

Introduction
The drive for the development of more cost-effective, expeditious, and environmentally benign synthetic transformations for the production of fine chemicals, pharmaceuticals, and agrochemicals provides the impetus for the development of new catalytic processes. A central theme in our program over the last few years has been the development of new metal-catalyzed cross-coupling and carbocyclization reactions.1–4 The major highlight of this work has been the realization that these transformations may be combined to furnish new multicomponent processes involving inter- and intramolecular components for the construction of polycyclic systems.5
Background
The allylic alkylation is a powerful synthetic transformation that has been extensively studied using a wide range of transition metal complexes.6,7 The enantioselective metal-catalyzed process is particularly pertinent; however, the major limitation with this transformation is the necessity to employ substrates that furnish symmetrical π-allyl intermediates, to circumvent problems associated with regiochemical infidelity. Although there are reports of regioselective allylic alkylation with acyclic 3-substituted propenyl derivatives, useful selectivity is, for the most part, limited to electronically biased aryl substituents.6,8 Furthermore, the alkylation of unsymmetrical chiral nonracemic allylic alcohols generally leads to considerable racemization through π-σ-π isomerization due to the fluxional or rapidly equilibrating nature of π-allyl intermediates.9
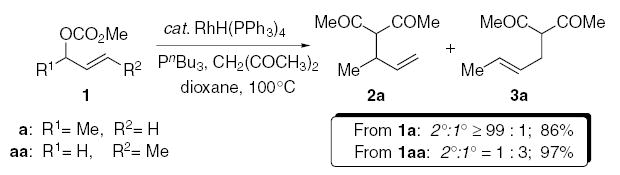
Tsuji and co-workers demonstrated the regiospecific rhodium-catalyzed allylic alkylation, using stabilized carbon nucleophiles (Eq. 1).10 The allylic alkylation of the branched allylic carbonate 1a with 2,4-pentanedione in the presence of a rhodium(I) complex, furnished the branched allylic alkylation product 2a in excellent yield (ds ≥99:1), whereas the isomeric carbonate 1aa, under analogous conditions, gave the alternative regioisomer 3a as the major product. In light of this unusual specificity, we anticipated that this process could also be stereospecific, and thereby circumvent many of the inherent problems associated with this type of transformation.
We demonstrated that the rhodium-catalyzed allylic substitution is indeed highly stereospecific, which resulted in the development of the most versatile allylic substitution reaction for acyclic unsymmetrical allylic alcohol derivatives described to date.11 The following details an account of the major advances in this area, the development of new carbocyclization reactions, and the manner in which we succeeded in combining these transformations in tandem reaction sequences.
Stereospecific Rhodium-Catalyzed Allylic Substitution Reactions
A general assessment of the mechanism of the metal-catalyzed allylic alkylation indicates that the reaction could, in principle, either proceed through a configurationally stable enyl12,13 ii or a distorted π-allyl iii organometallic species, as depicted in Figure 1. We expected that regioselectivity in this type of transformation could be improved by adjusting the electronic environment and/or coordination sphere of the metal, and that the stereospecificity could be maintained by suppressing the rate of isomerization of the initial enyl or metal-allyl intermediate relative to SN2’ substitution (i.e., for i k2 ≫ k1 and for ent-i k3 ≫ k−1). Although the dynamics of this isomerization are complex, the ability to control the rate of isomerization provides the key to the development of a stereospecific metal-catalyzed allylic substitution reaction.
Figure 1.
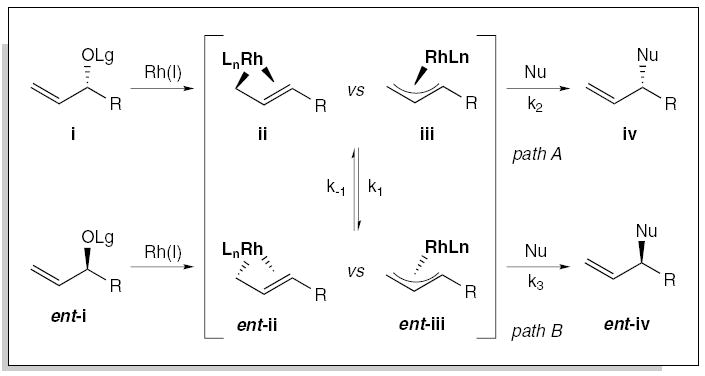
Mechanistic hypothesis for stereospecific metal-catalyzed allylic substitution.
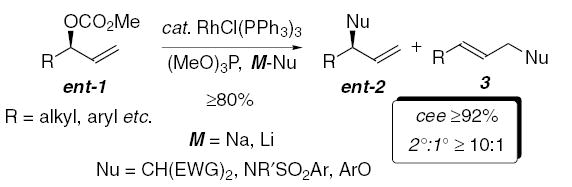
Preliminary studies demonstrated the in situ modification of Wilkinson’s catalyst with trimethylphosphite furnished a catalyst that facilitates the allylic substitution with excellent regioselectivity and retention of absolute configuration, for a variety of enantiomerically enriched acyclic allylic carbonates (Eq. 2).1–3 This transformation is consistent with the formation of a configurationally stable distorted π-allyl or enyl11 (σ + π) organorhodium intermediate.1b The allylic substitution is tolerant of substituents that have historically proven problematic; for example, linear and branched alkyl, aryl, and the hydroxymethyl group with various protecting groups may be utilized without significantly compromising selectivity. Moreover, the allylic substitutions are always highly enantiospecific, irrespective of the regioselectivity.2a,14
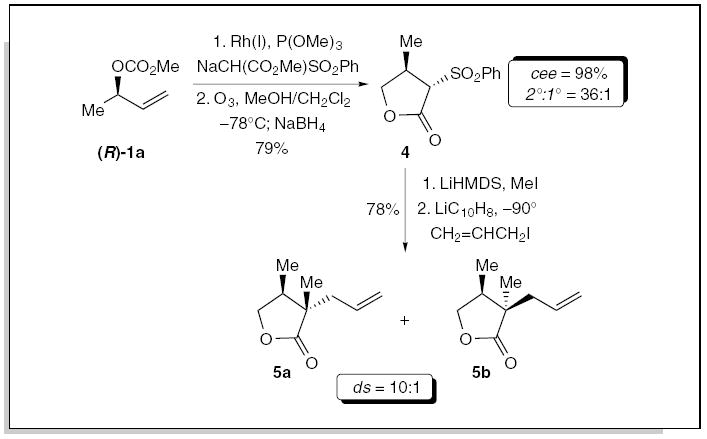
The stereospecific allylic substitution with stabilized carbon nucleophiles was highlighted by the development of a novel method for the construction of α,β-ternary-quaternary substituted carbon stereogenic centers, using a γ-lactone template (Scheme 1). The rhodium-catalyzed allylic alkylation with the sodium salt of methyl phenylsulfonylacetate, followed by reductive ozonolysis of the allylic alkylation product, afforded the γ-lactone 4 in 79% overall yield as the major diastereoisomer.1c Alkylation of 4 with methyl iodide, followed by the reductive formation of the enolate with lithium naphthalenide to facilitate the installation of the allyl group, furnished the γ-lactones 5a/b in 78% overall yield, as a 10:1 mixture of diastereoisomers favoring 5a. This provides the basis for a versatile method for the construction of α,β-ternary-quaternary stereogenic centers through the selective introduction of groups at the α- and β-positions of the γ-lactone.
Scheme 1.
The stereocontrolled construction of acyclic anti-1,3-carbon stereogenic centers, including C2-symmetrical fragments, represents a fundamentally important process for target directed synthesis. Although a variety of excellent synthetic strategies have been devised to address this problem, we were the first to demonstrate the ability to control 1,3-carbon stereogenic centers using sequential enantiospecific metal-catalyzed allylic substitution reactions with unsymmetrical acyclic chiral nonracemic secondary allylic alcohol derivatives.1d Furthermore, the desymmetrization of C2-symmetrical 1,3-carbon stereogenic centers provides an expeditious route to stereotetrads, as exemplified in Scheme 2.
Scheme 2.
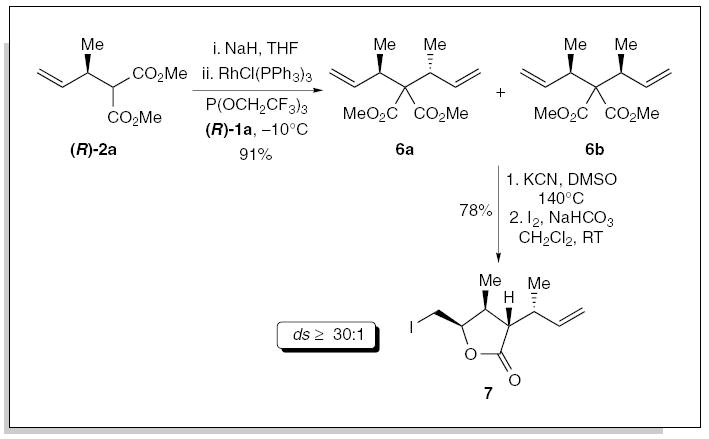
Preliminary studies focused on the selective formation of 6a. Rhodium-catalyzed allylic alkylation of (R)-2a (93% ee) using trifluoroethyl phosphite at −10°C, furnished the alkylation product 6a in 91% yield, with optimum selectivity (2°:1° = 24:1; ds = 26:1). The improved selectivity was tentatively attributed to the increased π-acidicity of the trifluoroalkyl phosphite ligand. Krapcho decarboxylation15 and in situ saponification of the diester 6a furnished the pseudo-C2-symmetrical carboxylic acid, which was then desymmetrized via an iodolactonization to furnish 7 in 78% yield (ds ≥30:1).16 We have also combined this type of allylic alkylation reaction, albeit using unsaturated α-substituted malonates, with ring-closing metathesis for the direct construction of carbocycles.1e,17
Rhodium-Catalyzed Allylic Amination
The development of the regio- and enantiospecific metal-catalyzed allylic amination of unsymmetrical acyclic allylic alcohol derivatives with alkyl and aryl sulfonamides provides a versatile and general allylic amination reaction.18 Preliminary studies demonstrated that while the nature of the alkali metal salt was important, the amination is tolerant to a wide array of substituents.2a This in turn prompted the development of a stereospecific approach to trans- and cis-2,5-disubstituted pyrrolines, as outlined in Scheme 3.2b
Scheme 3.
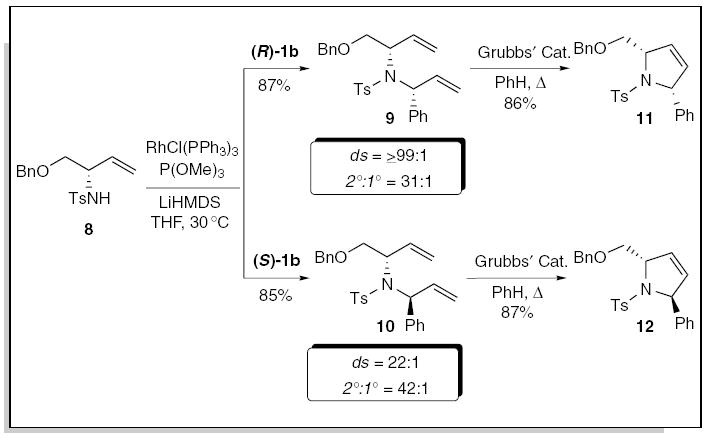
Diastereospecific rhodium-catalyzed allylic amination with the enantiomerically enriched allylic carbonates (S)- and (R)-1b (R = Ph) using the lithium anion of 8 furnished the dienes 9 and 10 in 85% and 87% yield, favoring the branched products. Interestingly, the matched alkylation with (R)-1b proceeded with excellent stereospecificity (ds ≥99:1), while the analogous alkylation with the mismatched allylic carbonate (S)-1b gave lower, but nonetheless useful selectivity (ds = 22:1). Treatment of the dienes 9 and 10 with Grubbs’ catalyst in refluxing benzene furnished the trans- and cis-2,5-disubstituted pyrrolines 11 and 12 in 86% and 87% yield, respectively.17 This study demonstrated that the rhodium-catalyzed allylic amination reaction combined with ring-closing metathesis provides a highly efficient method for the stereospecific construction of trans- and cis-2,5-disubstituted pyrrolines.
We have also demonstrated that N-aryl sulfonamides undergo regio- and enantiospecific rhodium-catalyzed allylic amination,2c providing important intermediates for the preparation of a variety of heterocycles. However, this type of nucleophile is less tolerant than the alkyl version, in that o, o-disubstitution and/or α-branched allylic alcohol derivatives are problematic. Nonetheless, the synthetic utility was highlighted in a stereoselective synthesis of the dihydrobenzo[b]indoline derivative 15a (Scheme 4).
Scheme 4.
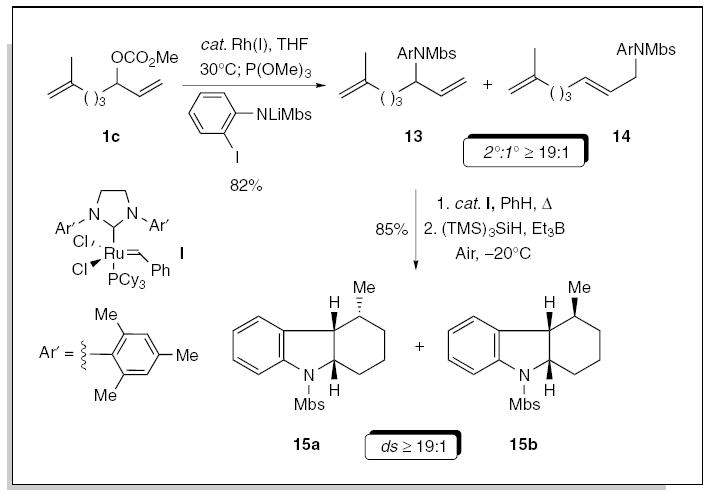
The rhodium-catalyzed allylic amination of 1c, with the lithium anion of 2-iodo-(N-4-methoxybenzenesulfonyl)aniline furnished the corresponding N-(arylsulfonyl)anilines13/14 in 82% yield, as a ≥19:1 mixture of regioisomers favoring 13. The diene 13 was then subjected to ring-closing metathesis with the second-generation Grubbs’ N-heterocyclic carbene catalyst,19 followed by a free radical cyclization, to furnish the dihydrobenzo[b]indolines 15a/b in 85% yield, favoring 15a (ds ≥19:1). Hence, the combination of the allylic amination with ring-closing metathesis, followed by a free radical cyclization provides an expeditious approach to the dihydrobenzo[b]indoline skeleton.
Rhodium-Catalyzed Allylic Etherification
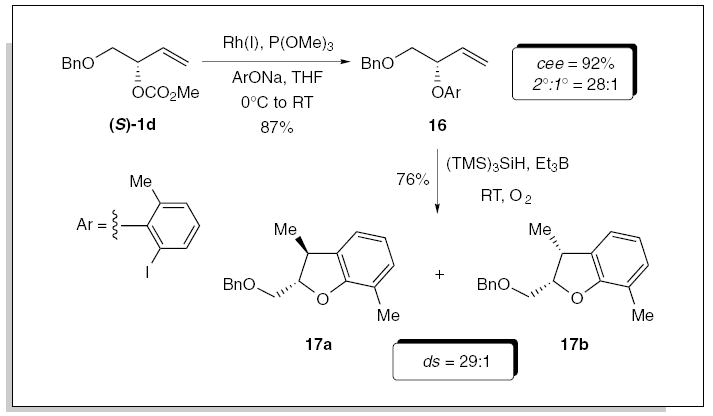
The rhodium-catalyzed allylic etherification was also examined, in which the trimethylphosphite modified Wilkinson’s catalyst again proved optimum for cross-coupling the sodium salt of various o- and o, o-substituted phenols, to afford the secondary allylic aryl ethers in excellent yield with retention of absolute configuration.3a This allylic etherification proved highly tolerant of sterically demanding alkyl, aryl, heteroatom, and halide substituents at the ortho-position within the phenolic nucleophile, making it a highly versatility transformation. The combination of the rhodium-catalyzed allylic etherification with a radical cyclization allowed the construction of the chiral nonracemic dihydrobenzo[b]furan derivatives, as outlined in Scheme 5. Rhodium-catalyzed allylic etherification of (S)-1d (≥99% ee), with the sodium anion of 2-iodo-6-methyl phenol, furnished the corresponding aryl allyl ether 16 in 87% yield, (2°:1° = 28:1; 92% cee),3a,14 which was subjected to a free radical cyclization to afford the dihydrobenzo[b]furans 17a/b in 76% yield, favoring 17a (ds = 29:1).
Scheme 5.
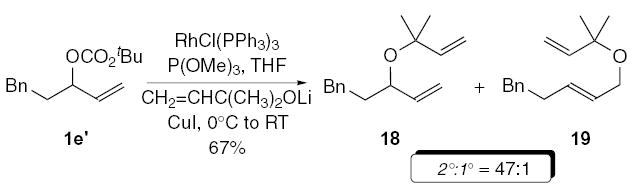
For alkyl alcohols, the intermolecular metal-catalyzed allylic etherification has, for the most part, been limited to primary alcohols due to the challenges associated with secondary and tertiary alcohols. This can again be attributed to poor regioselectivity, and the propensity for metal alkoxides to promote elimination of the metal-allyl intermediate and/or hydrolysis of the leaving group in the allylic alcohol precursor. We recently demonstrated that copper(I) alkoxides undergo highly regio- and enantiospecific rhodium-catalyzed allylic etherification reactions.3b Primary, secondary, and tertiary copper(I) alkoxides provide excellent nucleophiles, which have resulted in the development of the most general allylic etherification reaction described to date. Interestingly, although copper iodide (I > Br > Cl) proved optimal for regioselectivity, copper chloride was crucial for enantiospecificity (Cl ~ Br > I), providing another example of the effect of halide ions in transition metal-catalyzed reactions.20 The allylic etherification of allylic carbonate 1e’ with the copper(I) alkoxide derived from lithium anion of 3-methyl buten-3-ol, gave the allylic ethers 18/19 in 67% yield, favoring 18 (2°:1° = 47:1), illustrating the immense synthetic potential for this transformation (Eq. 3).
Hence, the rhodium-catalyzed version of the allylic substitution reaction circumvents many of the limitations associated with regioselectivity and retention of absolute configuration encountered using other transition metals. The broad range of stabilized carbon and heteroatom nucleophiles, coupled with the ability to utilize a wide array of substrates having substituents that have historically proven problematic, makes this transformation a powerful addition to the chemical literature.1–3
Rhodium-Catalyzed [4+2+2] Cycloaddition Reactions
Transition metal-catalyzed carbocyclization reactions represent powerful methods for the efficient construction of complex polycyclic systems.21 The intermolecular [4+4] cycloisomerization is representative of this class of transformations, in which acyclic dienes undergo formal cycloaddition reactions to furnish the corresponding eight-membered rings.22,23 Inspired by these studies, we developed the first metal-catalyzed intermolecular [4+2+2] cycloaddition of the heteroatom-tethered enyne derivatives with 1,3-butadiene for the construction of the bicyclic eight-membered heterocycles.4
A plausible catalytic cycle involves the reaction of the rhodium catalyst with a tethered enyne i resulting in the formation of the metallacycle iii (Fig. 2). Coordination of the 1,3-butadiene should then promote migratory insertion followed by a reductive elimination to give the [4+2+2] cycloaddition adduct v. The significant stereoelectronic differences between the enyne and diene component, make it possible to suppress the formation of the homocarbocyclization adduct 22 and/or oligomerization of the 1,3-butadiene.24
Figure 2.
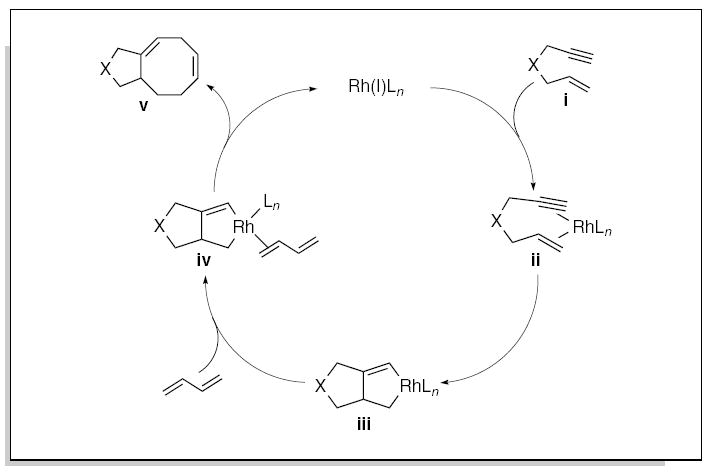
Mechanistic hypothesis for the development of the metal-catalyzed [4+2+2] carbocyclization reaction.
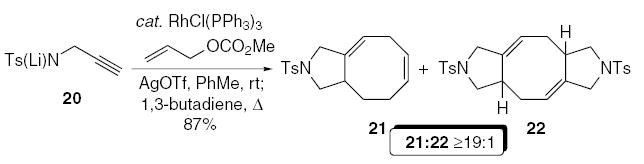
The synthetic versatility of this new carbocyclization reaction was highlighted in the development of a tandem three-component allylic amination/cycloaddition reaction (Eq. 4). Rhodium-catalyzed allylic amination of allyl methyl carbonate with lithium salt of N-tosylpropargylamine 20 furnished the enyne intermediate, which was subsequently heated at reflux under an atmosphere of 1,3-butadiene, to afford the cycloaddition adducts 21/22 in 87% yield, as a ≥19:1 mixture of products favoring 21.
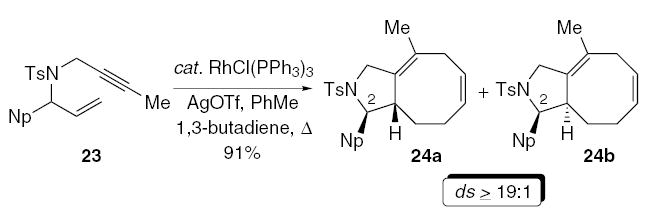
Additional studies examined the propensity for diastereocontrol in the rhodium-catalyzed [4+2+2] carbocyclization using a C-2 substituent. Preliminary attempts revealed that although the cycloaddition was indeed feasible, the carbocyclization of the enyne 23 required reduced concentration to suppress unwanted side reactions (Eq. 5). Nonetheless, the carbocyclization under the optimized conditions furnished the azabicycles 24a/b in 91% yield, favoring 24a (ds ≥19:1).25 Hence, the obvious advantage of this approach is the ability to directly increase molecular complexity of the carbocyclization derivative with minimal synthetic effort.
Tandem Rhodium-Catalyzed Allylic Alkylation/Pauson-Khand Annulation
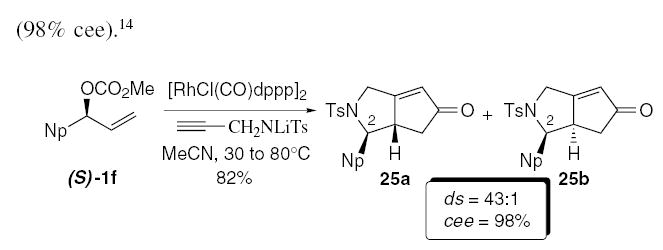
The Pauson-Khand (PK) carbocyclization is an important and widely studied transformation in which a tethered enyne undergoes a formal [2+2+1] reaction to furnish a bicyclic cyclopentenone.26 A significant limitation with this process is the necessity for substrate intramolecularity in order to suppress competitive intermolecular metal-catalyzed reactions.27 We envisioned an alternative approach to this problem that utilized a single metal catalyst to facilitate the formation of the enyne followed by the Pauson-Khand in a tandem sequence, using the reaction temperature to modulate catalytic activity.5 The advantage of this strategy is the ability to significantly increase the molecular complexity of the bicyclic adduct through the introduction of a stereogenic center at C-2, which we anticipated would control diastereoselectivity in the PK annulation. Indeed this strategy proved viable for carbon and heteroatom tethers, in which the ethers furnished optimal diastereoselectivity.
The chiral nonracemic allylic carbonate (S)-1f (≥99% ee) furnished the azabicycles 25a/b in 82% yield, as a 43:1 mixture of diastereoisomers favoring 25a (Eq. 6), in which the allylic amination is highly stereospecific (98% cee).14
The diastereoselectivity in the rhodium-catalyzed Pauson-Khand annulation is consistent with the mechanistic hypothesis outlined in Figure 3. Initial complexation of the enyne i presumably results in a mixture of diastereomeric complexes (ii/iii), where the relative population is influenced by the size of the substituent at C-2. Insertion of the metal then leads to the formation of the diastereomeric metallacycles iv/v, which are poised to undergo migratory insertion of metal-bound carbon monoxide, followed by reductive elimination to afford the bicyclic adducts vi/vii. Hence, provided the initial coordination is reversible, increasing the size of the substituent at C-2 should significantly improve the diastereoselectivity favoring the syn-diastereoisomer vi.
Figure 3.
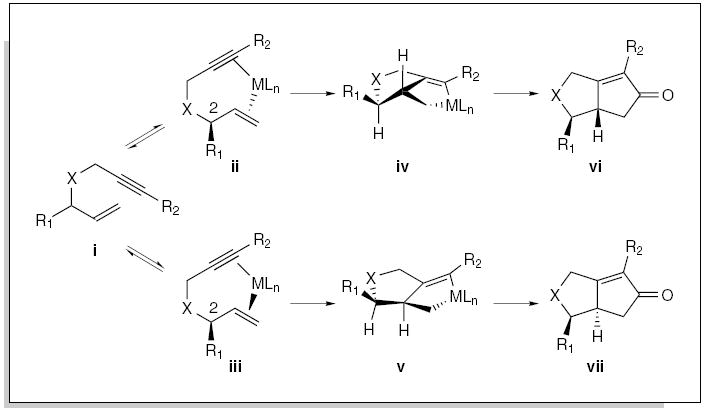
Origin of diastereocontrol for C-2-substituted tethered enynes in the Pauson-Khand reaction.
The regio-and diastereoselective tandem rhodium-catalyzed allylic substitution/Pauson-Khand annulation reaction demonstrated that excellent regioselectivity may be obtained with additional rhodium catalysts, provided there is a strong π-acidic ligand present on the metal center. The annulation reaction is also diastereoselective and highly enantiospecific, which is likely to have significant utility for asymmetric synthesis.
Conclusion and Future Challenges
In conclusion, this work has led to the development of a regio- and enantiospecific rhodium-catalyzed allylic substitution reaction that provides the most versatile and general method for the cross-coupling of various stabilized carbon and heteroatom nucleophiles with unsymmetrical allylic alcohol derivatives developed to date. The ability to modify a commercially available catalyst in situ, thus avoiding the handling of air- and moisture-sensitive catalysts, makes this an ideal protocol which is currently being utilized both in academic and industrial environments. Future challenges will be in the area of unstabilized nucleophiles and the development of diastereoselective variants. The rhodium-catalyzed [4+2+2] reaction provides a new carbocyclization, in which additional studies will focus on a broader understanding of the reaction mechanism and synthetic applications. The combination of the rhodium-catalyzed allylic substitution with other carbocyclization reactions provides rapid access to an array of important synthons present in pharmacologically important molecules. Future challenges in this area will entail the development and modification of catalysts to facilitate more complex variants of these and other carbocyclization reactions.
Acknowledgments
We would like to acknowledge the experimental and intellectual contributions of our co-workers in the catalysis subgroup. In particular, we would like to highlight the seminal contributions of our former colleagues John E. Robinson, Lawrence J. Kennedy, and Jade D. Nelson. We sincerely thank the National Institutes of Health (GM58877) and the Donors of The Petroleum Research Fund, administered by the American Chemical Society for generous financial support. We also thank the numerous pharmaceutical companies for unrestricted funds (PAE), and the Department of Chemistry at Indiana University for an E. M. Kratz Fellowship (DKL).
References
- 1.a Evans PA, Nelson JD. Tetrahedron Lett. 1998;39:1725. [Google Scholar]; b Evans PA, Nelson JD. J Am Chem Soc. 1998;120:5581. [Google Scholar]; c Evans PA, Kennedy LJ. Org Lett. 2000;2:2213. doi: 10.1021/ol005953g. [DOI] [PubMed] [Google Scholar]; d Evans PA, Kennedy LJ. J Am Chem Soc. 2001;123:1234. doi: 10.1021/ja005689m. [DOI] [PubMed] [Google Scholar]; e Evans PA, Kennedy LJ. Tetrahedron Lett. 2001;42:7015. [Google Scholar]
- 2.a Evans PA, Robinson JE, Nelson JD. J Am Chem Soc. 1999;121:6761. 12214. [Google Scholar]; b Evans PA, Robinson JE. Org Lett. 1999;1:1929. doi: 10.1021/ol991064l. [DOI] [PubMed] [Google Scholar]; c Evans PA, Robinson JE, Moffett KK. Org Lett. 2001;3:3269. doi: 10.1021/ol016467b. [DOI] [PubMed] [Google Scholar]
- 3.a Evans PA, Leahy DK. J Am Chem Soc. 2000;122:5012. [Google Scholar]; b Evans PA, Leahy DK. J Am Chem Soc. 2002;124:7882. doi: 10.1021/ja026337d. [DOI] [PubMed] [Google Scholar]
- 4.Evans PA, Robinson JE, Baum EW, Fazal AN. J Am Chem Soc. 2002;124:8782. doi: 10.1021/ja026351q. [DOI] [PubMed] [Google Scholar]
- 5.Evans PA, Robinson JE. J Am Chem Soc. 2001;123:4609. doi: 10.1021/ja015531h. [DOI] [PubMed] [Google Scholar]
- 6.For book chapters on metal-catalyzed allylic substitution, see: (a) Tsuji, J. In Palladium Reagents and Catalysts, Wiley: New York, 1996, ch. 4, pp. 290–404; (b) Trost, B.M., Lee, C. In Catalytic Asymmetric Synthesis, 2nd ed., Ojima, I., ed., Wiley-VCH: New York, 2000, ch. 8, pp. 593–649.
- 7.For lead references on other transition metal-catalyzed allylic alkylation reactions, see: (a) Co: Bhatia B, Reddy MM, Iqbal J.Tetrahedron Lett 1993346301 [Google Scholar]; (b) Fe: Xu Y, Zhou B.J Org Chem 198752974 [Google Scholar]; (c) Ir: Takeuchi R, Kashio M.Angew Chem Int Ed Engl 199736263 [Google Scholar]; (d) Ir: Helmchen G, Bartels B.J Chem Soc Chem Commun 1999741 [Google Scholar]; (e) Mo: Ward YD, Villanueva LA, Allred GD, Liebeskind LS.J Am Chem Soc 1996118897 [Google Scholar]; (f) Ni: Bricout H, Carpentier JF, Mortreux A.J Chem Soc Chem Commun 19951863 [Google Scholar]; (g) Pt: Brown JM, McIntyre JE.J Chem Soc Perkin Trans 21985961 [Google Scholar]; (h) Ru: Trost BM, Fraisse PL, Ball ZT.Angew Chem Int Ed Engl 2002411059. [DOI] [PubMed] [Google Scholar]; (i) W: Lloyd-Jones GC, Pfalz A.Angew Chem Int Ed Engl 199534462.and pertinent references cited therein. [Google Scholar]
- 8.a Trost BM, Hachiya I. J Am Chem Soc. 1998;120:1104. [Google Scholar]; b Bartels B, Helmchen G. Chem Commun. 1999:741. [Google Scholar]; c You S-L, Zhu X-Z, Luo Y-M, Hou X-L, Dai L-X. J Am Chem Soc. 2001;123:7471. doi: 10.1021/ja016121w. [DOI] [PubMed] [Google Scholar]; d Lopez F, Ohmura T, Hartwig JF. J Am Chem Soc. 2003;125:3426. doi: 10.1021/ja029790y. [DOI] [PubMed] [Google Scholar]; e Hayashi T, Okada A, Suzuka T, Kawatsura M. Org Lett. 2003;5:1713. doi: 10.1021/ol0343562. [DOI] [PubMed] [Google Scholar]
- 9.Hayashi T, Yamamoto A, Hagihara T. J Org Chem. 1986;51:723. [Google Scholar]
- 10.a Tsuji J, Minami I, Shimizu I. Tetrahedron Lett. 1984;25:5157. [Google Scholar]; b Minami I, Shimizu I, Tsuji J. J Organometallic Chem. 1985;296:269. [Google Scholar]
- 11.For related examples of rhodium-catalyzed allylic substitution reactions, see: (a) Takeuchi R, Kitamura N.New J Chem 1998659 [Google Scholar]; b Fagnou K, Lautens M. Org Lett. 2000;2:2319. doi: 10.1021/ol0060782. and pertinent references therein. [DOI] [PubMed] [Google Scholar]
- 12.Enyl complexes are, by definition, those that have a discrete σ- and π-metal carbon component within a single ligand. Sharp, P.R. In Comprehensive Organometallic Chemistry II, Abel, E.W., Stone, F.G.A., Wilkinson, G., eds., Pergamon Press: New York, 1995, ch. 2, p. 272.
- 13.Tanaka I, Jin-no N, Kushida T, Tsutsui N, Ashida T, Suzuki H, Sakurai H, Moro-oka Y, Ikawa T. Bull Chem Soc Jpn. 1983;56:657. [Google Scholar]
- 14.The term conservation of enantiomeric excess {cee = (product ee/starting material ee) × 100} provides a convenient method of describing the enantiospecificity of the reaction.
- 15.Krapcho AP. Synthesis. 1982:805. [Google Scholar]
- 16.Kurth MJ, Brown EG. J Am Chem Soc. 1987;109:6844. [Google Scholar]
- 17.For a recent review on ring-closing metathesis, see: Fürstner A.Angew Chem Int Ed 2000393012.and pertinent references cited therein. [Google Scholar]
- 18.For a recent review on allylic amination see: Johannsen M, Jørgensen KA.Chem Rev 1998981689. [DOI] [PubMed] [Google Scholar]
- 19.Scholl M, Ding S, Lee CW, Grubbs RH. Org Lett. 1999;1:953. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]
- 20.For a recent review on halide effects in transition metal catalysis see: Fagnou K, Lautens M.Angew Chem Int Ed 20024126. [DOI] [PubMed] [Google Scholar]
- 21.a Ojima I, Tzamarioudaki M, Li Z, Donavan RJ. Chem Rev. 1996;96:635. doi: 10.1021/cr950065y. [DOI] [PubMed] [Google Scholar]; b Aubert C, Buisine O, Malacria M. Chem Rev. 2002;102:813. doi: 10.1021/cr980054f. [DOI] [PubMed] [Google Scholar]
- 22.For recent reviews on metal-mediated cyclooctanoid construction see: (a) Mehta G, Singh V.Chem Rev 199999881. [DOI] [PubMed] [Google Scholar]; b Yet L. Chem Rev. 2000;100:2963. doi: 10.1021/cr990407q. [DOI] [PubMed] [Google Scholar]
- 23.For related transition metal-catalyzed carbocyclizations leading to 8-membered rings see: (a) Wender PA, Gamber GG, Hubbard RD, Zhang L.J Am Chem Soc 20021242876. [DOI] [PubMed] [Google Scholar]; b Gilbertson SR, DeBoef B. J Am Chem Soc. 2002;124:8784. doi: 10.1021/ja026536x. [DOI] [PubMed] [Google Scholar]
- 24.The rhodium-catalyzed cycloisomerization of 1 3-butadiene is known to favor oligomerization rather than the formation of cyclooctadiene see: Bosch M, Brookhart MS, Ilg K, Werner H.Angew Chem Int Ed 2000392304. [DOI] [PubMed] [Google Scholar]
- 25.The stereochemistry of the major diastereoisomer was reassigned based on a recent X-ray crystal structure. The rather unusual eight-membered ring conformation forces the protons pseudo-equational explaining the observed NOE enhancement (5.2%), despite their trans-relationship.
- 26.For a recent review on the Pauson-Khand reaction see: Brummond KM, Kent JL.Tetrahedron 2000563263 [Google Scholar]
- 27.For a related tandem allylic alkylation/Pauson-Khand carbocyclization see:Jeong N, Seo SD, Shin J-Y.J Am Chem Soc 200012210220 [Google Scholar]