

Abstract
We previously reported that the compound O-2093 is a selective inhibitor of the reuptake of the endocannabinoid anandamide (AEA). We have now re-examined the activity of O-2093 in vivo and synthesized four structural analogs (O-2247, O-2248, O-3246, and O-3262), whose activity was assessed in: (a) binding assays carried out with membranes from cells overexpressing the human CB1 and CB2 receptors; (b) assays of transient receptor potential of the vanilloid type-1 (TRPV1) channel functional activity (measurement of [Ca2+]i); (c) [14C]AEA cellular uptake and hydrolysis assays in rat basophilic leukaemia (RBL-2H3) cells; (d) the mouse ‘tetrad' tests (analgesia on a hot plate, immobility on a ‘ring', rectal hypothermia and hypolocomotion in an open field); and (e) the limb spasticity test in chronic relapsing experimental allergic encephalomyelitis (CREAE) mice, a model of multiple sclerosis (MS).
O-2093, either synthesized by us or commercially available, was inactive in the ‘tetrad' up to a 20 mg kg−1 dose (i.v.). Like O-2093, the other four compounds exhibited low affinity in CB1 (Ki from 1.3 to >10 μM) and CB2 binding assays (1.3<Ki< 8 μM), low potency and efficacy in a TRPV1 functional assay (EC50>10 μM), very low potency as fatty acid amide hydrolase (FAAH) inhibitors (IC50>25 μM) and were inactive in the ‘tetrad' up to a 30 mg kg−1 dose (i.v.).
While O-2247 and O-2248 were poor inhibitors of [14C]AEA cellular uptake (IC50>40 μM), O-3246 and O-3262 were quite potent in this assay. O-3246, which exhibits only a very subtle structural difference with O-2093, is the most potent inhibitor of AEA uptake reported in vitro under our experimental conditions (IC50=1.4 μM) and is 12-fold more potent than O-2093.
When injected intravenously O-3246 and O-3262, again like O-2093 and unlike O-2247 and O-2248, significantly inhibited limb spasticity in mice with CREAE.
These data confirm the potential utility of selective AEA uptake inhibitors as anti-spasticity drugs in MS and, given the very subtle chemical differences between potent and weak inhibitors of uptake, support further the existence of a specific mechanism for this process.
Keywords: Cannabinoid, endocannabinoid, 2-arachidonoylglycerol, membrane transport, spasticity, multiple sclerosis
Introduction
Endocannabinoids are defined as endogenous agonists of the G-protein-coupled receptors for Cannabis psychoactive principle Δ9-tetrahydrocannabinol. To date, two such receptors have been cloned, the CB1 and CB2 cannabinoid receptors (Howlett, 2002, for a review), and, together with endocannabinoids, they have been implicated in several physiological and pathological conditions in mammals (Di Marzo et al., 2004a; Howlett et al., 2004, for reviews). Like all endogenous mediators, the levels of the two major endocannabinoids, arachidonoylethanolamide (anandamide) (AEA) (Devane et al., 1992) and 2-arachidonoylglycerol (2-AG) (Mechoulam et al., 1995; Sugiura et al., 1995) are regulated by specific biosynthetic and degradative reactions catalyzed by enzymes that have been recently characterized and cloned (Di Marzo et al., 2004a). AEA and 2-AG are produced ‘on demand' following cell stimulation, immediately released to act upon cannabinoid receptors and ultimately inactivated via cellular reuptake and intracellular hydrolysis. The possible existence of a plasma membrane protein mediating both endocannabinoid release after de novo biosynthesis, and endocannabinoid reuptake following activation of CB1 and CB2 cannabinoid receptors has been investigated and is still controversial (Di Marzo et al., 1994; Glaser et al., 2003; Fegley et al., 2004; Ligresti et al., 2004; Oddi et al., 2005; and Hillard & Jarrahian, 2003; McFarland & Barker, 2004 for reviews). Nevertheless, many substances have been developed that are capable of selectively inhibiting the cellular reuptake of AEA (Lopez-Rodriguez et al., 2003; Ortar et al., 2003) without interfering with other proteins nor with the major enzyme involved in the degradation of this compound, fatty acid amide hydrolase (FAAH) (Cravatt et al., 1996). In some cases, the capability of a compound to inhibit AEA reuptake is dramatically impaired by introducing very subtle changes in its chemical structure, such as inversion of the stereochemistry of a chiral center (Piomelli et al., 1999; Di Marzo et al., 2004b; Ligresti et al., 2004), thus supporting the existence of a specific protein to facilitate this process. Among the potential therapeutic effects shared by ‘direct' agonists of CB1 cannabinoid receptors and inhibitors of endocannabinoid reuptake (which by elevating endocannabinoid levels, act as ‘indirect' agonists of these receptors) (Di Marzo et al., 2004a), one of the most promising is the capability of inhibiting limb spasticity in mice with chronic relapsing experimental allergic encephalomyelitis (CREAE), a model of multiple sclerosis (MS) (Baker et al., 2000; 2001; de Lago et al., 2004).
We have previously shown that the chemical modification of arvanil, a nonselective, albeit relatively potent, AEA uptake inhibitor with high potency against spasticity in CREAE mice (Melck et al., 1999; Brooks et al., 2002), yields compounds that still selectively inhibit the uptake process without exhibiting two other properties typical of the starting compound, that is activation of both CB1 cannabinoid receptors and, particularly, the transient receptor potential vanilloid type 1 (TRPV1) channel (Di Marzo et al., 2001; 2002). One of these compounds, O-2093 (Figure 1) served as the lead compound for the present study. Apart from inhibiting AEA uptake, O-2093 was inactive at CB1 and TRPV1 receptors and did not significantly inhibit FAAH. In the present study, we have designed and synthesized four structural analogs of O-2093 (Figure 1) with the aim of increasing its efficacy at the putative AEA membrane transporter. Through the chemical modification of O-2093, we have systematically increased property to inhibit AEA cellular uptake, thus obtaining the most potent inhibitor of AEA reuptake thus far reported under our experimental conditions. Finally, we confirm that potent inhibitors of AEA transport across the cell membrane are also very efficacious inhibitors of spasticity in the CREAE model of MS, with no apparent cannabimimetic side effects.
Figure 1.

Chemical structures of O-2093 and its four novel analogs.
Methods
Synthesis of compounds
The synthesis of analogs of O-2093 (MW=616.70) was carried out in the same manner as we reported for the synthesis of O-2093 (Di Marzo et al., 2002). For the synthesis of O-2247 and O-2248 (Figure 1, MW=619.15 and 605.12, respectively), the appropriate secondary amines were synthesized from 4-chlorobenzaldehyde by reductive alkylation with 2,4-dichlorobenzylamine or 2,4-dichloroaniline, respectively (triacetoxyborohydride/acetic acid/CH2CL2) in 56–62% yield (Abdel-Magid et al., 1996). Condensation of these secondary amines with the acid chloride of arachidonic acid formed O-2247 and O-2248. O-3246 and O-3262 (Figure 1, MW=616.70 and 547.81.12, respectively) were similarly synthesized from the corresponding secondary amines. In the case of O-3262, the (1-(3-dimethylaminopropyl)-3-ethylcarbodiimide·HCl) EDCI/4-dimethyl-amino-pyridine (DMAP) procedure was used for condensation with arachidonic acid rather than the acid chloride procedure. All compounds were characterized on the basis of their [1H] nuclear magnetic resonance spectra (run on a Jeol Eclipse 300 MHz) and elemental analyses (Figure 1). O-2093 was also purchased from Tocris, Bristol, U.K. After purification, the compounds were >95% pure on the basis of nuclear magnetic resonance (NMR), thin layer chromatography (TLC), mass spectrometric and elemental analyses. They were stable if kept under nitrogen and in the freezer (−20°C).
Assay of AEA cellular reuptake
The effect of compounds on the uptake of [14C]AEA by rat basophilic leukemia (RBL-2H3) cells was studied by using 2.4 μM (10,000 c.p.m.) of [14C]AEA as described previously (Bisogno et al., 1997). Cells were incubated with [14C]AEA for 5 min at 37°C, in the presence or absence of varying concentrations of the inhibitors. Residual [14C]AEA in the incubation medium after extraction with CHCl3/CH3OH 2 : 1 (by volume), determined by scintillation counting of the lyophilized organic phase, was used as a measure of the AEA that was taken up by cells (De Petrocellis et al., 2000). Previous studies (Bisogno et al., 1997) had shown that after a 5-min incubation the amount of [14C]AEA that disappeared from the medium of RBL-2H3 cells is found mostly (>90%) as unmetabolized [14C]AEA in the cell extract. Nonspecific binding of [14C]AEA to cells and plastic dishes was determined in the presence of 100 μM AEA and was never higher than 30%. Data are expressed as the concentration exerting 50% inhibition of AEA uptake (IC50) calculated by GraphPad®.
Assay of FAAH
The effect of compounds on the enzymatic hydrolysis of AEA was studied as described previously (Bisogno et al., 1997), using membranes prepared from RBL-2H3 cells, incubated with the test compounds and [14C]AEA (2.4 μM) in 50 mM Tris-HCl, pH 9, for 30 min at 37°C. [14C]Ethanolamine produced from [14C]AEA hydrolysis was measured by scintillation counting of the aqueous phase after extraction of the incubation mixture with two volumes of CHCl3/CH3OH 2 : 1 (by volume). Data are expressed as the concentration exerting 50% inhibition of AEA hydrolysis (IC50), calculated by GraphPad.
Assay of functional activity at TRPV1 receptors
Overexpression of human TRPV1 cDNA into human embryonic kidney (HEK) 293 cells was carried out as described previously (Hayes et al., 2000). Cells were grown as monolayers in minimum essential medium supplemented with nonessential amino acids, 10% fetal calf serum, and 0.2 mM glutamine and maintained under 95% O2/5% CO2 at 37°C. The effect of the substances on [Ca2+]i was determined by using Fluo-3 (Molecular Probes, Eugene, OR, U.S.A.), a selective intracellular fluorescent probe for Ca2+ (De Petrocellis et al., 2000; Smart et al., 2000). Cells were transferred into six-well dishes coated with poly-L-lysine (Sigma, Milan, Italy) 1 day prior to experiments and grown in the culture medium mentioned above. On the day of the experiment, the cells (50–60,000 per well) were loaded for 2 h at 25°C with 4 μM Fluo-3 methylester in dimethyl sulfoxide containing 0.04% Pluoronic. After loading, cells were washed with Tyrode's solution, pH=7.4, trypsinized, resuspended in Tyrode's solution, and transferred to the cuvette of the fluorescence detector (PerkinElmer LS50B) under continuous stirring. Experiments were carried out by measuring cell fluorescence at 25°C (λEX=488 nm, λEM=540 nm) before and after the addition of the test compounds at various concentrations. The efficacy of the effect was determined by comparing it to the analogous effect observed with 4 μM ionomycin. In some cases, the experiments were repeated in the presence of the TRPV1 antagonist capsazepine, at a concentration (20 nM) sufficient to block the effect of 100 nM capsaicin.
Assay of binding activity at CB1 and CB2 receptors
For the CB1 receptor, [3H]CP-55,940 (KD=690 pM) binding to P2 membranes was conducted as described previously, except whole rat brain (rather than cortex only) was used. Displacement curves were generated by incubating drugs with 1 nM of [3H]CP-55,940. The assays were performed in triplicate, and the results represent the combined data from three individual experiments. CB2 receptor binding was carried out in Chinese hamster ovary (CHO) cells that were maintained in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal clone II (HyClone, Logan UT, U.S.A.) and 5% CO2 at 37°C in a Forma incubator. Cell lines were created by transfection of CB2pcDNA3 into CHO cells by the Lipofectamine reagent (Life Technologies, Gaithersburg, MD, U.S.A.). The human CB2 cDNA was provided by Dr Sean Munro (MRC, Cambridge, U.K.). Stable transformants were selected in growth medium containing geneticin (1 mg ml−1, reagent, Life Technologies, Gaithersburg, MD, U.S.A.). Cells were harvested in phosphate-buffered saline containing 1 mM EDTA and centrifuged at 500 × g. The cell pellet was homogenized in 10 ml of solution A (50 mM Tris-HCl, 320 mM sucrose, 2 mM EDTA, 5 mM MgCl2, pH 7.4). The homogenate was centrifuged at 1600 × g (10 min), the supernatant saved, and the pellet washed three times in solution A with subsequent centrifugation. The combined supernatants were centrifuged at 100,000 × g (60 min). The (P2 membrane) pellet was resuspended in 3 ml of buffer B (50 mM Tris-HCl, 1 mM EDTA, 3 mM MgCl2, pH 7.4) to yield a protein concentration of approximately 1 mg ml−1. The tissue preparation was divided into equal aliquots, frozen on dry ice, and stored at −70°C. Binding was initiated by the addition of 40–50 μg membrane protein to silanized tubes containing [3H]CP-55,940 (102.9 Ci mmol−1) and a sufficient volume of buffer C (50 mM Tris-HCl, 1 mM EDTA, 3 mM MgCl2, and 5 mg ml−1 fatty acid free bovine serum albumin (BSA), pH 7.4) to bring the total volume to 0.5 ml. The addition of 1 μM unlabeled CP-55,940 was used to assess nonspecific binding. Following incubation (30°C for 1 h), binding was terminated by the addition of 2 ml of ice-cold buffer D (50 mM Tris-HCl, pH 7.4, plus 1 mg ml−1 BSA) and rapid vacuum filtration through Whatman GF/C filters (pretreated with polyethyleneimine [0.1%] for at least 2 h). Tubes were rinsed with 2 ml of ice-cold buffer D, which was also filtered, and the filters subsequently rinsed twice with 4 ml of ice-cold buffer D. Before radioactivity was quantitated by liquid scintillation spectrometry, filters were shaken for 1 h in 5 ml of scintillation fluid.
Pharmacological effects in mice
Cannabinoids were dissolved in a mixture of ethanol (5%), Emulphor (North American Chemicals, Cranbury, NJ, U.S.A.) (5%), and saline (90%) for i.v. administration. The analogs were administered to male ICR mice by tail-vein injection and evaluated for their ability to produce hypomotility, hypothermia, antinociception and ring immobility. This route of administration limits pharmacokinetic problems associated with first pass metabolism and lipophilicity of compounds. Prior to testing in any of the tetrad procedures, mice were acclimated to the experimental setting (ambient temperature 22–24°C) for at least 1 h. Subsequently, mice were injected with one of the compounds or with O-3262 or vehicle followed 5 min later by an injection of AEA (potentiation tests). Each mouse was tested in two procedures (either spontaneous activity and tail flick or rectal temperature and ring immobility). Tail-flick latency or rectal temperature was measured at 4 min after the last injection. At 1 min after measurement of antinociception or rectal temperature, mice were placed in individual activity chambers where spontaneous activity was measured for 10 min or they were placed on the ring immobility apparatus for 5 min. Preinjection control values were determined for rectal temperature and tail-flick latency (in s). Activity was measured as total number of interruptions of 16 photocell beams per chamber during the 10-min test and expressed as % inhibition of activity of the vehicle group. Maximum latency of 10 s was used for the tail flick test. Antinociception was calculated as percent of maximum possible effect (%MPE), as in formula below. Control latencies typically ranged from 1.5 to 4.0 s. Rectal temperature was measured with a telethermometer (Yellow Springs Instrument Co., Yellow Springs, OH, U.S.A.) and a thermistor probe (model YSI 400; Markson LabSales Inc., Hillsboro, OR, U.S.A.) inserted at a depth of 2 mm and was expressed as the difference between control temperature (before injection) and temperature following drug administration. During placement on the ring immobility apparatus, the total amount of time (in s) that the mouse remained motionless was measured. This value was divided by 300 s and multiplied by 100 to obtain a percent immobility rating. The criterion for ring immobility was the absence of all voluntary movement, including snout and whisker movement. Given their poor CB1 and CB2 cannabinoid receptor binding affinities, the four analogs of O-2093 were evaluated at a single dose of 30 mg kg−1 (n=5–6 mice per test). O-2093 was evaluated over a dose range of 3–20 mg kg−1.
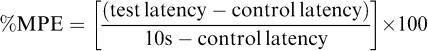 |
Assay of limb spasticity in CREAE mice
Spasticity was induced in ABH mice following the induction of experimental allergic encephalomyelitis (EAE) using syngenic spinal cord homogenate in Freund's adjuvant on day 0 and 7. Mice exhibited relapsing-remitting episodes of paralysis and spasticity developed typically after 2–3 relapses, at about 80–100 days postinduction as described previously (Baker et al., 2000). Spasticity was assessed by measuring the force required for hindlimb flexion against a strain gauge prior to and following the administration of compound (Baker et al., 2000). These were injected intravenously in the same vehicle as above, in order to limit pharmacokinetic problems associated with first pass metabolism and lipophilicity of cannabinoids, and observed when using other administration routes that may interfere with speed and level of efficacy, as has been reported previously (Baker et al., 2000). Spasticity, resulting from accumulating neurological deficit is associated with limb stiffness and was measured during remission from active paralytic attacks, where limbs lack functional movement and exhibit weak resistance to flexion (Baker et al., 2000). There is significant variation between the degree of spasticity between individual limbs and animals, which makes direct comparison between different groups difficult (Baker et al., 2000). Therefore, the forces from individual limbs were assessed, pairwise, using Analysis of Variance tests (Baker et al., 2001).
Results
Effect of O-2093 and its analogs on [14C]AEA uptake by RBL-2H3 cells
When coincubated with [14C]AEA and intact RBL-2H3 cells, where an active transport of AEA across the cell membrane has been described (Bisogno et al., 1997), O-2093 inhibited the cellular uptake of the radiolabeled compound with an IC50=17.3±2.0 μM (Figure 2), very similar to that previously reported by us (11.5±1.3 μM, Di Marzo et al., 2002). On the other hand, O-3246, an isomer of O-2093, was more than 10-fold more potent, exhibiting an IC50=1.4±0.2 μM and a 100% inhibition of uptake at 5 μM (Figure 2). Intriguingly, the analog with no chlorine atoms on both aromatic moieties, that is O-3262, exhibited an IC50 of 2.8±0.3 μM (Figure 2) that was intermediate between O-2093 and O-3246. Finally, the two analogs with chlorine atoms and no hydroxy group on both aromatic moieties, that is O-2247 and O-2248, only exhibited very weak activity against [14C]AEA uptake (IC50 >40 μM, Table 1).
Figure 2.

Effect of O-2093 and its four novel analogs on [14C]AEA uptake by RBL-2H3 cells. Data are means±s.e. of three different experiments.
Table 1.
Summary of the in vitro activity of the O-2093 and its four novel analogs
| Compound | [14C]AEA uptake by RBL-2H3 cells (IC50, μM) | [14C]AEA hydrolysis by RBL-2H3 cell membranes (IC50, μM) | rCB1 (Ki, μM) | hCB2 (Ki, μM) | [Ca2+]i in HEK cells expressing hTRPV1 (max. effect at 10 μM, as % of the effect of 4 μMionomycin) |
|---|---|---|---|---|---|
| O-2093 | 17.3±2.0 | >50 | 1.29±0.11 | 2.38±0.51 | 27.1±2.6* |
| O-3246 | 1.4±0.2 | >50 | 2.69±0.19 | 2.18±0.38 | 18.2±1.4* |
| O-3262 | 2.8±0.3 | >50 | 2.02±0.02 | 1.31±0.12 | 24.4±2.2* |
| O-2247 | 43.3±4.1 | >50 | >10 | 8.04±0.80 | 16.7±1.8 |
| O-2248 | >50 | 27.9±3.4 | >10 | 5.12±0.21 | 8.1±1.1 |
| OMDM-2 | 4.1±0.8 | >50 | 5.10±0.30 | 4.95±1.40 | 31.0±3.2* |
Data are reported as IC50 (μM) for the uptake and hydrolysis assays, Ki (μM) for the binding assays, and as maximal effect (i.e. as % of the effect on calcium observed with 4 μM ionomycin), for the [Ca2+]i assay, and are means±s.e.m. of 3 experiments. Asterisk denotes compounds that were tested in this assay also in the presence of 20 nM of the TRPV1 antagonist capsazepine, with no change in the effect observed. The previously reported (Ortar et al., 2003; de Lago et al., 2004) inhibitor of AEA uptake, OMDM-2, was used as reference compound.
Effect of O-2093 and its analogs on [14C]AEA hydrolysis by RBL-2H3 cells
As reported previously (Di Marzo et al., 2002), O-2093 was inactive (IC50>50 μM) on [14C]AEA hydrolysis, which in this study was tested using membranes from RBL-2H3 cells. All four new O-2093 analogs synthesized for this study were also inactive (IC50>50 μM), except for O-2248, which exhibited some weak activity (IC50=27.9±3.4 μM) (Table 1).
Effect of O-2093 and its analogs on human TRPV1-mediated enhancement of [Ca2+]i
As reported previously (Di Marzo et al., 2002), O-2093 was only very weakly active at enhancing [Ca2+]i in HEK cells transfected with the human recombinant TRPV1 (maximal stimulation at 10 μM was 24.2±2.0% of the effect of 4 μM ionomycin). Likewise, all four new O-2093 analogs synthesized for this study were also very weakly active (maximal stimulation at 10 μM ranged between 8 and 24% of the effect of 4 μM ionomycin) and, at least in the case of O-3246 and O-3262 (i.e. the two most potent compounds on AEA uptake), it was not sensitive to the TRPV1 antagonist, capsazepine, 20 nM) (Table 1).
Effect of O-2093 and its analogs in CB1 and CB2 receptor binding assays and in the mouse ‘tetrad'
Binding affinities for CB1 and CB2 cannabinoid receptors were notably low for O-2093 and for its four analogs. Affinity for the CB1 cannabinoid receptor ranged from 1.29 to >10 μM. As a group, affinities for CB2 cannabinoid receptors were only slightly better, ranging from 1.31 to 8.04 μM. Consistent with their poor binding affinities at CB1 cannabinoid receptors, none of the five compounds showed a complete cannabimimetic profile in the ‘tetrad' of in vivo mouse tests up to a dose of 30 mg kg−1 (data not shown). Unlike in our previous study, O-2093 did not produce antinociceptive or hypothermic effects in ICR mice, regardless of source of the drug (in house synthesis or Tocris Ltd., U.K.). Although O-2093 moderately suppressed spontaneous activity (68% inhibition), it did so only at the 20 mg kg−1 dose. Lack of overt ‘tetrad' effects were also evident in ABH mice when the compounds were tested at up to 5 mg kg−1 i.v. (not shown). In contrast to the lack of effect observed with each compound alone, potentiation of the effects of a submaximal dose of AEA by O-3262 (one of the most potent inhibitors of AEA uptake) was observed for locomotor inhibition and antinociception (Table 2).
Table 2.
Potentiation of cannabimimetic effects of sub-maximal doses of AEA by the AEA uptake inhibitor, O-3262 (30 mg kg−1, i.v.)
| Treatment condition | % Inhibition of locomotion | Antinociception (% MPE) |
|---|---|---|
| Vehicle+vehicle | 0±7.8 | 9±3.0 |
| Vehicle+O-3262, 30 mg kg−1 | 38±10.9 | 19±5.8 |
| Vehicle+AEA, 3 mg kg−1 | — | 32±14.2 |
| Vehicle+AEA, 10 mg kg−1 | 49±7.0 | — |
| O-3262, 30 mg kg−1+AEA, 3 mg kg−1 | — | 83±11.5** |
| O-3262, 30 mg kg−1+AEA, 10 mg kg−1 | 82±3.6* | — |
Data are reported as means±s.e.m.. Asterisks indicate significant difference from vehicle+10 mg kg−1 AEA (*) or from vehicle+3 mg kg−1 AEA (**) (i.e., doses of AEA that were sub-maximal for the measure) as well as from the vehicle+vehicle condition and the vehicle+O-3262 condition (P<0.05 as determined by one-way ANOVA with Newman–Kuels post hoc tests).
Effect of O-2093 and its analogs on limb spasticity in CREAE mice
All compounds except O-2247 and O-2248, tested at the dose of 1 mg kg−1 (i.v.), exerted a strong inhibition of the force required to bend individual spastic limbs to full flexion against a strain gauge. The effect was maximal (30.3–32.7%) after 30 min from administration (Table 3). As the basal level of spasticity can vary significantly among individual mice, and several (10–15) mice are therefore required to obtain reliable data even for one dose, it was not normally possible to perform a full dose–response curve for all compounds. However, a lower dose of O-2093 (0.05 mg kg−1 i.v.) remained active, thus suggesting that the higher doses used may have been supra-optimal and accounting for a relative lack of discrimination between O-3246, O-3242 O-2093 in this in vivo assay (Table 3).
Table 3.
Effect of O-2093 and its four novel analogs on limb spasticity in mice with CREAE
| Compound (dose) | n | Time from administration | Mean resistance to flexion force(N)±s.e. | P value |
|---|---|---|---|---|
| O–2247 (1 mg kg−1 i.v) | 7 | Baseline | 0.237±0.020 | |
| +10 min | 0.249±0.027 | NS | ||
| +30 min | 0.221±0.018 | NS | ||
| +60 min | 0.249±0.031 | NS | ||
| O–2248 (1 mg kg−1 i.v) | 10 | Baseline | 0.258±0.034 | |
| +10 min | 0.252±0.031 | NS | ||
| +30 min | 0.274±0.033 | NS | ||
| +60 min | 0.255±0.019 | NS | ||
| O–3246 (1 mg kg−1 i.v) | 15 | Baseline | 0.250±0.018 | |
| +10 min | 0.240±0.017 | NS | ||
| +30 min | 0.174±0.013 | P<0.001 | ||
| +60 min | 0.173±0.011 | P<0.001 | ||
| O–3262 (1 mg kg−1 i.v) | 14 | Baseline | 0.208±0.011 | |
| +10 min | 0.153±0.007 | P<0.001 | ||
| +30 min | 0.140±0.008 | P<0.001 | ||
| +60 min | 0.153±0.009 | P<0.001 | ||
| O–2093 (1 mg kg−1 i.v) | 13 | Baseline | 0.261±0.033 | |
| +10 min | 0.199±0.023 | P=0.005 | ||
| +30 min | 0.182±0.022 | P<0.001 | ||
| O–2093 (0.05 mg kg−1 i.v) | 13 | Baseline | 0.172±0.048 | |
| +10 min | 0.117±0.045 | P<0.001 | ||
| +30 min | 0.111±0.037 | P<0.001 | ||
| +60 min | 0.095±0.033 | P<0.001 |
Following the development of spasticity during the course of experimental allergic encephalomyelitis, the force required to bend individual spastic limbs to full flexion against a strain gauge was assessed. Compounds were injected at the dose indicated and recordings following drug administration were compared to baseline levels, using repeated measures Analysis of Variance. NS=non-significant (P>0.05).
Discussion
Previous investigations have shown that inhibitors of endocannabinoid cellular uptake are capable of exerting strong activity in each of the four tests of the mouse ‘tetrad' of behavioral actions (analgesia on a ‘hot plate', immobility on a ‘ring', rectal hypothermia and hypolocomotion in an ‘open field' (Martin et al., 1991)) only when they also exert strong functional activity at cannabinoid CB1 receptors or at vanilloid TRPV1 channels (Di Marzo et al., 2001; 2002; Wiley & Martin, 2003; de Lago et al., 2004). One exception appeared to be O-2093, an endocannabinoid uptake inhibitor previously developed by us, which exerted weak or no activity at cannabinoid and TRPV1 receptors and yet exhibited activity in the mouse ‘tetrad' (Di Marzo et al., 2002). In the present study, we modified the chemical structure of O-2093 with the aim of increasing its potency at the putative AEA membrane transporter. The first surprising finding of this study, however, was that O-2093, either obtained from a commercial source or resynthesized by us, did not produce cannabimimetic activity in the four behavioral tests carried out in mice, as it had in our previous study (Di Marzo et al., 2002). This new finding has now been confirmed several times since the publication of that original study. Since in all cases: (1) O-2093 was always >95% pure after its preparation, and stable for up to 4 months when kept at −20°; and (2) the assays were carried out exactly under the same conditions of administration, it is unlikely that this discrepancy is due to impurities present in the first batch and absent in the batches used in the present study, or to pharmacokinetic factors. A possible explanation is that in the previous study, where we assessed the activity of eight structurally similar compounds, all of which were active in the ‘tetrad' assays, an unfortunate exchange of samples may have occurred during the preparation of O-2093 aliquots for storage. Another possibility is that the batch used in the previous study may have been degraded to active compounds immediately before its testing. For these reasons, we repeated in the present study also the in vitro assays originally performed with O-2093 (Di Marzo et al., 2002). In this case, we found similar results, except for a slightly lower potency in the uptake assay. In conclusion, we can confirm here that the compound is an inhibitor of endocannabinoid uptake with little or no affinity for cannabinoid CB1 and CB2 receptors, nor activity at TRPV1 vanilloid receptors and FAAH. Importantly, the affinity of O-2093 at CB2 receptors had not been evaluated in our first study with this compound (Di Marzo et al., 2002).
The most interesting data from this study, however, concern four O-2093 analogs that we have synthesized here, some of which show striking pharmacological features. All new compounds share with O-2093 very weak, if any, activity as FAAH inhibitors, CB1 and CB2 ligands, TRPV1 agonists, or in the mouse ‘tetrad' of tests. However, two of these derivatives, O-3246 and O-3262, exhibited potent activity as inhibitors of AEA cellular uptake. Interestingly, in O-3246 the simple shift of the two chlorine atoms in the two aromatic moieties of O-2093 from orto to meta to the 4-hydroxy-groups, results in an 12-fold increase in potency in this assay, without causing any other significant change in the other assays carried out in this study. Indeed, O-3246, with an IC50 of 1.4 μM, is the most potent inhibitor of AEA cellular uptake ever found using our experimental conditions. In fact, it has been demonstrated by many authors that the potency and efficacy of endocannabinoid uptake inhibitors depends very much on the experimental set-up of the assay (see e.g. Bisogno et al., 2001; Fowler et al., 2004). Not only the position but also the presence of the chlorine atoms on the two aromatic moieties of O-2093 seems to be important for activity against the putative membrane transporter responsible for AEA uptake. In fact, O-3262, which differs from the previous two analogs by having no chlorine atoms, also exhibited potency sixfold higher than O-2093, although twofold lower than O-3246. Therefore, in order for these analogs to inhibit AEA uptake efficaciously, they must have the chlorine atoms meta to the 4-hydroxy groups, or not have them at all. On the other hand, the 4-hydroxy groups on the two aromatic moieties are necessary for activity since the two O-2093 analogs that lack these groups, and have chlorine atoms instead, that is O-2247 and O-2248, are significantly less active than O-2093. This observation is in agreement with previous structure-activity studies on arvanil (Di Marzo et al., 2001; 2002), which showed that the 4-hydroxy group in other analogs of this compound are fundamental for both their activity as AEA uptake inhibitors and as TRPV1 agonists.
The finding that, in a complex molecule such as O-2093, the simple structural change leading to O-3246 causes a dramatic increase in the potency for the inhibition of AEA uptake can be considered as further indirect evidence for the presence of one or more specific proteins facilitating this process, and AEA transport across the cell membrane in general. Furthermore, the observation that both O-3246 and O-3262 exert little, if any, inhibition of AEA intracellular hydrolysis, which in RBL-2H3 cells is catalyzed by FAAH, confirm recent evidence (Fegley et al., 2004; Ligresti et al., 2004; Ortega-Gutierrez et al., 2004) that this latter protein is not solely responsible for AEA cellular uptake, as instead had been proposed by earlier studies (Glaser et al., 2003). Clearly, by virtue of its selectivity for AEA uptake and its lack of ‘central' cannabimimetic activity (as assessed here in the mouse ‘tetrad' of tests), O-3246 and O-3262 can serve as templates for the development of a new generation of potent endocannabinoid uptake inhibitors with several potential therapeutic applications (see Di Marzo et al., 2004a for review). Importantly, when coadministered with inactive doses of AEA, O-3262 was capable of rendering this endocannabinoid active at inhibiting nociception and locomotor behavior, in agreement with the action of this compound as an inhibitor of AEA inactivation also in vivo.
Baker et al. (2001) discovered that inhibitors of endocannabinoid cellular uptake have the capability of inhibiting limb spasticity in mice with CREAE, a model of MS. This observation has been confirmed with six different uptake inhibitors, that is AM404 and arvanil (which may also act via TRPV1 receptors (Baker et al., 2001; Brooks et al., 2002)), VDM-11, OMDM1 and OMDM2 (de Lago et al., 2004), and UCM707 (de Lago et al., 2005). Here, we have also evaluated the effects of O-2093 and its four new analogs on limb spasticity, and found that only those compounds which inhibited AEA cellular uptake exerted a strong inhibitory action on spasticity. We did not observe any real significant difference in efficacy in this assay between the least and more potent uptake inhibitors studied here, that is O-2093 and O-3246. All three active compounds could yield similar effects, with maximal reductions in spasticity maximal after 30 min from administration ranging from 30.3 to 32.7% of control, and persistent up to 60 min. Although caution is required when comparing results in different groups of animals as the level of spasticity can vary significantly between animals (Baker et al., 2000), this level of inhibition is similar to that reported previously with cannabinoid receptor agonists (Baker et al., 2000; Brooks et al., 2002). Indeed, a lower dose of O-2093 (0.05 mg kg−1) still exerts a 34% reduction in spasticity 30 min after administration, thus indicating that these three compounds may be active as antispastic drugs also at lower doses. This suggests that the doses tested here for the other four compounds (1 mg kg−1, i.v.) may have been already at the plateau of the antispasticity effect of the active ones. However, although unlikely given their strong chemical similarities, there may be differences in the pharmacokinetic properties of the differing compounds, which could affect their in vivo efficacy compared to their in vitro potency and may account for the observation that O-3246 appeared to be slower in reducing spasticity compared to O-2093 or O-3262. While, based on the present data, we cannot (and do not intend to) establish a correlation between the potencies of the five compounds as AEA uptake inhibitors and their efficacy as antispastic agents, our findings do support further the use of such inhibitors as templates for the development of new anti-spastic medicaments to be used in MS. The recent finding that AEA uptake inhibitors are effective at decreasing the progress, and not only the signs, of this disorder in animal models (Mestre et al., 2005; Ortega-Gutierrez et al., 2005), widens even more their therapeutic potential in MS.
In conclusion, we have reported here the development of two potent and selective AEA uptake inhibitors and have demonstrated their activity as potential antispastic drugs in MS. Moreover, we have provided further strong, albeit still indirect, evidence for the existence of a specific process responsible for endocannabinoid transport across the cell membrane.
Acknowledgments
The authors acknowledge the Italian Ministry of University and Research (within the FIRB program to VDM), the Multiple Sclerosis Society of Great Britain and Northern Ireland (to DB), the Brain Research Trust (to DB), and the National Institute for Drug Abuse (Grants DA-09789 and DA-08904) for funding this work.
Abbreviations
- AEA
arachidonoylethanolamide (anandamide)
- 2-AG
2-arachidonoylglycerol
- BSA
bovine serum albumin
- CREAE
chronic relapsing experimental allergic encephalomyelitis
- DMAP
4-dimethyl-amino-pyridine
- DMEM
Dulbecco's modified Eagle's medium
- EDCI
1-(3-dimethylaminopropyl)-3-ethylcarbodiimide·HCl
- FAAH
fatty acid amide hydrolase
- HEK
human embryonic kidney
- MPE
maximum possible effect
- MS
multiple sclerosis
- NMR
nuclear magnetic resonance
- RBL-2H3
rat basophilic leukaemia
- TLC
thin layer chromatography
- TRPV1
transient receptor potential of the vanilloid type-1
References
- ABDEL-MAGID A.F., CARSON K.G., HARRIS B.D., MARYANOFF C.A., SHAH R.D. Reductive amination of aldehydes and ketones with sodium triacetoxyborohydride. Studies on direct and indirect reductive amination procedures. J. Org. Chem. 1996;61:3849–3862. doi: 10.1021/jo960057x. [DOI] [PubMed] [Google Scholar]
- BAKER D., PRYCE G., CROXFORD J.L., BROWN P., PERTWEE R.G., HUFFMAN J.W., LAYWARD L. Cannabinoids control spasticity and tremor in a multiple sclerosis model. Nature. 2000;404:84–87. doi: 10.1038/35003583. [DOI] [PubMed] [Google Scholar]
- BAKER D., PRYCE G., CROXFORD J.L., BROWN P., PERTWEE R.G., MAKRIYANNIS A., KHANOLKAR A., LAYWARD L., FEZZA F., BISOGNO T., DI MARZO V. Endocannabinoids control spasticity in a multiple sclerosis model. FASEB J. 2001;15:300–302. doi: 10.1096/fj.00-0399fje. [DOI] [PubMed] [Google Scholar]
- BISOGNO T., MAURELLI S., MELCK D., DE PETROCELLIS L., DI MARZO V. Biosynthesis, uptake, and degradation of anandamide and palmitoylethanolamide in leukocytes. J. Biol. Chem. 1997;272:3315–3323. doi: 10.1074/jbc.272.6.3315. [DOI] [PubMed] [Google Scholar]
- BISOGNO T., MACCARRONE M., DE PETROCELLIS L., JARRAHIAN A., FINAZZI-AGRO A., HILLARD C., DI MARZO V. The uptake by cells of 2-arachidonoylglycerol, an endogenous agonist of cannabinoid receptors. Eur. J. Biochem. 2001;268:1982–1989. doi: 10.1046/j.1432-1327.2001.02072.x. [DOI] [PubMed] [Google Scholar]
- BROOKS J.W., PRYCE G., BISOGNO T., JAGGAR S.I., HANKEY D.J., BROWN P., BRIDGES D., LEDENT C., BIFULCO M., RICE A.S., DI MARZO V., BAKER D. Arvanil-induced inhibition of spasticity and persistent pain: evidence for therapeutic sites of action different from the vanilloid VR1 receptor and cannabinoid CB(1)/CB(2) receptors. Eur. J. Pharmacol. 2002;439:83–92. doi: 10.1016/s0014-2999(02)01369-9. [DOI] [PubMed] [Google Scholar]
- CRAVATT B.F., GIANG D.K., MAYFIELD S.P., BOGER D.L., LERNER R.A., GILULA N.B. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- DE LAGO E., FERNANDEZ-RUIZ J., ORTEGA-GUTIERREZ S., CABRANES A., PRYCE G., BAKER D., LOPEZ-RODRIGUEZ RAMOS J.A.UCM707, an inhibitor of the anandamide uptake, behaves as a symptom control agent in models of Huntington's disease and multiple sclerosis, but fails to delay/arrest the progression of different motor-related disorders Eur. Neuropsychopharmacol. 2005. in press [DOI] [PubMed]
- DE LAGO E., LIGRESTI A., ORTAR G., MORERA E., CABRANES A., PRYCE G., BIFULCO M., BAKER D., FERNANDEZ-RUIZ J., DI MARZO V. In vivo pharmacological actions of two novel inhibitors of anandamide cellular uptake. Eur. J. Pharmacol. 2004;484:249–257. doi: 10.1016/j.ejphar.2003.11.027. [DOI] [PubMed] [Google Scholar]
- DE PETROCELLIS L., BISOGNO T., DAVIS J.B., PERTWEE R.G., DI MARZO V. Overlap between the ligand recognition properties of the anandamide transporter and the VR1 vanilloid receptor: inhibitors of anandamide uptake with negligible capsaicin-like activity. FEBS Lett. 2000;483:52–56. doi: 10.1016/s0014-5793(00)02082-2. [DOI] [PubMed] [Google Scholar]
- DEVANE W.A., HANUS L., BREUER A., PERTWEE R.G., STEVENSON L.A., GRIFFIN G., GIBSON D., MANDELBAUM A., ETINGER A., MECHOULAM R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- DI MARZO V., BIFULCO M., DE PETROCELLIS L.The endocannabinoid system and its therapeutic exploitation Nat. Rev. Drug. Discov. 2004a3771–784.Review [DOI] [PubMed] [Google Scholar]
- DI MARZO V., BISOGNO T., DE PETROCELLIS L., BRANDI I., JEFFERSON R.G., WINCKLER R.L., DAVIS J.B., DASSE O., MAHADEVAN A., RAZDAN R.K., MARTIN B.R. Highly selective CB(1) cannabinoid receptor ligands and novel CB(1)/VR(1) vanilloid receptor ‘hybrid' ligands. Biochem. Biophys. Res. Commun. 2001;281:444–451. doi: 10.1006/bbrc.2001.4354. [DOI] [PubMed] [Google Scholar]
- DI MARZO V., FONTANA A., CADAS H., SCHINELLI S., CIMINO G., SCHWARTZ J.C., PIOMELLI D. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature. 1994;372:686–691. doi: 10.1038/372686a0. [DOI] [PubMed] [Google Scholar]
- DI MARZO V., GRIFFIN G., DE PETROCELLIS L., BRANDI I., BISOGNO T., WILLIAMS W., GRIER M.C., KULASEGRAM S., MAHADEVAN A., RAZDAN R.K., MARTIN B.R. A structure/activity relationship study on arvanil, an endocannabinoid and vanilloid hybrid. J. Pharmacol. Exp. Ther. 2002;300:984–991. doi: 10.1124/jpet.300.3.984. [DOI] [PubMed] [Google Scholar]
- DI MARZO V., LIGRESTI A., MORERA E., NALLI M., ORTAR G. The anandamide membrane transporter. Structure-activity relationships of anandamide and oleoylethanolamine analogs with phenyl rings in the polar head group region. Bioorg. Med. Chem. 2004b;12:5161–5169. doi: 10.1016/j.bmc.2004.07.026. [DOI] [PubMed] [Google Scholar]
- FEGLEY D., KATHURIA S., MERCIER R., LI C., GOUTOPOULOS A., MAKRIYANNIS A., PIOMELLI D. Anandamide transport is independent of fatty-acid amide hydrolase activity and is blocked by the hydrolysis-resistant inhibitor AM1172. Proc. Natl. Acad. Sci. U.S.A. 2004;101:8756–8761. doi: 10.1073/pnas.0400997101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FOWLER C.J., TIGER G., LIGRESTI A., LOPEZ-RODRIGUEZ M.L., DI MARZO V. Selective inhibition of anandamide cellular uptake versus enzymatic hydrolysis – a difficult issue to handle. Eur. J. Pharmacol. 2004;492:1–11. doi: 10.1016/j.ejphar.2004.03.048. [DOI] [PubMed] [Google Scholar]
- GLASER S.T., ABUMRAD N.A., FATADE F., KACZOCHA M., STUDHOLME K.M., DEUTSCH D.G. Evidence against the presence of an anandamide transporter. Proc. Natl. Acad. Sci. U.S.A. 2003;100:4269–4274. doi: 10.1073/pnas.0730816100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAYES P., MEADOWS H.J., GUNTHORPE M.J., HARRIES M.H., DUCKWORTH D.M., CAIRNS W., HARRISON D.C., CLARKE C.E., ELLINGTON K., PRINJHA R.K., BARTON A.J., MEDHURST A.D., SMITH G.D., TOPP S., MURDOCK P., SANGER G.J., TERRETT J., JENKINS O., BENHAM C.D., RANDALL A.D., GLOGER I.S., DAVIS J.B. Cloning and functional expression of a human orthologue of rat vanilloid receptor-1. Pain. 2000;88:205–215. doi: 10.1016/S0304-3959(00)00353-5. [DOI] [PubMed] [Google Scholar]
- HILLARD C.J., JARRAHIAN A.Cellular accumulation of anandamide: consensus and controversy Br. J. Pharmacol. 2003140802–808.Review [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOWLETT A.C.The cannabinoid receptors Prostaglandins Other Lipid Mediat. 200268–69619–631.Review [DOI] [PubMed] [Google Scholar]
- HOWLETT A.C., BREIVOGEL C.S., CHILDERS S.R., DEADWYLER S.A., HAMPSON R.E., PORRINO L.J.Cannabinoid physiology and pharmacology: 30 years of progress Neuropharmacology 200447(Suppl 1)345–358.Review [DOI] [PubMed] [Google Scholar]
- LIGRESTI A., MORERA E., VAN DER STELT M., MONORY K., LUTZ B., ORTAR G., DI MARZO V. Further evidence for the existence of a specific process for the membrane transport of anandamide. Biochem. J. 2004;380:265–272. doi: 10.1042/BJ20031812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOPEZ-RODRIGUEZ M.L., VISO A., ORTEGA-GUTIERREZ S., FOWLER C.J., TIGER G., DE LAGO E., FERNANDEZ-RUIZ J., RAMOS J.A. Design, synthesis, and biological evaluation of new inhibitors of the endocannabinoid uptake: comparison with effects on fatty acid amidohydrolase. J. Med. Chem. 2003;46:1512–1522. doi: 10.1021/jm0210818. [DOI] [PubMed] [Google Scholar]
- MARTIN B.R., COMPTON D.R., THOMAS B.F., PRESCOTT W.R., LITTLE P.J., RAZDAN R.K., JOHNSON M.R., MELVIN L.S., MECHOULAM R., WARD S.J. Behavioral, biochemical, and molecular modeling evaluations of cannabinoid analogs. Pharmacol. Biochem. Behav. 1991;40:471–478. doi: 10.1016/0091-3057(91)90349-7. [DOI] [PubMed] [Google Scholar]
- MCFARLAND M.J., BARKER E.L.Anandamide transport Pharmacol. Ther. 2004104117–135.Review [DOI] [PubMed] [Google Scholar]
- MECHOULAM R., BEN-SHABAT S., HANUS L., LIGUMSKY M., KAMINSKI N.E., SCHATZ A.R., GOPHER A., ALMOG S., MARTIN B.R., COMPTON D.R. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem. Pharmacol. 1995;50:83–90. doi: 10.1016/0006-2952(95)00109-d. [DOI] [PubMed] [Google Scholar]
- MELCK D., BISOGNO T., DE PETROCELLIS L., CHUANG H., JULIUS D., BIFULCO M., DI MARZO V. Unsaturated long-chain N-acyl-vanillyl-amides (N-AVAMs): vanilloid receptor ligands that inhibit anandamide-facilitated transport and bind to CB1 cannabinoid receptors. Biochem. Biophys. Res Commun. 1999;262:275–284. doi: 10.1006/bbrc.1999.1105. [DOI] [PubMed] [Google Scholar]
- MESTRE L., CORREA F., AREVALO-MARTIN A., MOLINA-HOLGADO E., VALENTI M., ORTAR G., DI MARZO V., GUAZA C. Pharmacological modulation of the endocannabinoid system in a viral model of multiple sclerosis. J. Neurochem. 2005;92:1327–1339. doi: 10.1111/j.1471-4159.2004.02979.x. [DOI] [PubMed] [Google Scholar]
- ODDI S., BARI M., BATTISTA N., BARSACCHI D., COZZANI I., MACCARRONE M. Confocal microscopy and biochemical analysis reveal spatial and functional separation between anandamide uptake and hydrolysis in human keratinocytes. Cell. Mol. Life Sci. 2005;62:386–395. doi: 10.1007/s00018-004-4446-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ORTAR G., LIGRESTI A., DE PETROCELLIS L., MORERA E., DI MARZO V. Novel selective and metabolically stable inhibitors of anandamide cellular uptake. Biochem. Pharmacol. 2003;65:1473–1481. doi: 10.1016/s0006-2952(03)00109-6. [DOI] [PubMed] [Google Scholar]
- ORTEGA-GUTIERREZ S., HAWKINS E.G., VISO A., LOPEZ-RODRIGUEZ M.L., CRAVATT B.F. Comparison of anandamide transport in FAAH wild-type and knockout neurons: evidence for contributions by both FAAH and the CB1 receptor to anandamide uptake. Biochemistry. 2004;43:8184–8190. doi: 10.1021/bi049395f. [DOI] [PubMed] [Google Scholar]
- ORTEGA-GUTIERREZ S., MOLINA-HOLGADO E., ARÉVALO-MARTÍN A., CORREA F., VISO A., LOPEZ-RODRIGUEZ M.L., DI MARZO V., GUAZA C. Activation of the endocannabinoid system as therapeutic approach in a murine model of multiple sclerosis. FASEB J. 2005;19:1338–1340. doi: 10.1096/fj.04-2464fje. [DOI] [PubMed] [Google Scholar]
- PIOMELLI D., BELTRAMO M., GLASNAPP S., LIN S.Y., GOUTOPOULOS A., XIE X.Q., MAKRIYANNIS A. Structural determinants for recognition and translocation by the anandamide transporter. Proc. Natl. Acad. Sci. U.S.A. 1999;96:5802–5807. doi: 10.1073/pnas.96.10.5802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SMART D., GUNTHORPE M.J., JERMAN J.C., NASIR S., GRAY J., MUIR A.I., CHAMBERS J.K., RANDALL A.D., DAVIS J.B. The endogenous lipid anandamide is a full agonist at the human vanilloid receptor (hVR1) Br. J. Pharmacol. 2000;129:227–230. doi: 10.1038/sj.bjp.0703050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUGIURA T., KONDO S., SUKAGAWA A., NAKANE S., SHINODA A., ITOH K., YAMASHITA A., WAKU K. 2-Arachidonoylglycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem. Biophys. Res. Commun. 1995;215:89–97. doi: 10.1006/bbrc.1995.2437. [DOI] [PubMed] [Google Scholar]
- WILEY J.L., MARTIN B.R. Cannabinoid pharmacological properties common to other centrally acting drugs. Eur. J. Pharmacol. 2003;471:185–193. doi: 10.1016/s0014-2999(03)01856-9. [DOI] [PubMed] [Google Scholar]