

Abstract
Chemokines have been implicated in the pathogenesis of a wide variety of diseases. This report describes the generation of transgenic mice that conditionally express M3, a herpesvirus protein that binds and inhibits chemokines. In response to doxycycline, M3 expression was induced in a variety of tissues and M3 was detectable in the blood by Western blotting. No gross or histological abnormalities were seen in mice expressing M3. To determine whether M3 expression could modify a significant pathophysiological response, we examined its effect on the development of intimal hyperplasia in response to femoral arterial injury. Intimal hyperplasia is thought to play a critical role in the development of restenosis after percutaneous transluminal coronary angioplasty and in the progression of atherosclerosis. Induction of M3 expression resulted in a 67% reduction in intimal area and a 68% reduction in intimal/medial ratio after femoral artery injury. These data support a role for chemokines in regulating intimal hyperplasia and suggest that M3 may be effective in attenuating this process. This transgenic mouse model should be a valuable tool for investigating the role of chemokines in a variety of pathological states.
Chemokines are small 8- to 11-kd secreted proteins that act as chemoattractants and activators of leukocytes through their interaction with G-protein-coupled receptors. More than 40 chemokines have been identified and grouped according to the relative positions of their cysteine residues. CC chemokines have the first two cysteine residues adjacent to each other, whereas CXC chemokines have one amino acid separating the first two cysteine residues. Two chemokines fit into neither category. CL1 (lymphotactin) contains only one cysteine residue and CX3CL1 (fractalkine) has three amino acids separating the first two cysteine residues.1,2
Chemokines can be divided broadly into those that are inducible at sites of diseased or damaged tissues, and those that are constitutively expressed. The latter are responsible for basal lymphoid trafficking and the structuring of lymphoid organs. Soluble chemokines and their G-protein-coupled receptors have been linked to cardiovascular diseases, such as atherosclerosis and cardiac allograft rejection; inflammatory diseases, such as asthma and rheumatoid arthritis; cancer; and infectious diseases, such as AIDS.3
Poxviruses and the herpes-viruses secrete proteins that bind and inhibit chemokine activity. These chemokine-binding proteins are thought to be used to evade the host immune system and allow viral proliferation and dissemination.4,5 The murine γ-herpesvirus 68 (MHV-68) secretes a 44-kd protein that is encoded by the open reading frame M3. The M3 protein binds C, CC, CXC, and CX3C chemokines.6,7 Cross-linking assays using 125I-labeled CXCL8 [interleukin (IL)-8] and CCL3 (MIP-1α) have demonstrated high M3 affinity for many human and murine CC chemokines, such as CCL2 (MCP-1), CCL13 (MCP-4), CCL5 (RANTES), and CCL21 (SLC). High M3 affinity has also been reported for CXC chemokines, such as human CXCL1 (GRO-α) and CXCL10 (IP-10), for murine CXCL13 (BLC), and for human CL1 (lymphotactin) and CX3CL1 (fractalkine). Binding of M3 seems specific for chemokines, because cytokines such as interleukin-1β, interleukin-18, interferon-γ, and tumor necrosis factor-α are not bound by M3.6
Binding of M3 to chemokines is associated with inhibition of chemokine activity. In vitro studies have demonstrated that M3 inhibits chemokine-mediated increases in intracellular calcium by blocking the interaction between chemokines and their cellular receptors.6,8 M3 also inhibits chemotaxis of a CCR7-transfected cell line induced by CCL19 (MIP-3β) and CCL21 (SLC) in vitro and CCL21 (SLC)-mediated recruitment of lymphocytes to the pancreas in vivo.9
Because of the broad range of diseases that are mediated by chemokines and the therapeutic potential of chemokine blockade suggested by a variety of animal studies,10–16 we generated a transgenic mouse in which high serum levels of M3 can be induced by administration of doxycycline (DOX). We examined the effect of M3 induction on the development of intimal hyperplasia in response to femoral arterial injury. Intimal hyperplasia is thought to play a critical role in the development of restenosis after percutaneous transluminal coronary angioplasty and in the progression of atherosclerosis.17 Induction of M3 resulted in a 67% reduction in intimal area and a 68% reduction in intimal/medial ratio (I/M ratio). These data demonstrate that the levels of M3 induced in this mouse model are sufficient to interfere with an important pathophysiological response and strengthen the hypothesis that chemokines play an important role in the response to arterial injury.
Materials and Methods
Generation of M3 Transgenic Animals
A bi-directional responder transgene was constructed by cloning the cDNA for M3 into pBI-G (Clontech, Palo Alto, CA). Construction of the activator transgene was previously described.18 Separation of the transgenes from vector sequences after digestion with restriction enzymes was accomplished by zonal sucrose gradient centrifugation as described.19 Fractions containing the transgene were pooled, microcentrifuged through Microcon-100 filters (Amicon, MA), and washed five times with microinjection buffer (5 mmol/L Tris-HCl, pH 7.4, 5 mmol/L NaCl, 0.1 mmol/L ethylenediaminetetraacetic acid, Amicon Corp., Danvers, MO).
DNA containing the responder transgene was microinjected into mouse eggs (C57BL/6J × DBA/2 F2; Charles River Laboratories, Wilmington, MA), which were then transferred into oviducts of CD1 (Charles River Laboratories) foster mothers, according to published procedures.20 Genotyping of transgenic mice was performed by polymerase chain reaction analysis of DNA isolated from tails. The activator transgenic mice were identified using the following primers: 5′-CGGGTCTACCATCGAGGGCCTGCT-3′ and 5′-CCCGGGGAATCCCCGTCCCCCAAC-3′. Responder transgenic mice were identified using the following primers: 5′-ACCAGCGAATACCTGTTCCGTCATAGC-3′ and 5′-AGTAAGGCGGTCGGGATAGTTTTCTTGC-3′. The endogenous low-density lipoprotein receptor gene, used as an internal control, was amplified with the following primers: 5′-ACCCCAAGACGTGCTCCCAGGATG-3′ and 5′-CGCAGTGCTCCTCATCTGACTTGT-3′. Polymerase chain reaction conditions were: 95°C, 30 seconds; 60°C, 30 seconds; 72°C, 60 seconds for 30 cycles. All transgenic mice were kept under pathogen-free conditions. Experiments were performed following protocols approved by the Animal Care and Use Committee of Schering-Plough and the Mount Sinai School of Medicine.
β-Galactosidase Activity Assay and Histochemistry
Kidneys were collected 48 hours after a single intraperitoneal injection of 500 μg of DOX in saline and homogenized in lysis buffer [100 mmol/L Tris-Cl, pH 7.5, with 400 mmol/L NaCl, 0.05% sodium azide, 1% Triton X-100, 1 mmol/L phenylmethyl sulfonyl fluoride, 3 mmol/L ethylenediaminetetraacetic acid, and proteinase inhibitor cocktail (Roche Molecular Biochemicals, Indianapolis, IN)]. The homogenate was centrifuged 10 minutes at 8000 rpm, the supernatant collected, and total protein concentration was determined using Bradford reagent (Bio-Rad, Hercules, CA). β-Galactosidase activity was measured in a volume of supernatant containing 20 μg of protein using a chemiluminescence-based assay (Clontech) according to the manufacturer’s recommendations. For histochemistry, tissues were harvested from mice, embedded in tissue compound (Sakura Finetek Co., Torrence, CA) and frozen in 2-methylbutane cooled with dry ice. Ten-μm-thick frozen sections were fixed with 2% paraformaldehyde in phosphate buffered saline (PBS) and incubated overnight in the presence of 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-gal, Roche Molecular Biochemicals) as previously described.21 Nuclear fast red was used for counterstaining.
M3 Western Blotting and Gel Shift Assay
Serum from mice collected 48 hours after a single intraperitoneal injection of 500 μg of DOX was electrophoresed on 4 to 12% NuPage Novex Bis-Tris gels with 2-(N-morpholino)-ethanesulfonic acid-sodium dodecyl sulfate buffer (Invitrogen, Grand Island, NY) and transferred to a polyvinylidene difluoride membrane (Bio-Rad) using a discontinuous buffer system (Bio-Rad bulletin 2134). The membranes were incubated with a rabbit antiserum raised against M3 expressed in Escherichia coli (kindly provided by J. Pedro Simas, Faculdade de Medecina de Lisboa, Portugal) for 1 hour followed by incubation with horseradish peroxidase-labeled goat anti-rabbit antibody for 1 hour. Immunoreactive bands were detected using the BM chemiluminescence blotting kit (Roche Molecular Biochemicals). For the chemokine cross-linking gel shift assay, serum was incubated with 0.5 nmol/L 125I-CXCL8 (IL-8) (Amersham, Arlington Heights, IL) for 2 hours at room temperature in the presence of 0, 5, 50, 250, or 1000 nmol/L unlabeled IL-8 (R&D, Minneapolis, MN). Samples were then incubated with 5 mmol/L of the cross-linker bis-(sulfosuccinnimidy)-suberate (BS3; Pierce, Rockford, IL) for 30 minutes at room temperature and then electrophoresed on a 12% polyacrylamide gel (Bio-Rad). The gel was developed with biomax film (Eastman-Kodak, Rochester, NY) for 1 hour.
Flow Cytometry and Blood Chemistry
After red blood cell lysis, 106 cells were incubated with 5 μg/ml Fc block (BD Pharmingen, San Diego, CA) and 300 μg/ml mouse IgG (Pierce). Cells were then stained with the directly conjugated primary monoclonal antibodies Gr-1 (RB6-8C5), B220 (RA3-6B2), CD3 (145-2c11), and MAC-1 (M1/70) (BD Pharmingen) in PBS, 1% bovine serum albumin, and 0.1% sodium azide for 20 minutes at 4°C in the dark. To determine viability, samples were subsequently stained with 20 μl of 5 μg/ml of propidium iodide (Calbiochem, San Diego, CA). Events were acquired on a Becton-Dickinson FACScan (Mountain View, CA) and analyzed using the CellQuest software.
Blood chemistry was performed by Ani Lytics (Gaithersburg, MD). The following parameters were measured: alanine aminotransferase, alkaline phosphatase, gamma glutamyl transferase, uric acid, amylase, creatine phosphokinase, lactate dehydrogenase, glucose, blood urea nitrogen, creatinine, cholesterol, high-density lipoprotein, triglyceride, bilirubin, albumin, total protein, and globulin.
Vascular Injury
Mice were housed at the Center for Laboratory Animal Sciences at the Mount Sinai Medical Center, New York, NY. Protocols and animal care were approved by the Institutional Animal Care and Use Committee and were in agreement with the Guide for the Care and Use of Laboratory Animals. Two days before the arterial injury, mice received a single injection of 500 μg of DOX intraperitoneally and then 2 mg/ml of DOX in the drinking water. DOX treatment was continued 10 days after the injury. Mice drink ∼3 ml of water per day. This should result in an intake of ∼6 mg of DOX/day. Femoral artery injury was performed using a modification of techniques described previously.22 Mice (22 to 40 g) were anesthetized with inhaled isoflurane. A 2-mm segment of both femoral arteries immediately distal to the epigastric branch was dissected and isolated with two surgical ties. An arteriotomy was performed and a 0.010-inch angioplasty guide-wire (Guidant Advanced Cardiovascular Systems, CA) was introduced into the isolated segment. The proximal tie was released and a 1.5-cm segment of the guide wire was advanced and retrieved three times. The guide wire was removed, the arteriotomy repaired with 11-0 suture and the proximal and distal ties were released to restore blood flow. Before the removal of the surgical ties, the animals were given one dose of intravenous heparin (100 U/kg) to prevent acute occlusive thrombosis.
Morphometric and Immunohistochemical Analysis
Mice were sacrificed 4 weeks after arterial injury. The femoral arteries were perfusion-fixed in 4% paraformaldehyde and embedded in paraffin. Five 5-μm-thick sections proximal to the arteriotomy site were cut at 50-μm intervals, and stained with combined Mason’s trichrome elastin and hematoxylin-eosin (CME). Each section was digitized and analyzed by computerized morphometry using Image Pro Plus software (Media Cybernetics, Silver Springs, MD) and mean values were obtained for each artery. Measurements included luminal area, medial area, intimal area, vessel area, and the lengths of the internal elastic lamina and external elastic lamina. The I/M ratio was obtained by dividing the intimal area by the medial area.
For immunohistochemical analysis, representative sections were stained for smooth muscle cells (alkaline phosphatase-conjugated monoclonal anti-smooth muscle α-actin, 1:100 dilution; Sigma, St. Louis, MO), macrophages (MOMA-2, rat anti-mouse macrophages/monocytes, 1:400 dilution; Biosource International, Camarillo, CA). Negative controls were prepared by substitution of the primary antibody with an irrelevant antibody of the same isotype.
Statistical Analysis
Data are expressed as mean ± SEM. Comparisons were made using an unpaired Student’s t-test assuming unequal variances. Statistical significance was assigned to P values <0.05.
Results
Generation of a Conditional Transgenic System for Expression of M3
A tetracycline-dependent gene expression system previously described23 was used to generate transgenic mice expressing M3 conditionally. This bigenic system comprises two transgenes: the activator transgene containing the reverse tetracycline-controlled transactivator (rtTA) expressed constitutively and the responder transgene containing the gene of interest under transcriptional control of the tetracycline responsive promoter element (TRE). Transcription of the gene of interest is induced in the presence of tetracycline or DOX. To generate the responder transgenic mice, a bi-directional responder transgene was constructed containing the M3 gene and the LacZ gene encoding β-galactosidase (β-gal) (Figure 1). Nine transgenic founder mice were isolated from which four transgenic lines were derived. No β-gal activity was detected in frozen sections from kidney, liver, muscle, pancreas, and spleen of transgenic mice carrying the responder transgene only, indicating that responder transgene expression was silent in the absence of rtTA.
Figure 1.

Bi-transgenic system for conditional expression of M3. Activator and responder transgenes for conditional expression of the M3 and LacZ genes. CMV/β-actin, CMV enhancer/chicken β-actin promoter; rtTA, reverse tetracycline-controlled transactivator; r β-globin pA, rabbit β-globin polyadenylation signal; SV40, SV40 polyadenylation signal; Pmin, CMV minimal promoter; β-globin pA, β-globin polyadenylation signal.
We have previously described the development of transgenic mice carrying an activator transgene driven by the CMV enhancer/β-actin promoter. These transgenic mice express the activator transgene in multiple tissues.18 Transgenic mice from each of the responder lines described above were crossed to these activator transgenic mice to obtain mice carrying both the activator and responder transgenes (in the following referred to as transgenic mice). β-gal activity was detected by a chemiluminescence-based assay in the kidneys of transgenic mice derived from all four-responder lines 48 hours after a single intraperitoneal injection of 500 μg of DOX. The line with the highest level of induction was selected for further analysis. Tissues from mice receiving DOX were analyzed by histochemical staining. Highest levels of β-galactosidase activity were noted in the liver and kidney, with an intermediate level in the heart. No activity was noted in the noninduced mice, demonstrating that transgenic expression was dependent on DOX (Figure 2).
Figure 2.

DOX-dependent expression of β-gal in tissues from transgenic mice. β-gal staining of fresh frozen tissues prepared from untreated transgenic mice (−DOX) or from transgenic mice 48 hours after a single dose of DOX (500 μg i.p.) (+DOX). β-gal activity is detected in the presence but not the absence of DOX in heart, liver, and kidney. Original magnifications: ×40 [A and B (heart)]; ×100 [C and D (liver), E and F (kidney)].
DOX Treatment Results in High Levels of Circulating M3
Because M3 is abundantly secreted from infected cells in vitro,8 we hypothesized that transgenic expression of M3 in multiple tissues would lead to its presence in the circulation. To test this hypothesis, we collected blood from animals treated with DOX (500 μg i.p.). M3 was detected in the serum of DOX-treated mice by Western blot analysis (Figure 3A). Incubation of serum with radioactive labeled CXCL8 (IL-8), a chemokine known to be bound by M3 with high affinity,6 resulted in a gel shift typical of an M3-IL-8 complex, indicating that serum M3 retains its chemokine-binding activity. The gel shift was inhibited in the presence of increasing concentration of unlabeled CXCL8 (IL-8) (Figure 3B).
Figure 3.
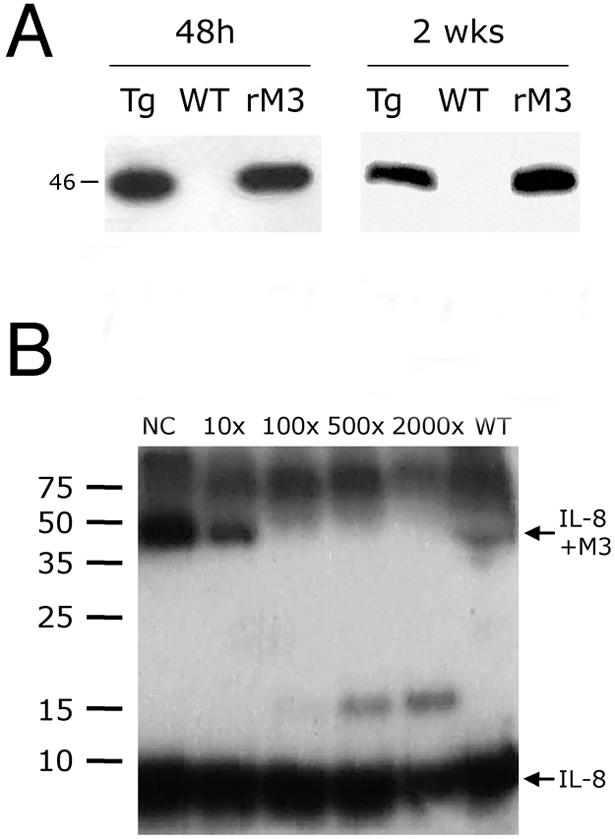
Expression analysis of M3 in blood. A: Western blot of M3 in blood from M3 transgenic mice (Tg) and wild-type littermates (WT). Blood was collected 48 hours after a single dose of DOX (500 μg i.p.) and 2 weeks after a single dose of DOX (500 μg i.p.) and 2 mg/ml in drinking water. rM3, 50 ng of recombinant M3. Molecular mass is shown in kd. B: Chemokine-binding activity of blood from M3 transgenic mice. Blood collected from transgenic mice after DOX treatment was incubated with 125I-IL-8 in the absence (NC) or presence of increasing concentrations of unlabeled IL-8 and a cross-linking agent. An autoradiography of the sodium dodecyl sulfate-polyacrylamide gel electrophoresis analysis with molecular masses in kd is shown.
Short-Term Expression of M3 Is Not Associated with Significant Toxicity
To examine potential M3 toxicity, transgenic mice were treated with DOX (2 mg/ml in drinking water) for 1 month. During treatment, animals appeared normal and maintained normal weight. At necropsy, no gross changes were observed in any organ. No abnormalities were detected on light microscopy of hematoxylin and eosin-stained sections of brain, heart, intestine, kidney, liver, lung, pancreas, peripheral lymph nodes, spleen, and thymus. After 1 month, no abnormality was detected in routine blood chemistries (Table 1). In addition by flow cytometry, the relative and absolute numbers of peripheral blood leukocytes, stained with Gr-1, Mac-1, B220, and CD3 from animals expressing M3 did not differ from that of control animals treated with DOX (Table 2). These results suggest that short-term (1 month) expression of M3 is not overtly toxic to the mice.
Table 1.
Serum Chemistries of Transgenic Mice after DOX Induction
| TAM3 (n = 7) | Reference range | |
|---|---|---|
| ALT | 36.0 ± 33.2 U/L | 24–140 |
| ALP | 171.4 ± 36.2 U/L | 25–222 |
| GGT | 1.3 ± 1.1 U/L | 0–2 |
| Uric acid | 2.2 ± 0.6 mg/L | 2.2–4.6 |
| Amylase | 2058.1 ± 227.3 U/L | 602–2311 |
| CK | 76.1 ± 59.9 U/L | 0–800 |
| LDH | 293.3 ± 135.3 U/L | 260–680 |
| Glucose | 257.6 ± 20.2 mg/L | 124–262 |
| BUN | 34.1 ± 4.7 mg/L | 9–28 |
| Creatinine | 0.37 ± 0.05 mg/L | 0.2–0.7 |
| Cholesterol | 104.0 ± 15.0 mg/L | 46–100 |
| HDL | 83.4 ± 9.1 mg/L | 25–100 |
| Triglyceride | 209.4 ± 85.0 mg/L | 115–155 |
| Bilirubin | 0.19 ± 0.09 mg/L | 0.0–0.9 |
| Albumin | 3.8 ± 0.2 g/L | 2.6–4.6 |
| Protein | 5.1 ± 1.4 g/L | 4.0–6.2 |
| Globulin | 1.7 ± 0.2 g/L | 1.9–3.2 |
Peripheral blood was collected from transgenic (Tg) mice treated with DOX in drinking water (2 mg/mL) for 1 month. All serum chemistries performed were within reference range for mice.
Table 2.
Peripheral Blood Leukocyte Differential of Tg and WT Mice after DOX Induction
| Tg (n = 5) | WT (n = 3) | |
|---|---|---|
| B220 | 38.9 ± 2.6 | 43.1 ± 1.4 |
| CD3 | 31.9 ± 2.7 | 26.9 ± 3.6 |
| Gr-1 | 14.2 ± 1.9 | 12.1 ± 1.4 |
| Mac-1 | 20.5 ± 1.3 | 18.3 ± 1.7 |
Peripheral blood was collected from transgenic (Tg) and wild-type (WT) mice treated with DOX in drinking water (2 mg/mL) for 1 month. By flow cytometry, no difference (P > .05) in peripheral leukocyte numbers (relative numbers) was noted by staining for B220 (B-cell marker), CD3 (T-cell marker), Gr-1 (granulocyte marker), and Mac-1 (granulocyte/macrophage marker).
M3 Inhibits Intimal Hyperplasia after Femoral Arterial Injury
To determine whether the expression of M3 interfered with a physiological response, mice were subjected to bilateral femoral arterial injury. DOX+ mice (n = 8) received DOX 2 days before injury and daily through postoperative day 10, whereas DOX− mice (n = 6) received vehicle only. Similar to previous studies, the injury protocol resulted in complete endothelial denudation in both groups, determined by ICAM-1 staining (data not shown). Fourteen DOX+ and eleven DOX− arteries were used for analysis.
Arterial injury produced a substantial expansion of the intima at 4 weeks in DOX− mice, resulting in an I/M ratio of 0.9 (±0.23), similar to that reported for other studies.16,22 In contrast, DOX treatment resulted in a 67% reduction in intimal area (P < 0.01) and a 68% reduction in I/M ratio (P < 0.05) (Figure 4, A and C). Typical femoral artery sections of DOX+ and DOX− mice 4 weeks after injury are shown in Figure 5, A and B. There were no significant differences in medial area or in total vessel area (Figure 4, B and D). As expected, DOX− arteries demonstrated no β-gal activity. Despite the diminished intimal response to injury, DOX+ arteries showed no β-gal staining, but the surrounding skeletal muscle demonstrated β-gal staining (Figure 5, C and D). As previously described the media and intima stained diffusely for α-actin at 4 weeks in DOX− mice, consistent with the predominance of smooth muscle cells, whereas no macrophages were found in either layer (Figure 5, E and F).
Figure 4.

Morphometric analysis of DOX− (−) and DOX+ (+) femoral arteries 4 weeks after injury. A: Intimal areas in μm2 of DOX− (9308 ± 2116) and DOX+ (3048 ± 913) femoral arteries. B: Medial areas in μm2 of DOX− (11,998 ± 672) and DOX+ (13,339 ± 944) femoral arteries. C: I/M ratio of DOX− (0.90 ± 0.23) and DOX+ (0.29 ± 0.10) femoral arteries. D: Vessel size in μm2 of DOX− (75,093 ± 4755) and DOX+ (66,319 ± 4223). Bars represent mean and SEM (*, P < 0.05; **, P < 0.01).
Figure 5.

Histological analysis of femoral arteries. Combined Mason’s trichrome elastin and hematoxylin-eosin staining of DOX− femoral artery (A) and DOX+ artery (B) 4 weeks after injury. White bars indicate the intimal area. The DOX+ artery shows substantially less intimal hyperplasia when compared to the DOX− artery. β-galactosidase staining of DOX− femoral artery (C) and DOX+ femoral artery (D) 2 days after induction with DOX. Neither DOX− nor DOX+ femoral artery show staining. Note the staining of skeletal muscle in the DOX+ artery. E: DOX− artery 4 weeks after injury showing actin staining in the intima and media (medial area underneath and adjacent to the intimal area shown by the white bar). F: DOX− artery 4 weeks after injury show absence to MOMA staining for macrophages. Original magnifications, ×400 (A, B).
To test if DOX treatment alone affects intimal hyperplasia, we performed arterial injury on littermates missing either the rtTa or the M3 transgene. Five of these mice induced with DOX as per the protocol described previously were injured and sacrificed at 4 weeks. Four other mice not induced with DOX were injured and sacrificed at 4 weeks. Morphometric analysis of seven arteries from the DOX+ group and seven arteries from the DOX− group was performed as described previously. There was no significant difference in media area (DOX+, 10,016 ± 3912; DOX−, 11,258 ± 4404; P = 0.69) or in the I/M ratio (DOX+, 0.84± 0.28; DOX−, 0.88± 0.35; P = 0.78) when the two groups were compared.
Discussion
Chemokines have been increasingly implicated in the pathogenesis of a wide array of diseases.2 In this report, we describe the generation of transgenic mice that conditionally express the chemokine-binding protein M3. Treatment of these animals with DOX resulted in the presence of M3 in the blood. This circulating pool was probably produced by organs such as the liver and kidney, in which β-gal activity was demonstrated. We also show that induction of M3 was associated with a significant blunting of a physiological response, a marked reduction in intimal hyperplasia in the setting of femoral arterial injury. We postulate that this effect was caused by the circulating M3, because the transgene was not expressed in the vascular wall.
M3 has been found to bind C, CC, CXC, and CX3C chemokines with variable affinity.6,7 The molecular interaction of M3 with chemokines has been recently determined.24 The crystal structure of M3 bound to CCL2 (MCP-1) shows that M3 dimerizes and generates a binding site for chemokines that interacts with the N-terminal region of CCL2 (MCP-1) that is involved in binding to CCR2, thus mimicking the chemokine-receptor interaction and masking the receptor-binding site on CCL2 (MCP-1). An independent study using a panel of CXCL8 (IL-8) analogs shows that M3 interacts with the N-terminal region of CXCL8 (IL-8) involved in binding to chemokine receptors and that M3 blocks the interaction of chemokines with glycosaminoglycans, an interaction required for the correct presentation of chemokines to the passing leukocytes and for chemokine activity in vivo.25 Although there are no comprehensive studies on the effects of M3 binding to chemokines, a number of studies have demonstrated that M3 binding is associated with inhibition of chemokine functions, such as mobilization of intracellular calcium, chemotaxis, and leukocyte recruitment.6,7,9
Coronary artery disease is a leading cause of death in the United States and other industrialized nations.26,27 Percutaneous transluminal coronary angioplasty alone or together with implantable stents is now the most commonly used approach for treating occlusive coronary artery disease.28 Restenosis, however, limits the long-term success of percutaneous transluminal coronary angioplasty, necessitating second procedures or coronary bypass surgery. Intimal hyperplasia, because of smooth muscle cell proliferation and migration, is thought to be a major cause of restenosis17 and may also play an important role in the progression of atherosclerotic plaques.29,30
A variety of animal models of arterial injury have been developed to examine the molecular events underlying intimal hyperplasia and to test pharmacological and molecular biological approaches to inhibiting this process.31–35 Many studies have focused on the role of growth factors, cytokines, and intracellular signaling pathways in mediating intimal hyperplasia.36–45 In contrast, only a few have examined the role of chemokines. Targeted deletion of the CCL2 (MCP-1) receptor, CCR2, resulted in an ≈60% reduction in intimal hyperplasia at 28 days in a mouse model of femoral arterial injury.16 In an identical injury model, targeted deletion of CCL2 (MCP-1) resulted in ≈29% reduction in intimal hyperplasia.46 In a rat model of balloon arterial injury, antibodies to CCL2 (MCP-1) reduced intimal hyperplasia by 29 to 40% at 14 days and 56% at 5 weeks.47 These studies suggest that CCL2 (MCP-1) and its receptor mediate in part the intimal response to arterial injury.
In the present study, induction of M3, a nonspecific chemokine-binding protein, decreased intimal hyperplasia by ≈67%. Similarly, intravenous administration of M-T7, a chemokine-binding protein produced by the myxoma virus, resulted in an ≈80% reduction in a rabbit model of intimal hyperplasia.48 These studies support a role for chemokines in mediating intimal hyperplasia. Both M3 and M-T7 bind to CCL2 (MCP-1) and recent data suggests that M3 masks the binding site of CCL2 (MCP-1)24 and may thus inhibit binding to its receptor, CCR2. Although it is likely that part of the inhibitory effects of M3 and M-T7 are mediated by CCL2 (MCP-1), the more pronounced effect on intimal hyperplasia seen with these nonspecific inhibitors as well as in the CCR2-deficient mice suggests that other chemokines may be involved. Such chemokines may include ligands for CCR2 other than CCL2 (MCP-1). We think that it is unlikely that DOX itself contributed to the decreased intimal hyperplasia observed in mice expressing M3. In two studies, DOX treatment had no effect on the development of intimal hyperplasia.49,50 In a recent study, DOX treatment was associated with a modest decrease in I/M ratio 28 days after rat aortic injury (1.67 to 1.36).51 However, in these animals, there was no significant decrease in the intimal area. In this study, we did not find any effect of DOX treatment on intimal area or on I/M ratio.
In contrast to a previous report,22 we did not observe significant inflammatory cell adherence to the vessel lumen 1 hour after injury (data not shown). In this study, we used a model of vascular injury that restores normal blood flow to the injured segment and may thus simulate a more physiological condition. In the previous model, interruption or at least significant reduction of blood flow in the injured segment of the vessel caused by surgical ligation may have promoted inflammatory cell adherence to the vessel wall.
Macrophages were not seen in the vessel wall 28 days after injury in either DOX+ or DOX− animals. This is consistent with previously published reports of murine femoral arterial injury models, in which leukocyte infiltration was not seen 2 hours, 1 day, 5 days, and 28 days after injury.22,31 Although it remains possible that transient influx and efflux of leukocytes may play a role, the effect of chemokines on intimal hyperplasia may be independent of their ability to attract inflammatory cells to sites of injury. Although several studies have suggested that CCL2 (MCP-1) causes smooth muscle cell proliferation,52–54 one study has suggested that it may be inhibitory.55 We have recently found that I-30956 and eotaxin (R Kodali, W Kim, M Gazdiou, and MB Taubman; unpublished observations) cause chemotaxis of human aortic smooth muscle cells. In addition, CCL2 (MCP-1), CCL4 (MIP-1β), and CXCL12 (SDF-1α) induce expression of the procoagulant molecule tissue factor (TF) in arterial smooth muscle cells by a protein kinase C-dependent mechanism.57–59 Thus, chemokines may have direct effects on arterial smooth muscle cells that contribute to intimal hyperplasia. TF has also been implicated in mediating intimal hyperplasia,60 raising the possibility that the effect of chemokines may be secondary to their ability to induce TF. Although TF antigen is abundant in the intima 28 days after arterial injury, no difference was seen by immunostaining in DOX+ and DOX− mice (data not shown). It remains possible that important differences in TF expression might be present at earlier time points. Additional studies will be necessary to establish the mechanism(s) underlying the effect of M3 and the role of chemokines in intimal hyperplasia.
It is also possible that chemokines mediate intimal hyperplasia through effects on cells distant from the site of injury. Recent studies have suggested that cells derived from the bone marrow contribute to intimal hyperplasia in models of vascular injury.61,62 It is interesting to speculate that chemokines may direct bone marrow-derived cells to the vascular wall. There are precedents that support this notion. In models of ischemia-reperfusion, bone-marrow stem cells can be mobilized to the heart where they differentiate into myocytes.63 These stem cells, which express chemokine receptors,64 may be directed to the heart by a chemokine-dependent mechanism. Furthermore using a rat model of vascular injury, Fujiyama and colleagues65 suggest that bone marrow cells of the monocyte-lineage are directed in a CCL2 (MCP-1)-dependent manner to the vascular wall where they differentiate into endothelial cells. Similarly, chemokines that are up-regulated in the vessel wall after injury may mobilize bone marrow-derived cells to sites of injury, and differentiation of these cells into a smooth muscle cell phenotype may contribute to the neointima. Thus, although speculative at this point, M3 may inhibit intimal hyperplasia by blocking the targeting of bone marrow-derived cells to sites of vascular injury.
The induction of M3 was not associated with obvious toxic effects, such as change in coat appearance, social behavior, eating pattern, or weight loss. In addition, there were no major effects on peripheral blood counts or basic chemistries. M3 thus has the potential to be an effective short-term therapeutic agent with minimal toxicity.
In conclusion, we have generated a conditional transgenic system for the evaluation of animals expressing M3. In this model, production of M3 did not result in overt toxicity throughout a 30-day induction but generated levels of M3 that inhibited a pathophysiological response. Chemokines are important regulators of the immune system and have been linked to a variety of cardiovascular, inflammatory, pulmonary, malignant, and infectious diseases. Many of these diseases involve ensembles of chemokines and therefore may not be responsive or may be suboptimally responsive to blockade of a single chemokine or its receptor. For the same reason, studies involving targeted deletions of a single chemokine or its receptor in disease models may fail to establish a role for chemokines. By producing a protein that binds a wide variety of chemokines, our transgenic mouse model provides an important new resource for examining the role of chemokines and the efficacy of chemokine blockade in a broad variety of diseases.
Acknowledgments
We thank L. Johnson, B. Wilburn, and P. Zalamea for expert technical assistance.
Footnotes
Address reprint requests to Sergio A. Lira, Mount Sinai School of Medicine, Immunobiology Center, 1425 Madison Ave., Box 1630, New York, NY 10029-6574. E-mail: sergio.lira@mssm.edu.
Supported in part by the National Institutes of Health (grant HL61818 to M.B.T.) and the Irene Diamond Fund (to S.A.L., an Irene Diamond Associate Professor of Immunology).
R.P. and K.K.J. contributed equally to this work.
References
- Rossi D, Zlotnik A. The biology of chemokines and their receptors. Annu Rev Immunol. 2000;18:217–242. doi: 10.1146/annurev.immunol.18.1.217. [DOI] [PubMed] [Google Scholar]
- Luster AD. Chemokines—chemotactic cytokines that mediate inflammation. N Engl J Med. 1998;338:436–445. doi: 10.1056/NEJM199802123380706. [DOI] [PubMed] [Google Scholar]
- Gerard C, Rollins BJ. Chemokines and disease. Nat Immunol. 2001;2:108–115. doi: 10.1038/84209. [DOI] [PubMed] [Google Scholar]
- Lalani AS, Barrett JW, McFadden G. Modulating chemokines: more lessons from viruses. Immunol Today. 2000;21:100–106. doi: 10.1016/s0167-5699(99)01556-x. [DOI] [PubMed] [Google Scholar]
- Seet BT, McFadden G. Viral chemokine-binding proteins. J Leukoc Biol. 2002;72:24–34. [PubMed] [Google Scholar]
- Parry CM, Simas JP, Smith VP, Stewart CA, Minson AC, Efstathiou S, Alcami A. A broad spectrum secreted chemokine binding protein encoded by a herpesvirus. J Exp Med. 2000;191:573–578. doi: 10.1084/jem.191.3.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Berkel V, Barrett J, Tiffany HL, Fremont DH, Murphy PM, McFadden G, Speck SH, Virgin HI. Identification of a gammaherpesvirus selective chemokine binding protein that inhibits chemokine action. J Virol. 2000;74:6741–6747. doi: 10.1128/jvi.74.15.6741-6747.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Berkel V, Preiter K, Virgin HWT, Speck SH. Identification and initial characterization of the murine gammaherpesvirus 68 gene M3, encoding an abundantly secreted protein. J Virol. 1999;73:4524–4529. doi: 10.1128/jvi.73.5.4524-4529.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen KK, Chen SC, Hipkin RW, Wiekowski MT, Schwarz MA, Chou CC, Simas JP, Alcami A, Lira SA. Disruption of CCL21-induced chemotaxis in vitro and in vivo by M3, a chemokine-binding protein encoded by murine gammaherpesvirus 68. J Virol. 2003;77:624–630. doi: 10.1128/JVI.77.1.624-630.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narumi S, Kaburaki T, Yoneyama H, Iwamura H, Kobayashi Y, Matsushima K. Neutralization of IFN-inducible protein 10/CXCL10 exacerbates experimental autoimmune encephalomyelitis. Eur J Immunol. 2002;32:1784–1791. doi: 10.1002/1521-4141(200206)32:6<1784::AID-IMMU1784>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM. Chemokines in neurological trauma models. Ann NY Acad Sci. 2002;961:346–349. doi: 10.1111/j.1749-6632.2002.tb03120.x. [DOI] [PubMed] [Google Scholar]
- Lohmann T, Laue S, Nietzschmann U, Kapellen TM, Lehmann I, Schroeder S, Paschke R, Kiess W. Reduced expression of Th1-associated chemokine receptors on peripheral blood lymphocytes at diagnosis of type 1 diabetes. Diabetes. 2002;51:2474–2480. doi: 10.2337/diabetes.51.8.2474. [DOI] [PubMed] [Google Scholar]
- Nicoletti F, Conget I, Di Mauro M, Di Marco R, Mazzarino MC, Bendtzen K, Messina A, Gomis R. Serum concentrations of the interferon-gamma-inducible chemokine IP-10/CXCL10 are augmented in both newly diagnosed type I diabetes mellitus patients and subjects at risk of developing the disease. Diabetologia. 2002;45:1107–1110. doi: 10.1007/s00125-002-0879-5. [DOI] [PubMed] [Google Scholar]
- Riffo-Vasquez Y, Spina D. Role of cytokines and chemokines in bronchial hyperresponsiveness and airway inflammation. Pharmacol Ther. 2002;94:185–211. doi: 10.1016/s0163-7258(02)00217-6. [DOI] [PubMed] [Google Scholar]
- Hofmann N, Lachnit N, Streppel M, Witter B, Neiss WF, Guntinas-Lichius O, Angelov DN. Increased expression of ICAM-1, VCAM-1, MCP-1, and MIP-1a by spinal perivascular phagocytes during experimental allergic encephalomyelitis in rats. BMC Immunol. 2002;3:11. doi: 10.1186/1471-2172-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roque M, Kim WJ, Gazdoin M, Malik A, Reis ED, Fallon JT, Badimon JJ, Charo IF, Taubman MB. CCR2 deficiency decreases intimal hyperplasia after arterial injury. Arterioscler Thromb Vasc Biol. 2002;22:554–559. doi: 10.1161/hq0402.105720. [DOI] [PubMed] [Google Scholar]
- Hoffmann R, Mintz GS, Haager PK, Bozoglu T, Grube E, Gross M, Beythien C, Mudra H, vom Dahl J, Hanrath P. Relation of stent design and stent surface material to subsequent in-stent intimal hyperplasia in coronary arteries determined by intravascular ultrasound. Am J Cardiol. 2002;89:1360–1364. doi: 10.1016/s0002-9149(02)02347-0. [DOI] [PubMed] [Google Scholar]
- Wiekowski MT, Chen SC, Zalamea P, Wilburn BP, Kinsley DJ, Sharif WW, Jensen KK, Hedrick JA, Manfra D, Lira SA. Disruption of neutrophil migration in a conditional transgenic model: evidence for CXCR2 desensitization in vivo. J Immunol. 2001;167:7102–7110. doi: 10.4049/jimmunol.167.12.7102. [DOI] [PubMed] [Google Scholar]
- Yang TY, Chen SC, Leach MW, Manfra D, Homey B, Wiekowski M, Sullivan L, Jenh CH, Narula SK, Chensue SW, Lira SA. Transgenic expression of the chemokine receptor encoded by human herpesvirus 8 induces an angioproliferative disease resembling Kaposi’s sarcoma. J Exp Med. 2000;191:445–454. doi: 10.1084/jem.191.3.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan B, Constantini F, Lacy L. Cold Spring Harbor NY: Cold Spring Harbor Laboratory Press,; Manipulating the Mouse Embryo. 1986 [Google Scholar]
- Soares HD, Chen SC, Morgan JI. Differential and prolonged expression of Fos-lacZ and Jun-lacZ in neurons, glia, and muscle following sciatic nerve damage. Exp Neurol. 2001;167:1–14. doi: 10.1006/exnr.2000.7558. [DOI] [PubMed] [Google Scholar]
- Roque M, Fallon JT, Badimon JJ, Zhang WX, Taubman MB, Reis ED. Mouse model of femoral artery denudation injury associated with the rapid accumulation of adhesion molecules on the luminal surface and recruitment of neutrophils. Arterioscler Thromb Vasc Biol. 2000;20:335–342. doi: 10.1161/01.atv.20.2.335. [DOI] [PubMed] [Google Scholar]
- Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci USA. 1992;89:5547–5551. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander JM, Nelson CA, van Berkel V, Lau EK, Studts JM, Brett TJ, Speck SH, Handel TM, Virgin HW, Fremont DH. Structural basis of chemokine sequestration by a herpesvirus decoy receptor. Cell. 2002;111:343–356. doi: 10.1016/s0092-8674(02)01007-3. [DOI] [PubMed] [Google Scholar]
- Webb LM, Clark-Lewis I, Alcami A. The gammaherpesvirus chemokine binding protein binds to the N terminus of CXCL8. J Virol. 2003;77:8588–8592. doi: 10.1128/JVI.77.15.8588-8592.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusuf S, Reddy S, Ounpuu S, Anand S. Global burden of cardiovascular diseases: part I: general considerations, the epidemiologic transition, risk factors, and impact of urbanization. Circulation. 2001;104:2746–2753. doi: 10.1161/hc4601.099487. [DOI] [PubMed] [Google Scholar]
- Yusuf S, Reddy S, Ounpuu S, Anand S. Global burden of cardiovascular diseases: part II: variations in cardiovascular disease by specific ethnic groups and geographic regions and prevention strategies. Circulation. 2001;104:2855–2864. doi: 10.1161/hc4701.099488. [DOI] [PubMed] [Google Scholar]
- Ishihara M, Inoue I, Kawagoe T, Shimatani Y, Kurisu S, Nishioka K, Kouno Y, Umemura T, Nakamura S. Fifteen-year trend in the treatment and outcome of acute myocardial infarction in Japan. Circ J. 2002;66:178–181. doi: 10.1253/circj.66.178. [DOI] [PubMed] [Google Scholar]
- Zhdanov VS, Sternby NH, Drobkova IP, Galakhov IE. Hyperplasia of coronary intima in young males in relation to development of coronary heart disease in adults. Int J Cardiol. 2000;76:57–64. doi: 10.1016/s0167-5273(00)00369-7. [DOI] [PubMed] [Google Scholar]
- Krus S, Turjman MW, Flejka E. Comparative morphology of the hepatic and coronary artery walls. Part II. The relation between the internal elastic membrane, non-atherosclerotic intimal thickening and atherosclerosis. Med Sci Monit. 2000;6:249–252. [PubMed] [Google Scholar]
- Sata M, Maejima Y, Adachi F, Fukino K, Saiura A, Sugiura S, Aoyagi T, Imai Y, Kurihara H, Kimura K, Omata M, Makuuchi M, Hirata Y, Nagai R. A mouse model of vascular injury that induces rapid onset of medial cell apoptosis followed by reproducible neointimal hyperplasia. J Mol Cell Cardiol. 2000;32:2097–2104. doi: 10.1006/jmcc.2000.1238. [DOI] [PubMed] [Google Scholar]
- Walsh K. Building a better mouse model. J Mol Cell Cardiol. 2000;32:1921–1922. doi: 10.1006/jmcc.2000.1248. [DOI] [PubMed] [Google Scholar]
- van Erven L, Velema E, Bos AN, Post MJ, Borst C. Thrombogenicity and intimal hyperplasia after conventional and thermal balloon dilation in normal rabbit iliac arteries. J Vasc Res. 1992;29:426–434. doi: 10.1159/000158961. [DOI] [PubMed] [Google Scholar]
- Seto M, Yano K, Sasaki Y, Azuma H. Intimal hyperplasia enhances myosin phosphorylation in rabbit carotid artery. Exp Mol Pathol. 1993;58:1–13. doi: 10.1006/exmp.1993.1001. [DOI] [PubMed] [Google Scholar]
- Bonan R, Paiement P, Leung TK. Swine model of coronary restenosis: effect of a second injury. Cathet Cardiovasc Diagn. 1996;38:44–49. doi: 10.1002/(SICI)1097-0304(199605)38:1<44::AID-CCD10>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Bauters C, Meurice T, Hamon M, McFadden E, Lablanche JM, Bertrand ME. Mechanisms and prevention of restenosis: from experimental models to clinical practice. Cardiovasc Res. 1996;31:835–846. [PubMed] [Google Scholar]
- Newby AC, Zaltsman AB. Molecular mechanisms in intimal hyperplasia. J Pathol. 2000;190:300–309. doi: 10.1002/(SICI)1096-9896(200002)190:3<300::AID-PATH596>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Rectenwald JE, Moldawer LL, Huber TS, Seeger JM, Ozaki CK. Direct evidence for cytokine involvement in neointimal hyperplasia. Circulation. 2000;102:1697–1702. doi: 10.1161/01.cir.102.14.1697. [DOI] [PubMed] [Google Scholar]
- Ding H, Wang R, Marcel R, Fisher DZ. Adenovirus-mediated expression of a truncated PDGFbeta receptor inhibits thrombosis and neointima formation in an avian arterial injury model. Thromb Haemost. 2001;86:914–922. [PubMed] [Google Scholar]
- Leppanen O, Janjic N, Carlsson MA, Pietras K, Levin M, Vargeese C, Green LS, Bergqvist D, Ostman A, Heldin CH. Intimal hyperplasia recurs after removal of PDGF-AB and -BB inhibition in the rat carotid artery injury model. Arterioscler Thromb Vasc Biol. 2000;20:E89–E95. doi: 10.1161/01.atv.20.11.e89. [DOI] [PubMed] [Google Scholar]
- Trieu VN, Narla RK, Myers DE, Uckun FM. EGF-genistein inhibits neointimal hyperplasia after vascular injury in an experimental restenosis model. J Cardiovasc Pharmacol. 2000;35:595–605. doi: 10.1097/00005344-200004000-00013. [DOI] [PubMed] [Google Scholar]
- Feldman LJ, Aguirre L, Ziol M, Bridou JP, Nevo N, Michel JB, Steg PG. Interleukin-10 inhibits intimal hyperplasia after angioplasty or stent implantation in hypercholesterolemic rabbits. Circulation. 2000;101:908–916. doi: 10.1161/01.cir.101.8.908. [DOI] [PubMed] [Google Scholar]
- Smith JD, Bryant SR, Couper LL, Vary CP, Gotwals PJ, Koteliansky VE, Lindner V. Soluble transforming growth factor-beta type II receptor inhibits negative remodeling, fibroblast transdifferentiation, and intimal lesion formation but not endothelial growth. Circ Res. 1999;84:1212–1222. doi: 10.1161/01.res.84.10.1212. [DOI] [PubMed] [Google Scholar]
- Sidway AN, Hakim FS, Jones BA, Norberto JM, Neville RF, Korman LY. Insulin-like growth factor-I binding in injury-induced intimal hyperplasia of rabbit aorta. J Vasc Surg. 1996;23:308–313. doi: 10.1016/s0741-5214(96)70275-6. [DOI] [PubMed] [Google Scholar]
- Hayashi K, Takahata H, Kitagawa N, Kitange G, Kaminogo M, Shibata S. N-acetylcysteine inhibited nuclear factor-kappaB expression and the intimal hyperplasia in rat carotid arterial injury. Neurol Res. 2001;23:731–738. doi: 10.1179/016164101101199252. [DOI] [PubMed] [Google Scholar]
- Kim WJ, Chereshnev I, Gazdoiu M, Fallon JT, Rollins BJ, Taubman MB. MCP-1 deficiency is associated with reduced intimal hyperplasia after arterial injury. Biochem Biophys Res Commun. 2003;310:936–942. doi: 10.1016/j.bbrc.2003.09.088. [DOI] [PubMed] [Google Scholar]
- Furukawa Y, Matsumori A, Ohashi N, Shioi T, Ono K, Harada A, Matsushima K, Sasayama S. Anti-monocyte chemoattractant protein-1/monocyte chemotactic and activating factor antibody inhibits neointimal hyperplasia in injured rat carotid arteries. Circ Res. 1999;84:306–314. doi: 10.1161/01.res.84.3.306. [DOI] [PubMed] [Google Scholar]
- Liu L, Lalani A, Dai E, Seet B, Macauley C, Singh R, Fan L, McFadden G, Lucas A. The viral anti-inflammatory chemokine-binding protein M-T7 reduces intimal hyperplasia after vascular injury. J Clin Invest. 2000;105:1613–1621. doi: 10.1172/JCI8934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mano T, Luo Z, Suhara T, Smith RC, Esser S, Walsh K. Expression of wild-type and noncleavable Fas ligand by tetracycline-regulated adenoviral vectors to limit intimal hyperplasia in vascular lesions. Hum Gene Ther. 2000;11:1625–1635. doi: 10.1089/10430340050111287. [DOI] [PubMed] [Google Scholar]
- Petrik PV, Gelabert HA, Moore WS, Quinones-Baldrich W. The effect of vitamin E and doxycycline on the development of intimal hyperplasia. J Surg Res. 1996;60:279–283. doi: 10.1006/jsre.1996.0043. [DOI] [PubMed] [Google Scholar]
- Bendeck MP, Conte M, Zhang M, Nili N, Strauss BH, Farwell SM. Doxycycline modulates smooth muscle cell growth, migration, and matrix remodeling after arterial injury. Am J Pathol. 2002;160:1089–1095. doi: 10.1016/S0002-9440(10)64929-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viedt C, Vogel J, Athanasiou T, Shen W, Orth SR, Kubler W, Kreuzer J. Monocyte chemoattractant protein-1 induces proliferation and interleukin-6 production in human smooth muscle cells by differential activation of nuclear factor-kappaB and activator protein-1. Arterioscler Thromb Vasc Biol. 2002;22:914–920. doi: 10.1161/01.atv.0000019009.73586.7f. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Pakala R, Katagiri T, Benedict CR. Monocyte chemotactic protein 1 amplifies serotonin-induced vascular smooth muscle cell proliferation. J Vasc Res. 2001;38:341–349. doi: 10.1159/000051065. [DOI] [PubMed] [Google Scholar]
- Porreca E, Di Febbo C, Reale M, Castellani ML, Baccante G, Barbacane R, Conti P, Cuccurullo F, Poggi A. Monocyte chemotactic protein 1 (MCP-1) is a mitogen for cultured rat vascular smooth muscle cells. J Vasc Res. 1997;34:58–65. doi: 10.1159/000159202. [DOI] [PubMed] [Google Scholar]
- Ikeda U, Okada K, Ishikawa S, Saito T, Kasahara T, Shimada K. Monocyte chemoattractant protein 1 inhibits growth of rat vascular smooth muscle cells. Am J Physiol. 1995;268:H1021–H1026. doi: 10.1152/ajpheart.1995.268.3.H1021. [DOI] [PubMed] [Google Scholar]
- Haque NS, Fallon JT, Pan JJ, Taubman MB, Harpel PC. Chemokine receptor-8 (CCR8) mediates human vascular smooth muscle cell chemotaxis and metalloproteinase-2 secretion. Blood. 2003;15:1296–1304. doi: 10.1182/blood-2002-05-1480. [DOI] [PubMed] [Google Scholar]
- Schecter AD, Rollins BJ, Zhang YJ, Charo IF, Fallon JT, Rossikhina M, Giesen PL, Nemerson Y, Taubman MB. Tissue factor is induced by monocyte chemoattractant protein-1 in human aortic smooth muscle and THP-1 cells. J Biol Chem. 1997;272:28568–28573. doi: 10.1074/jbc.272.45.28568. [DOI] [PubMed] [Google Scholar]
- Schecter AD, Calderon TM, Berman AB, McManus CM, Fallon JT, Rossikhina M, Zhao W, Christ G, Berman JW, Taubman MB. Human vascular smooth muscle cells possess functional CCR5. J Biol Chem. 2000;275:5466–5471. doi: 10.1074/jbc.275.8.5466. [DOI] [PubMed] [Google Scholar]
- Schecter AD, Berman AB, Yi L, Mosoian A, McManus CM, Berman JW, Klotman ME, Taubman MB. HIV envelope gp120 activates human arterial smooth muscle cells. Proc Natl Acad Sci USA. 2001;98:10142–10147. doi: 10.1073/pnas.181328798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roque M, Reis ED, Fuster V, Padurean A, Fallon JT, Taubman MB, Chesebro JH, Badimon JJ. Inhibition of tissue factor reduces thrombus formation and intimal hyperplasia after porcine coronary angioplasty. J Am Coll Cardiol. 2000;36:2303–2310. doi: 10.1016/s0735-1097(00)01018-4. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Sata M, Hirata Y, Nagai R. Diverse contribution of bone marrow cells to neointimal hyperplasia after mechanical vascular injuries. Circ Res. 2003;93:783–790. doi: 10.1161/01.RES.0000096651.13001.B4. [DOI] [PubMed] [Google Scholar]
- Sata M, Saiura A, Kunisato A, Tojo A, Okada S, Tokuhisa T, Hirai H, Makuuchi M, Hirata Y, Nagai R. Hematopoietic stem cells differentiate into vascular cells that participate in the pathogenesis of atherosclerosis. Nat Med. 2002;8:403–409. doi: 10.1038/nm0402-403. [DOI] [PubMed] [Google Scholar]
- Orlic D, Kajstura J, Chimenti S, Limana F, Jakoniuk I, Quaini F, Nadal-Ginard B, Bodine DM, Leri A, Anversa P. Mobilized bone marrow cells repair the infarcted heart, improving function and survival. Proc Natl Acad Sci USA. 2001;98:10344–10349. doi: 10.1073/pnas.181177898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapidot T, Petit I. Current understanding of stem cell mobilization: the roles of chemokines, proteolytic enzymes, adhesion molecules, cytokines, and stromal cells. Exp Hematol. 2002;30:973–981. doi: 10.1016/s0301-472x(02)00883-4. [DOI] [PubMed] [Google Scholar]
- Fujiyama S, Amano K, Uehira K, Yoshida M, Nishiwaki Y, Nozawa Y, Jin D, Takai S, Miyazaki M, Egashira K, Imada T, Iwasaka T, Matsubara H. Bone marrow monocyte lineage cells adhere on injured endothelium in a monocyte chemoattractant protein-1-dependent manner and accelerate reendothelialization as endothelial progenitor cells. Circ Res. 2003;93:980–989. doi: 10.1161/01.RES.0000099245.08637.CE. [DOI] [PubMed] [Google Scholar]