis were consistently observed in all animals. Sections were evaluated for the presence or absence of the vas deferens; and if present for evidence of pathological changes in the vas deferens. We also assessed the epididymis and its luminal content, and staging of the seminiferous tubules.
Morphometric Assessment of Lung Interstitium
Random digital images of the lung alveolar wall from all animals were acquired at ×10,000 magnification with a digital charge-coupled device camera in the TEM (AMT Corp., Danvers, MA). A minimum of 50 fields was collected from each animal and the thickness of the interstitium was measured from five points within each image. Linear measures were expressed as mean and SE and significance was determined by multivariate analysis of variance analysis.
Volumetric Determination of Exocrine Pancreas
Quantitative morphometry was performed on images captured by a digital charge-coupled device camera (Cool Snaps; Roper Scientific) attached to a light microscope. Images were all captured at a nominal magnification (×250) on at least four different slides of toluidine blue-stained pancreas using three different blocks from each animal. Pancreatic acinar volume (mm3), volume density (vol of acini/vol of exocrine pancreas), and numerical density (number of acini/mm3) were measured according to Weible’s principles of stereology.24 An image analysis program (Image Pro Plus; Iowa) used the following three formulae:
1. Acinar volume
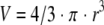 |
where V is the average volume of an acinus and r is the average acinar radius.
2. Acinar numerical density
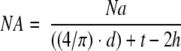 |
where NA is the number of acini per unit volume of tissue (number/mm3), Na is the number of acini per area, d is the average diameter measured, t is the section thickness (0.5 μm), and h is the smallest recognizable acinar diameter (15 μm).
3. Acinar volume density percent
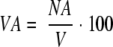 |
where VA is the total volume of the acini in a unit volume of tissue (mm3/mm3), NA is the acinar numerical density, and V is the average volume of a single pancreatic acinar.
Data from each animal group was expressed as a mean and SE and significance was calculated using multivariate analysis of variance analysis.
Results
Lung
Light Microscopy
As previously reported in younger animals,22 sections of lung demonstrated widespread pathology. In comparison with age-matched wild-type littermates, alveolar architecture of the Cftr−/− mice was compromised (Figure 1, A and B). Patchy areas of acinar dilation, typical of obstructive lung disease, were observed in all Cftr−/− animals at all ages. Interstitial disease, which was detected as early as 1 month of age, showed progression up to 6 months of age. Sections stained with either Masson’s trichrome or reticular stain demonstrated focal areas of intense interstitial staining indicative of an increase in collagen (Figure 1, A and B). After 6 months of age, there was little progression of lung pathology, although many of the alveoli showed greater variation in size. Inflammatory cells were frequently seen in the interstitium in animals less than 6 months of age, as were alveolar macrophages. When compared to wild-type animals of the same age, interstitial connective tissue was markedly increased.
Figure 1.

A: SEM of the surface of the respiratory epithelium from a terminal bronchiole in an 11-month-old wild-type animal. Note the numerous ciliated and nonciliated cells. B: Terminal bronchiole from a Cftr−/− littermate. Respiratory epithelium is encrusted in mucus-like material. C: Alveoli from the wild-type animal. D: Alveoli from the affected animal. Distal airways were caked with mucus-like material. Original magnifications: ×1000 (A, B); ×650 (C, D).
Immunoperoxidase staining for vimentin of some sections demonstrated an increase in the number of interstitial fibroblasts in the Cftr−/− animals up to 5 months of age. In older animals a marked decrease in the number of these cells was observed. Surprisingly, the number of inflammatory cells and macrophages in both the proximal and distal airways of the affected animals appeared to decrease with age. All of the affected animals showed positive PASD staining for acidic mucosubstances throughout the surface of the bronchioles and some alveoli also showed increased intensity and distribution.
Electron Microscopy
As previously demonstrated in young Cftr−/− mice,23 the entire proximal and distal airways were diffusely encrusted in a thick coating of mucus-like material at all ages, which completely enveloped the ciliated surface. From 1 month of age, SEM of the surface of the bronchiolar epithelial cells of affected animals demonstrated that the cilia were embedded in this material, as were the alveolar walls (Figure 1; A to D). Morphometric determination of alveolar interstitial thickness demonstrated a significant age-related increase in the affected animals (Figure 2) up to 5 months of age. After this age no further changes occurred in the interstitium except for the lack of fibroblasts. After 3 to 5 months of age, alveolar interstitial thickening of the Cftr−/− animals was significantly greater than their age-matched wild-type littermates (Figure 2B). Type II pneumocytes were flatter than their wild-type counterparts and many lacked normal looking lamellar bodies.
Figure 2.

A: TEM of alveoli from a lung of a 5-month-old Cftr−/− animal. Note the numerous lipid-containing fibroblasts (arrows) and abundant collagen (arrowheads) in the interstitium. B: Summary histogram comparing the interstitial thickness between wild-type and Cftr−/− animals as well as between age groups. Note the marked increase in interstitial thickness in the 3- to 5-month-old and older groups in the Cftr−/− animals. Scale bar, 2 μm. Original magnification, ×12,500.
Liver
Light Microscopy
All affected animals showed focal and progressive hepatobiliary disease (Figures 3A to 7D). Varying degrees of progressive bile duct proliferation, typical of obstruction, was observed in all Cftr−/− animals but not in their wild-type littermates. Focal portal tracts, in animals as young as 1 month of age, showed acute and chronic cholangitis. By 2 months of age polymorphonuclear leukocytes and chronic inflammatory cells were seen both within and surrounding focal biliary canaliculi and bile ducts. By 3 months of age there were varying degrees of periportal and bridging fibrosis, which progressed with age (Figure 3, B and C). Most animals sacrificed at 12 to 24 months of age developed focal biliary cirrhosis and some animals showed areas of advanced lobular cirrhosis (Figure 3D). Hepatic steatosis, which was common in the Cftr−/− animals, increased in severity with age. These changes could not be attributed to the liquid diet because steatosis was not evident in the wild-type littermates fed the same diet. Surprisingly little inspissating material was detectable in the common bile duct, the interlobular bile ducts, or biliary canaliculi when evaluated by light microscopy using H&E or PAS staining.
Figure 3.

A: H&E from an 11-month-old wild-type animal maintained on the liquid diet showing no pathological abnormalities. Portal tracts are evident in the field of view (asterisks). B: Cftr−/− littermate shows portal tract inflammation, ductular cell proliferation, and some portal-to-portal bridging (arrowheads). Parenchymal and sinusoidal cells that surround the central vein (CV) appear normal. C: Higher power of B. Portal-to-portal bridging is evident. Arrowheads indicate ductular and inflammatory cells. D: Low power of liver from a 22-month-old Cftr−/− animal that has developed foci of advanced lobular cirrhosis. Original magnifications: ×250 (A, B); ×400 (C); ×200 (D).
Figure 4.

A: TEM of a portal tract from a 22-month-old wild-type animal containing a bile duct. The lumen (asterisk) appears void of any material. B: TEM of a portal tract from a 12-month-old Cftr−/− animal that has developed severe cholangitis showing macrophages and polymorphs both within the lumen of one of the ducts (asterisks) and in the periportal spaces. One of the ducts is filled with inspissated material. C: TEM of several hepatocytes from a 3-month-old Cftr−/− animal filled with lipid droplets, which is consistent with steatosis. D: Periportal fibrosis and duct proliferation. Bundles of collagen fibrils (arrowheads) and several bile ducts are seen in the field (asterisks). Original magnifications: ×2500 (A, B, D); ×5000 (C).
Figure 5.

A: Toluidine blue-stained epoxy section of pancreas from a wild-type 9-month-old animal. Note the uniform size of all of the acini and the near absence of any material in any of the ducts (arrows). B: TEM of an interlobular duct from the same animal in Figure 3A. Note the absence of any material in the lumen of the duct (asterisk). An islet (IS) is seen at the bottom right. C: Toluidine blue-stained section of pancreas from a Cftr−/− littermate of the animal seen in Figure 3, A and B. Note the inspissated material found in the interlobular duct (asterisk) and intralobular ducts (arrows). Many of the lumina of the acini appear dilated (arrowheads). D: TEM of dilated acinar lumen (asterisk) from pancreas of animal in Figure 3C. Note the fibrillar material in the lumen. E: TEM of interlobular duct in Figure 3C. Lumen (asterisk) is filled with inspissated material. Note the flattened appearance of the ductular cells. Original magnifications: ×450 (A); ×3500 (B); ×825 (C); ×5000 (D); ×3200 (E).
Figure 6.

A: Toluidine blue-stained section of pancreas from a 12-month-old Cftr−/− animal showing periacinar and periductal (asterisk) inflammation (arrowheads) and acinar loss and atrophy. B: Histogram of acinar volume. Acinar volume increased with age in the Cftr−/− age groups. C: Histogram of percentage of acinar volume to tissue volume. Unlike the acinar volumethere was a progressive decrease. D: Histogram of acinar numerical density. Density also decreased, which is suggestive of acinar cell drop-out in the older Cftr−/− animals. Original magnification, ×400.
Figure 7.

A: Toluidine blue-stained section from a 12-month-old Cftr−/− submaxillary gland. Note the periductal inflammation (arrowheads). The duct is filled with inspissated material (asterisk). B: TEM of material found in the lumen of a parotid duct from a 5-month-old Cftr−/− animal. In all of the affected animals many of the acinar and ductular lumina were filled with this material. Original magnifications: ×800 (A); ×10,000 (B).
Electron Microscopy
Many of the affected portal tracts contained inflammatory cells. These were found surrounding and within the bile duct lumina (Figure 4, B and C). Some of the bile ducts contained fibrillar material (Figure 4B). By 2 months of age, the majority of hepatocytes in the Cftr−/− animals contained lipid droplets consistent with steatosis (Figure 4C). Most biliary canaliculi of animals older than 9 months contained the same material and there was evidence of severe periportal fibrosis (Figure 4D).
Pancreas
Light Microscopy
As young as 2 months of age the intercalated, intralobular, and interlobular pancreatic ducts of the Cftr−/− animals were increased in diameter in comparison with their wild-type littermates. Intraluminal obstruction with inspissated material that progressed with age was observed in the Cftr−/− animals but not in the wild-type littermates (Figure 5, A and C). After 6 months, foci of inflammatory cells and macrophages were evident in the exocrine pancreas of the Cftr−/− mice (Figure 6A). Affected animals older than 12 months of age showed subjective evidence of acinar atrophy. Pathological changes were not observed in the islets of Langerhans of the affected mice or in the wild-type animals. Furthermore none of the affected animals showed clinical evidence of diabetes mellitus.
The results of the morphometric studies of the pancreas are summarized in (Figure 6; B, C, and D). Acinar volume of the wild-type animals, which reflects intraluminal volume, showed no significant alteration with aging. However, there was a modest age-related increase in mean acinar volume in the 3- to 6-month-old Cftr−/− animals and by 15 to 24 months of age, the acinar volume was significantly greater than that determined in the 1- to 2-month-old Cftr−/− animals (Figure 6B). Percentage of acinar volume expressed per unit pancreatic tissue volume showed no age-related changes in the Cftr+/+ mice. Also at 1 to 2 and 3 to 5 months of age the percentage of acinar to tissue volume of the exocrine pancreas in the Cftr−/− mice did not differ significantly from measurements in the wild-type, age-matched littermates. In contrast, the percentage of acinar volume to tissue volume in the Cftr−/− mice demonstrated a progressive decrease from 9 to 12 months of age, which became statistically significant at 15 to 24 months of age(Figure 6C). These morphometric results reflect intraluminal distention.
The number of acini, expressed per unit volume of pancreatic tissue, was also determined. Although the wild-type animals showed no age-related change in acinar number, the Cftr−/− animals showed an age-dependent decrease that was statistically significant from 3 to 5 months of age in comparison with the 1- to 2-month-old Cftr−/− mice (Figure 6D). The number of pancreatic acini of the Cftr−/− animals was also significantly lower than that determined in the 9- to 12-month-old and 15- to 24-month-old wild-type littermates. These data provide objective evidence of progressive age-related acinar cell drop out in the Cftr−/− mice.
Electron Microscopy
Acinar lumina of the affected animals were filled with fibrillar material as young as 1 month of age and the zymogen granules were not as numerous or dense in comparison with acini from the wild-type littermates. The majority of the acinar lumina in the Cftr−/− animals were dilated and plugged with inhomogenous material (Figure 5D). Unlike, the wild-type animals the intralobular and interlobular ducts of the Cftr−/− animals were plugged with inspissated material (Figure 5, B and E) and the epithelial cells were flattened (Figure 4E).
Parotid, Submaxillary, and Sublingual Glands
Light Microscopy
The parotid and submaxillary glands of the Cftr−/− animals showed histological abnormalities, whereas the sublingual glands were normal in appearance. The pathological features of the parotid and submaxillary glands were similar to those observed in the exocrine pancreas (Figure 6A and Figure 7). Unlike the exocrine pancreas of the Cftr−/− mice, the light microscopic appearance of the serous-type acini and ducts showed obvious inspissation with fibrillar material and greatly dilated lumina. This was particularly evident with toluidine blue-stained sections (Figure 7A). In the 9 to 12 month and older Cftr−/− animals, foci of acute and chronic inflammatory cells were present surrounding the ducts and progressive acinar loss was observed. Mineralized sludge was seen in several of the ducts from the parotids of the 15 to 24 month Cftr−/− animals.
Electron Microscopy
Lumina of the acini and ducts of the parotid and submaxillary glands were filled with material that was more amorphous and electron opaque than the typical secretions found in other pathologically affected organs (Figure 7B). As was observed with electron microscopy images of the plugged exocrine pancreatic ducts of the Cftr−/− mice, the affected ductal epithelial cells were flattened in comparison to their wild-type littermates.
Ileum
Histology
Consistent with reports in other murine CF strains, features of chronic intestinal obstruction were observed even in the youngest Cftr−/− mice. Typically the lumen of the ileum was filled with PASD-positive material, a feature not observed in any of the wild-type littermates (Figure 8, A and B). PASD staining of older animals demonstrated excessive amounts of acidic mucosubstances in the crypts. There was a clear increase in the number of goblet cells per villus in the affected animals. Three Cftr−/− animals developed abdominal distention between 9 to 12 months of age and were sacrificed because of a suspicion of intestinal obstruction. At necropsy the ileum was found to be totally obstructed with adherent mucofeculent material, which could not be removed from the intestinal epithelium with a scalpel blade. Intraluminal blockage in the ileo-cecal region of the intestine closely resembled distal intestinal obstruction syndrome, a unique late intestinal complication observed in the human form of CF. The proximal small intestine and stomach were filled with the liquid feed and were swollen several times their normal size.
Figure 8.

A: Periodic acid-Schiff (PAS) staining of a cross-section of ileum from a 3-month-old wild-type animal. Note the intense staining of the goblet cells on the villi and in the crypts (arrows). Some of the crypts were filled with PAS-stained material (arrowheads). B: Cftr−/− littermate showing crypts filled and distended with PAS-positive material (arrows) and an increase of PAS-positive material within the lumen (asterisk). C: TEM of crypt filled with inspissated material (asterisk). Original magnifications: ×300 (A, B); ×2500 (C).
Electron Microscopy
The most obvious electron microscopic feature observed in all of the Cftr−/− animals was the presence of excessive fibrillar mucus-like material in the lumina of the ileum (Figure). Most of the crypts were distended and filled with this material (Figure 8C).
Male Reproductive Tract
Histology
Several unsuccessful attempts were made at breeding the Cftr−/− males with wild-type females. This suggested that the affected males were functionally infertile. The vas deferens of six wild-type mice, ranging in age from 3 to 15 months, could be readily seen and the architecture was normal. However, in 8 of 10 Cftr−/− males (3 to 15 months of age) no vas deferens could be identified. In fact, the vas deferens could only be identified in two of the youngest Cftr−/− males 3 to 5 months of age. In these animals the lumen diameter was smaller than the age-matched wild-type littermates and the surrounding smooth muscle layer was thicker (Figure 9, A and B). The epididymus of the Cftr−/− males contained fewer spermatids and the majority were immature. Many had failed to develop an acrosome. Most of the vacant ducts of the epididymus were filled with extracellular material. In the affected males, very few of the seminiferous tubules contained late spermatids and the majority of the tubules were stage 3 or stage 4 of development.25
Figure 9.

A: H&E-stained section of a median section through the testes (T), epididymis (E), and vas deferens from a 3-month-old wild-type mouse. Arrow indicates the lumen of the vas deferens and an arrowhead indicates the surrounding smooth muscle layer. B: Similar section from an age-matched affected animal. Although the vas deferens was not detected in any of the animals older than 3 months, two animals in the 3-month group had a visible vas deferens. In comparison with the wild-type littermates the vas deferens had a reduction in luminal diameter (arrows) and thickening of the smooth muscle layer. The lumen also contained accumulation of viscous material. Original magnifications, ×100.
Discussion
We demonstrate, for the first time, that long-lived congenic C57BL/6J Cftr−/− mice develop the hallmarks of CF-like disease in all organs pathologically affected by the human form of cystic fibrosis. This is in striking contrast to previous observations of other murine models of CF bred in a mixed genetic background,15–20 which showed severe intestinal disease, but mild or no pathological changes in other organs affected by CF disease in humans. However, in most published studies the CF mice were sacrificed for evaluation at an early age. In the present report, progressive age-related changes in CF-like pathology became evident after 3 months of age, especially in the liver, exocrine pancreas, and parotid and submaxillary glands. In addition, the reproductive tract of the males showed clear histological evidence of severe obstructive azoospermia. The loss of identifiable vas deferens after 3 months of age closely resembles the human form of CF disease in males.
Because organ systems in mouse and man differ somewhat, both in anatomical and physiological terms, it is not surprising that the nature and course of the pathological changes are not identical to each other. For example, CFTR expression in the human bronchus is highest in submucosal glands.26 Therefore, the lack of tracheal and bronchial submucosal glands in the murine airway may account for the milder pathological changes in the murine airways.27,28 Nevertheless, as previously described,23 development of pathological changes in the lung of this congenic murine model of CF is unique and shares several characteristics in common with the early form of CF bronchopulmonary disease in human. This includes acinar and alveolar hyperinflation, presumably because of obstructive small airways disease, and extensive thickening with focal areas of fibrosis.3,29,30 Lung hyperinflation, which has also been observed in the early stages of evolving lung disease in human with CF, has been attributed to small airway obstruction.29 Presumably, defective mucociliary clearance results in ball valve blockage of the distal airways because of intraluminal accumulation of acid mucopolysaccharide substances.31 While the thickness of the interstitium increased up to 6 months of age we were unable to detect any further progression with age. There was excessive mucus production within the bronchi and bronchioles and as has been previously demonstrated in younger animals,22 SEM demonstrated which was diffusely distributed over the distal and proximal airway surfaces. This caked viscid material, which completely obliterated the surface cilia (Figure 1, E and F), has previously been shown to contain PAS-positive acid polysaccharide material.22
Environmental factors may also contribute to differences in pulmonary disease between the murine form of the disease and human CF. For example, although these animals were not housed in sterile conditions, they were in filter top cages and protected from many environmental pathogens. Nonetheless, the Cftr−/− mice failed to show histological evidence of spontaneous chronic bacterial infections at any age. Even the 2-year-old Cftr−/− mice showed no evidence of inflammation of the airways, which in the human form of CF is presumed to result from chronic bronchopulmonary infection. Several studies of other murine CF models derived from a mixed genetic background, exposed to the known CF pathogen Pseudomonas aeruginosa have significantly impaired clearance of bacteria, a more severe inflammatory reaction, and develop severe, pathogen-specific lung pathology when compared with age-matched wild-type littermates.32–34 Similarly, when exposed to common CF pathogens such as Burkholderia cepacia, murine models of CF develop significant airway inflammation and succumb to pulmonary disease at a faster rate than other CFTR knockout strains.35 Thus, the difference in manifestations of airway disease in this knockout CF mouse model in comparison with the human form of CF disease may, in part, be because of a lower susceptibility to spontaneous acquisition of chronic bronchopulmonary infections.
Hepatobiliary changes in this congenic Cftr−/− murine model showed remarkably similar pathology to the characteristics of human CF3,10–12 and there was considerable progression in the severity of the disease with aging. Focal periportal obstructive disease with bile duct proliferation and cholangitis was consistently apparent by 3 to 5 months of age and by 12 months of age many animals had developed patchy areas of biliary cirrhosis. By 2 years of age, these findings were universal. Another common observation in the human form of CF liver disease, hepatocyte vacuolization with evidence of microdroplet and macrodroplet steatosis, was seen in all of the Cftr−/− animals.13 Because these changes were not observed in the wild-type littermates, a treatment effect of the liquid diet is unlikely.
In its severest form, humans with CF develop progressive pancreatic damage, beginning in utero,5 and this leads to signs and symptoms of maldigestion because of pancreatic insufficiency at birth or in infancy.6,7 Typically, postmortem examination of older patients with end-stage pancreatic disease reveals complete loss of exocrine pancreatic tissue, identifiable with islets of Langerhans embedded in fibrous tissue and fatty stroma.3 However, postmortem studies of CF infants dying within the first 4 months of life show strikingly similar changes to those observed in this murine model of CF.5 There is a lack of development of pancreatic acinar tissue. The duct luminal volume, expressed as a proportion of total pancreatic volume, exceeds that found in age-matched non-CF patients and the ratio of acinar to connective tissue diminishes with age. In particular, premature infants dying from CF show duct dilatation with secretory material obstructing the small pancreatic ducts.5 This early obstructive feature of pancreatic disease in CF almost certainly leads to progressive atrophy of acinar tissue. Our morphological measurements of these CF mice show that they develop a form of pancreatic disease, which is consistent with chronic and progressive obstruction of small ducts and acini. There is progressive ductal and acinar dilatation with progressive loss of acinar tissue with aging. Thus, pancreatic disease in this murine CF model only differs from the human form of CF in terms of age of onset rate of progression and severity. Whether or not complete exocrine pancreatic atrophy develops with further aging remains to be determined.
Similar CF-like characteristics were observed in the salivary glands,3,8,9 but we were surprised to note that only the parotid and the serous component of the submaxillary glands were affected. Typically, the acini and ducts in these glands were distended and obstructed, changes that were not apparent in their wild-type littermates. Foci of periacinar and acinar loss were evident, and in the older animals periductal inflammation was present. Why were the pathological changes limited to serous secreting glands? Serous glands seem to be more susceptible to the consequences of intraluminal desiccation by reduced fluid secretion, which in turn increases the concentration of protein and mucus and induces ductal plugging and obstruction. Sublingual glands are mucus secreting and produce more viscous secretions that may make them more susceptible to the pathological consequences of absent CFTR-related ion and fluid flow. Little is known about the extent and distribution of CFTR expression within specific epithelial cells of the salivary glands. Ultrastructural examination of immunogold-labeling density shows a threefold greater expression of CFTR in the human parotid duct in comparison to the human sublingual duct (C.A. Ackerley, unpublished observation). Alternatively, differential expression of other chloride channels or ion channels that may increase flow, such as the chloride-bicarbonate exchanger of the nonserous glands, may account for the presence or absence of CF pathology in the ducts of specific glands.
It is worthwhile speculating that all serous-secreting glands are more vulnerable to pathological sequelae of impaired function of CFTR than other glands affected by CF disease. In this regard, the submucosal glands of the trachea and intrapulmonary bronchi of the human respiratory tree, the laryngeal glands of the mouse, and the exocrine pancreas of both the human and the mouse are serous glands and, in functional terms, appear to be highly dependent on CFTR-mediated secretion of ion and fluid for maintaining intraluminal macromolecules in solution. Thus, in keeping with disease pathology in humans this knockout CF mouse model exhibits defective serous gland function as a primary manifestation of pathological disease.
Varying degrees of severe obstructive intestinal disease, which have been observed in all murine CF knockout models, show identical characteristics to meconium ileus, an intestinal complication observed in 15 to 25% of humans born with CF.14 CF mice are susceptible to intestinal disease at all ages.21 A large percentage of affected pups die in utero or at birth from complications of meconium ileus. Impacted mucofeculent material within the ileum and cecum usually causes death from peritonitis after intestinal perforation. This C57B2/6 congenic strain is particularly susceptible to obstructive intestinal disease at weaning, and if the animals are fed solid chow, they succumb after complications of intestinal obstruction within days.21 Although introduction of a liquid diet at weaning ameliorates the intestinal complications, some animals still die of intestinal obstruction.23 At postmortem all of the affected animals showed varying degrees of intestinal disease with patchy areas of villus atrophy and mucosal hypertrophy. The goblet cells of the villi and crypts and intestinal lumina are filled with PASD-positive material. Some of the older animals developed distal intestinal obstruction requiring euthanasia. Obstructed tenacious, adherent viscous mucofeculent secretions within the ileum and cecum were typical of distal intestinal obstruction syndrome, a unique form of intermittent intestinal disease observed during adolescence and adulthood in humans with CF.36
To conclude, this congenic, knockout murine model of CF has most of the hallmarks of human CF disease making it a valuable model for the study of CF pathobiology and as a test bed for targeted therapies. Nevertheless, to allow significant pathological changes to occur in CF-affected organs, it is necessary to breed the animals to a much older age than had been previously appreciated. To maintain viability, intestinal obstruction can be ameliorated by nourishing the animals on a liquid diet. Alternatively, intestinal complications can be prevented by breeding murine models expressing intestinal tract human CFTR cDNA under the control of the rat intestinal fatty-acid binding protein gene promoter.37
Footnotes
Address reprint requests to Cameron A Ackerley, Department of Pathology, The Hospital for Sick Children, 555 University Ave., Toronto, Ontario, M5G 1X8, Canada. E-mail: cameron.ackerley@sickkids.ca.
Supported by the National Institutes of Health (Specialized Centers of Research (SCOR) grants NIDDK-2P50 and DK49096-09), the Canadian Cystic Fibrosis Foundation, and the Sellers Foundation.
Current address of G.K.: Director, Animal Care and Veterinary Services, University of Western Ontario, Room 510 D, Medical Sciences Building, London, Ontario, N6A 5C1, Canada.
References
- Rommens JM, Iannuzzi MC, Kerem B, Drumm ML, Melmer G, Dean M, Rozmahel R, Cole JL, Kennedy D, Hidaka N. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989;245:1059–1065. doi: 10.1126/science.2772657. [DOI] [PubMed] [Google Scholar]
- Anderson MP, Rich DP, Gregory RJ, Smith AE, Welsh MJ. Generation of cAMP-activated chloride currents by expression of CFTR. Science. 1991;251:679–682. doi: 10.1126/science.1704151. [DOI] [PubMed] [Google Scholar]
- Oppenheimer EH, Esterly JR. Pathology of cystic fibrosis review of the literature and comparison with 146 autopsied cases. Perspect Pediatr Pathol. 1975;2:241–278. [PubMed] [Google Scholar]
- Scriver CR, Fujiwara TM. Cystic fibrosis genotypes and views on screening are both heterogeneous and population related. Am J Hum Genet. 1992;51:943–950. [PMC free article] [PubMed] [Google Scholar]
- Imrie JR, Fagan DG, Sturgess JM. Structural and developmental abnormalities of the exocrine pancreas in cystic fibrosis. Am J Pathol. 1979;95:697–707. [PMC free article] [PubMed] [Google Scholar]
- Waters DL, Dorney SF, Gaskin KJ, Gruca MA, O’Halloran M, Wilcken B. Pancreatic function in infants identified as having cystic fibrosis in a neonatal screening program. N Engl J Med. 1990;322:303–308. doi: 10.1056/NEJM199002013220505. [DOI] [PubMed] [Google Scholar]
- Cleghorn G, Benjamin L, Corey M, Forstner G, Dati F, Durie P. Age-related alterations in immunoreactive pancreatic lipase and cationic trypsinogen in young children with cystic fibrosis. J Pediatr. 1985;107:377–381. doi: 10.1016/s0022-3476(85)80510-2. [DOI] [PubMed] [Google Scholar]
- Blomfield J, Rush AR, Allars HM, Brown JM. Parotid gland function in children with cystic fibrosis and child control subjects. Pediatr Res. 1976;10:574–578. doi: 10.1203/00006450-197606000-00004. [DOI] [PubMed] [Google Scholar]
- Kolberg H, Danielsson A, Glitterstam K, Henriksson R, Marklund S. Studies on parotid saliva in cystic fibrosis. Acta Paediatr Scand. 1982;71:321–322. doi: 10.1111/j.1651-2227.1982.tb09422.x. [DOI] [PubMed] [Google Scholar]
- di Sant’Agnese PA, Blanc WA. A distinctive type of biliary cirrhosis of the liver associated with cystic fibrosis of the pancreas. Pediatrics. 1956;188:387–409. [PubMed] [Google Scholar]
- Craig JM, Haddad H, Shwachman H. The pathologic changes in the liver in cystic fibrosis of the pancreas. Am Dis Child. 1957;93:357–369. doi: 10.1001/archpedi.1957.02060040359002. [DOI] [PubMed] [Google Scholar]
- Sokol RJ, Durie P. Recommendations for management of liver and biliary tract disease in cystic fibrosis. J Pediatr Gastroenterol Nutr. 1999;28:S1–S13. doi: 10.1097/00005176-199900001-00001. [DOI] [PubMed] [Google Scholar]
- Griscom NT, Capitanio MA, Wagoner ML, Culham G, Morris L. The visibly fatty liver. Radiology. 1975;117:385–389. doi: 10.1148/117.2.385. [DOI] [PubMed] [Google Scholar]
- Kerem E, Corey M, Kerem B, Durie P, Tsui LC, Levison H. Clinical and genetic comparisons of patients with cystic fibrosis, with or without meconium ileus. J Pediatr. 1989;114:767–773. doi: 10.1016/s0022-3476(89)80134-9. [DOI] [PubMed] [Google Scholar]
- Snouwaert JN, Brigman KK, Latour AM, Malouf NN, Boucher RC, Smithies O, Koller BH. An animal model for cystic fibrosis made by gene targeting. Science. 1992;257:1083–1088. doi: 10.1126/science.257.5073.1083. [DOI] [PubMed] [Google Scholar]
- Dorin JR, Dickinson P, Alton EW, Smith SN, Geddes DM, Stevenson BJ. Cystic fibrosis in the mouse by targeted insertional mutagenesis. Nature. 1992;359:211–215. doi: 10.1038/359211a0. [DOI] [PubMed] [Google Scholar]
- Delaney SJ, Alton EW, Smith SN, Lunn DP, Farley R, Lovelock PK, Thomson SA, Hume DA, Lamb D, Porteous DJ, Dorin JR, Wainwright BJ. Cystic fibrosis mice carrying the missense mutation G551D replicate human genotype-phenotype correlations. EMBO J. 1996;15:955–963. [PMC free article] [PubMed] [Google Scholar]
- Colledge WH, Abella BS, Southern KW, Ratcliff R, Jiang C, Cheng SH, MacVinish LJ, Anderson JR, Cuthbert AW, Evans MJ. Generation and characterization of a delta F508 cystic fibrosis mouse model. Nat Genet. 1995;10:445–452. doi: 10.1038/ng0895-445. [DOI] [PubMed] [Google Scholar]
- Zeiher BG, Eichwald E, Zabner J, Smith JJ, Puga AP, McCray PB, Jr, Capecchi MR, Welsh MJ, Thomas KR. A mouse model for the delta F508 allele of cystic fibrosis. J Clin Invest. 1995;96:2051–2064. doi: 10.1172/JCI118253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson P. Generation of a CF mutant mouse possessing the G480C mutation. Berlin: Berlin Book of Abstracts,; 22nd European CF Conference. 1998:p 143. [Google Scholar]
- Rozmahel R, Wilschanski M, Matin A, Plyte S, Oliver M, Auerbach W, Moore A, Forstner J, Durie P, Nadeau J, Bear C, Tsui LC. Modulation of disease severity in cystic fibrosis transmembrane conductance regulator deficient mice by a secondary genetic factor. Nat Genet. 1996;12:280–287. doi: 10.1038/ng0396-280. [DOI] [PubMed] [Google Scholar]
- Kent G, Iles R, Bear CE, Huan LJ, Griesenbach U, McKerlie C, Frndova H, Ackerley C, Gosselin D, Radzioch D, O’Brodovich H, Tsui LC, Buchwald M, Tanswell AK. Lung disease in mice with cystic fibrosis. J Clin Invest. 1997;100:3060–3069. doi: 10.1172/JCI119861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent G, Oliver M, Foskett JK, Frndova H, Durie P, Forstner J, Forstner GG, Riordan JR, Percy D, Buchwald M. Phenotypic abnormalities in long-term surviving cystic fibrosis mice. Pediatr Res. 1996;40:233–241. doi: 10.1203/00006450-199608000-00008. [DOI] [PubMed] [Google Scholar]
- Weible ER, editor. New York: Academic Press,; Point counting methods. Methods for Biological Morphometry. 1979:pp 101–159. [Google Scholar]
- Ham AW, Cormack DH. Philadelphia: J.B. Lippincott,; Histology. (ed 8) 1979:pp 877–893. [Google Scholar]
- Engelhardt JF, Yankaskas JR, Ernst SA, Yang Y, Marino CR, Boucher RC, Cohn JA, Wilson JM. Submucosal glands are the predominant site of CFTR expression in the human bronchus. Nat Genet. 1992;2:240–248. doi: 10.1038/ng1192-240. [DOI] [PubMed] [Google Scholar]
- Innes BA, Dorin JR. Submucosal gland distribution in the mouse has a genetic determination localized on chromosome 9. Mamm Genome. 2001;12:124–128. doi: 10.1007/s003350010244. [DOI] [PubMed] [Google Scholar]
- Pack RJ, Al-Ugaily LH, Morris G, Widdicombe JG. The distribution and structure of cells in the tracheal epithelium of the mouse. Cell Tissue Res. 1980;208:65–84. doi: 10.1007/BF00234174. [DOI] [PubMed] [Google Scholar]
- Lamarre A, Reilly BJ, Bryan AC, Levison H. Early detection of pulmonary function abnormalities in cystic fibrosis. Pediatrics. 1972;50:291–298. [PubMed] [Google Scholar]
- Bedrossian CW, Greenberg SD, Singer DB, Hansen JJ, Rosenberg HS. The lung in cystic fibrosis. A quantitative study including prevalence of pathologic findings among different age groups. Hum Pathol. 1976;7:195–204. doi: 10.1016/s0046-8177(76)80023-8. [DOI] [PubMed] [Google Scholar]
- Sharp JT, Hammond MD. Pressure-volume relationships. Crystal RG, West JB, editors. New York: Raven Press,; The LungScientific Foundations. 1991:pp 839–854. [Google Scholar]
- Davidson DJ, Dorin JR, McLachlan G, Ranaldi V, Lamb D, Doherty C, Govan J, Porteous DJ. Lung disease in the cystic fibrosis mouse exposed to bacterial pathogens. Nat Genet. 1995;9:351–357. doi: 10.1038/ng0495-351. [DOI] [PubMed] [Google Scholar]
- Thomas GR, Costelloe EA, Lunn DP, Stacey KJ, Delaney SJ, Passey R, McGlinn EC, McMorran BJ, Ahadizadeh A, Geczy CL, Wainwright BJ, Hume DA. G551D cystic fibrosis mice exhibit abnormal regulation of inflammation in lungs and macrophages. J Immunol. 2000;164:3870–3877. doi: 10.4049/jimmunol.164.7.3870. [DOI] [PubMed] [Google Scholar]
- Zahm JM, Gaillard D, Dupuit F, Hinnrasky J, Porteous D, Dorin JR, Puchelle E. Early alterations in airway mucociliary clearance and inflammation of the lamina propria in CF mice. Am J Physiol. 1997;272:C853–C859. doi: 10.1152/ajpcell.1997.272.3.C853. [DOI] [PubMed] [Google Scholar]
- Sajjan U, Thanassoulis G, Cherapanov V, Lu A, Sjolin C, Steer B, Wu YJ, Rotstein OD, Kent G, McKerlie C, Fortner J, Downey GP. Enhanced susceptibility to pulmonary infection with Burkholderia cepacia in Cftr(−/−) mice. Infect Immun. 2001;69:5138–5150. doi: 10.1128/IAI.69.8.5138-5150.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koletzko S, Stringer DA, Cleghorn GJ, Durie PR. Lavage treatment of distal intestinal obstruction syndrome in children with cystic fibrosis. Pediatrics. 1989;83:727–733. [PubMed] [Google Scholar]
- Zhou L, Dey CR, Wert SE, DuVall MD, Frizzell RA, Whitsett JA. Correction of lethal intestinal defect in a mouse model of cystic fibrosis by human CFTR. Science. 1994;266:1705–1708. doi: 10.1126/science.7527588. [DOI] [PubMed] [Google Scholar]