

Abstract
While the interferon (IFN)–inducible double-stranded RNA (dsRNA)–dependent protein kinase PKR is reported to initiate apoptosis in some instances, the mechanism by which diverse stress stimuli activate PKR remains unknown. Now we report that RAX, the only known cellular activator for PKR, initiates PKR activation in response to a broad range of stresses including serum deprivation, cytotoxic cytokine or chemotherapy treatment, or viral infection. Thus, knock-down of RAX expression by 80% using small interfering RNA (siRNA) prevents IFNγ/tumor necrosis factor α (TNFα)–induced PKR activation and eIF2α phosphorylation, IκB degradation, IRF-1 expression, and STAT1 phosphorylation, resulting in enhanced murine embryonic fibroblast (MEF) cell survival. In contrast, expression of exogenous RAX, but not of the nonphosphorylatable, dominant-negative RAX(S18A) mutant, sensitizes cells to IFNγ/TNFα, mitomycin C (MMC), or serum deprivation in association with increased PKR activity and apoptosis. Furthermore, RAX(S18A) expression in Fanconi anemia complementation group C–null MEF cells not only prevents PKR activation but also blocks hypersensitivity to IFNγ/TNFα or mitomycin C that results in enhanced apoptosis. In addition, reduced RAX expression facilitates productive viral infection with vesicular stomatitis virus (VSV) and promotes anchorage-independent colony growth of MEF cells. Collectively, these data indicate that RAX may function as a negative regulator of growth that is required to activate PKR in response to a broad range of apoptosis-inducing stress.
Introduction
Previously, we discovered that interleukin-3 (IL-3) withdrawal from factor-dependent myeloid cells activated the interferon-inducible double-stranded RNA (dsRNA)–dependent protein kinase, PKR, in a mechanism that leads to translation inhibition and induction of apoptosis.1 This finding and latter observations by others that platelet-derived growth factor (PDGF) and tumor necrosis factor α (TNFα) signaling require PKR, underscore the notion that PKR is not only involved in host antiviral defense, but also in cellular growth signaling.2,3 Furthermore, reports indicate that PKR functions in the cellular response to inflammatory cytokines interferon γ (IFNγ) and TNFα, and in Toll-like receptor signaling pathways, indicating that PKR may play a broader role in mediating both cellular stress and the innate immune responses.4-13 It has been proposed that PKR serves as a “molecular clock” during stress that sequentially promotes cell survival then induces apoptosis.14 In this regard, PKR is thought to initiate nuclear factor κB (NF-κB)–mediated gene expression and survival by a kinase-independent mechanism but, following prolonged stress, can become catalytically activated to inhibit translation and induce apoptosis. Thus, determining the molecular mechanism(s) by which this Jekyll-and-Hyde–like transformation of PKR may occur during the course of cellular stress is paramount to our understanding of how stress-activated signal transduction pathways are regulated.
Recently, we identified RAX, the only known cellular activator of PKR, and reported that RAX phosphorylation on serine 18 is required for PKR activation, translation inhibition, and apoptosis following IL-3 deprivation.15,16 RAX and its independently discovered human ortholog PACT are ubiquitously expressed, 98% identical, and consist of 3 dsRNA-binding domains.15,17,18 The N-terminal first and second dsRNA-binding domains are necessary for association with dsRNA and PKR while the third, C-terminal domain is not required for dsRNA or PKR binding but is required for PKR kinase activation.19,20 Interestingly, expression of the nonphosphorylatable RAX(S18A) mutant promotes a dominant-negative phenotype when IL-3 is withdrawn from factor-dependent cells characterized by failure to activate PKR, delayed translation inhibition, and enhanced cell survival.16 However, it remains uncertain whether only specific cellular stresses such as growth factor deprivation, or rather various stresses, including cytotoxic drug or cytokine treatment and viral infection, coordinate apoptosis signaling through a RAX-dependent mechanism.
PKR is required for the cytotoxic response to the inflammatory cytokine TNFα, the IFNγ-mediated up-regulation of IRF1, and synergy between IFNγ and TNFα to promote IκBβ degradation and activate NF-κB.13,21-23 Thus, murine embryonic fibroblasts (MEFs) derived from Pkr-null mice are unable to degrade IκB and activate NF-κB or to properly regulate the IRF1 promoter following treatment with either IFNγ or TNFα.13,21 Also, Stat1 phosphorylation on Ser727 and its transactivation activity are defective, and c-myc mRNA is induced, not suppressed, following IFNγ treatment of Pkr-null cells.24 Reducing PKR expression blocks TNFα-induced apoptosis in promonocytic U937 cells and inhibits IFNγ-dependent activation of NF-κB in fibrosarcoma cells.3 In addition, the synergy between IFNγ and TNFα to promote NF-κB activation is defective in Pkr-null cells, and 2-aminopurine (2-AP), a frequently used PKR inhibitor, can block IFNγ/TNFα synergism in normal MEFs.13 Overexpression of the catalytically inactive, dominant-negative PKR (K296R) mutant or treatment with 2-AP inhibits IFNγ/TNFα-induced degradation of IκBβ, NF-κB nuclear translocation, and synergistic activation of NF-κB-dependent gene expression in neuronal-derived cells.25 Collectively, these findings suggest that the cytotoxic signal(s) generated by IFNγ/TNFα cotreatment require PKR kinase activity to elicit enhanced NF-κB activity by initiating IκB degradation.
Since PKR plays a critical role in regulating cell growth and apoptosis, it is not surprising that PKR has been associated with the molecular phenotype of several diseases, including Fanconi anemia (FA).26 Cells from FA patients are hypersensitive to DNA crosslinking agents such as mitomycin C and exhibit increased apoptosis induced by inflammatory cytokines. These phenotypes may ultimately lead to bone marrow failure and leukemia in FA patients.27 Interestingly, forced expression of the dominant-negative PKR mutant PKR (K296R) in primary FA lymphoblasts potently suppresses apoptosis induced by IFNγ and TNFα.28 Furthermore, overexpression of PKR in Fancc-null MEF cells promotes increased cell death following treatment with either IFNγ and TNFα or dsRNA.26 Thus, inappropriate regulation of PKR activation may contribute to the FA phenotype.26,29-31
To examine the role of RAX during the cellular response to various cytotoxic stress applications, we generated MEF cell lines that express either markedly reduced (ie, > 80%) levels of RAX, exogenous RAX, or the nonphosphorylatable RAX(S18A) mutant. Results indicate that RAX functions as the necessary direct upstream regulator of PKR in a stress → RAX → PKR sequential signaling axis that leads to stress-induced cell death.
Materials and methods
Cell lines and reagents
Simian virus 40 (SV40) large T-antigen–immortalized normal and Fancc–/– MEFs derived from C57BL and 129Sv were from G. C. Bagby (Oregon Health & Science University [OHSU] Cancer Institute, Portland, OR). The retroviral vector pBMN-Z-I-blasto and Phoenix retrovirus producer cell line were from G. P. Nolan (Stanford University, CA). Cells were maintained in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and cultured in a humidified incubator at 37°C and 5% CO2 (Invitrogen, Carlsbad, CA). BHK-21 cells were maintained in F11 medium supplemented with 1% nonessential amino acids, 1% penicillin-streptomycin solution, 2% 200 mM glutamine, 0.7% glucose, and 10% FBS (Invitrogen).
To generate retroviral RAX and RAX(S18A) expression vectors, N-terminal FLAG-tagged RAX or RAX(S18A) were cloned into pBMN-Z-I-blasto as BamHI fragments, thereby replacing the LacZ gene. The resulting vectors, called pBBFlagRAX and pBBFlagRAX(S18A), express either FLAG-tagged RAX or RAX(S18A) from the Moloney murine leukemia virus (MoMLV) 5′ long terminal repeat (LTR), respectively. Phoenix packaging cells were transfected using Lipofectamine (Invitrogen, Carlsbad, CA). Approximately 24 hours after transfection, 3 mL of medium from the Phoenix cells was used to infect 2 × 105 MEF cells. After infection (24 hours), the medium was replaced, and 48 hours after infection, cells were selected with 4 μg/mL blasticidin S (no. 203 350; Calbiochem, La Jolla, CA). Immunoblotting using antibody to RAX or FLAG (no. F1804; Sigma-Aldrich, St Louis, MO) confirmed expression.
Antibodies to IκBα (sc-371), STAT1 (sc-346), IRF1 (sc-640), IFN-Rα (sc-700), TNF-R1 (sc-8436), and actin (sc-1615) from Santa Cruz Biotechnology (SCB; Santa Cruz, CA), and antibodies specific for phosphoserine-727 STAT1 (no. 9177; Cell Signaling Technology, Beverly, MA) were used for immunoblotting with cell lysates. For immunofluorescent staining of cell-surface receptors, 2 × 104 cells were cultured on chamber slides and fixed with acetone-methanol (1:1).
Generation of cell lines expressing RAX siRNA
The pSUPER RNAi system (Oligoengine, Seattle, WA) was used to produce vectors expressing RAX siRNAs. The Oligoengine design tool (www.oligoengine.com) and the Dharmacon siDESIGN tool (www.dharmacon.com/sidesign/; Dharmacon, Lafayette, CO) were used to devise 13 siRNA oligonucleotides to RAX. A negative control siRNA (nc siRNA) was designed by scrambling the sequence of si100. A basic local alignment search tool (BLAST) search (http://www.ncbi.nlm.nih.gov/) confirmed that each siRNA had no significant homology to any gene other than RAX. The 5′ to 3′ target sequences were: si100, CACCGATTCAGGTATTGCA; si164, AAGATCCGATGTGCAAGTA; si165, AGATCCGATGTGCAAGTAC; si166, GATCCGATGTGCAAGTACA; si210, ACCGTTGGTGACATAACTT; si211, CCGTTGGTGACATAACTTG; si310, GTATTTGCTTTGCAGTTCC; si340, TGCCTGATCCATCCAAACA; si467, AGAGGGAATACACCACGAT; si581, TATTTCTCCAGAGAACCAC; si610, CGAACGTGGTTGGACATTC; si937, AGTAGGCTTGGAGCGACTT; si938, GTAGGCTTGGAGCGACTTA; and nc siRNA, GACGAAAGCAGTTCGTCTA. For each target sequence, 64-base complementary oligonucleotides were purchased (Sigma-Genosys, St Louis, MO), hybridized, and ligated into pSUPER.retro.puro according to Oligoengine's protocol. DNA sequence analysis confirmed constructs (University of Florida, Gainesville, FL).
Phoenix cells were transfected with siRNA plasmids. After 48 to 72 hours, the resulting media was used to infect MEF cells as described, and cells were selected in 5 μg/mL puromycin. Cells were lysed in GLB (20 mM Tris-Cl [pH 7.6], 137 mM NaCl, 5 mM EDTA, 1% triton X-100, 15% glycerol, and 1 × protease inhibitor cocktail I [no. 539131; Calbiochem])16 and expression of PKR and RAX was determined by immunoblotting using antibody to PKR (1:1000, sc-1702, M-515, SCB) or RAX (1:500 dilution)15 and secondary horseradish peroxidase (HRP)–conjugated antibody (SCB) with enhanced chemiluminescence (ECL) reagents (GE Healthcare, Piscataway, NJ). Western blots were scanned at 400 dots per inch (dpi) and saved as TIFF files. Individual bands were quantified by densitometry analysis with background correction using Quantity One (Bio-Rad Laboratories, Hercules, CA).
Viability and apoptosis measurement
Viability during normal growth was determined by trypan blue dye exclusion. To measure viability and apoptosis under stress conditions, cells were plated at 5 × 105 cells/well in a 6-well plate. Where indicated, cells were treated for 24 hours with 10 or 20 ng/mL IFNγ (no. 485-MI-100; R & D Systems, Minneapolis, MN) followed by 24 hours of treatment with TNFα (no. 11088939001; Roche Applied Science, Indianapolis, IN) or with mitomycin C (no. 101 074 09 001, Roche Applied Science) for 24 hours. TUNEL (transferase-mediated dUTP nick-end labeling) assay was performed using an APO-BRDU kit (no. 556405; BD Biosciences, San Jose, CA) at the University of Florida flow cytometry facility (Gainesville, FL).
PKR and eIF2α phosphorylation assays
PKR autophosphorylation activity at the indicated times was assayed by 32P incorporation after immunoprecipitation.16 Briefly, PKR antibodies M-515 and B-10 (SCB) were used to immunoprecipitate and detect total PKR, respectively, from 250 μg cell lysate, and kinase reactions using immunoprecipitated PKR were performed for 15 minutes at 30°C with 32P-labeled ATP as previously described.16 Autoradiographs and immunoblots were scanned, saved as TIFF files, and quantified as described. The average change in phosphorylation level relative to any change in total protein level at each time point following stress treatment was calculated and compared with untreated cells. For all figures, the mean of 3 separate experiments was graphed with error bars depicting the standard deviation.
The level of eIF2α and eIF2α phosphorylation were monitored by immunoblotting with antibodies to eIF2α15 and specific for phosphoserine 51 eIF2α (1:1000, no. 9721; Cell Signaling Technology). Immunoblots were scanned and individual bands quantified as described. The change in phosphorylation relative to any change in total protein level during stress treatment was averaged from 3 separate experiments and graphed.
Statistical significance (P < .1) of the differences observed between cell lines or time points for viability, PKR activity, and eIF2α phosphorylation after stress was measured by paired t test using Excel (Microsoft, Redmond, WA).
Vesicular stomatitis virus infection, titering, and immunoblotting
MEF cell lines were infected with vesicular stomatitis virus (Indiana serotype) at a multiplicity of infection (MOI) of 1 or 5 for 1 hour at 37°C. Viability was measured 0, 8, and 24 hours after infection using trypan blue dye exclusion. After 24 hours of infection, culture medium was removed and saved while the infected cells were lysed, as described, to monitor viral protein levels by immunoblotting with antibody R1. Virus titer of the medium was determined on monolayers of BHK-21 cells at 37°C.
Anchorage-independent growth assay
Colony formation in soft agar medium after 14 days was quantified to measure cell transformation of MEF cells stably expressing RAX constructs. Briefly, 1 × 104 cells were mixed with complete medium containing 0.4% agarose and plated in a 60-mM dish over a base layer of solidified 0.8% agarose in complete medium. Plates were incubated at 37°C and medium replenished every 3 to 4 days. Crystal violet–stained plates were scanned at 300 dpi using a flatbed scanner (Pixma MP 780; Canon, Lake Success, NY) and enlarged printouts were used to count colonies (groups of approximately 50 or more cells).
Results
Cells expressing reduced RAX display increased survival under low-serum conditions
MEF cell lines with reduced RAX expression were created using siRNAs. Thirteen siRNAs were tested that extensively cover the RAX sequence. Following retroviral packaging, siRNA vectors were delivered to MEF cells, and cells were selected for stable expression. Three siRNA-expressing cell lines, si164, si165, and si166, displayed 81%, 54%, and 32% reduced RAX expression, respectively, compared with a scrambled nc siRNA (Figure 1A). In addition, MEFs stably expressing either an N-terminal FLAG-tagged RAX or the dominant-negative nonphosphorylatable RAX(S18A) mutant were also generated. Importantly, RAX or RAX siRNA expression did not alter the expression of PKR, TNFα-R1 receptor, IFNγ-Rα receptor, and actin (Figure 1A). Furthermore, immunofluorescent staining confirmed that cell-surface expression of TNFα-R1 and IFNγ-Rα was approximately equal for all cell lines (data not shown). All cell lines displayed similar growth kinetics under serum-replete conditions (ie, 10% serum; Figure 1B). However, after 24 hours in medium with low serum (0.1%), the control siRNA or vector-only control cells were approximately 73% viable compared with the si164-expressing cells (ie, only 19% endogenous RAX–expressed) that were approximately 91% viable (P = .002, n = 5), while those cells expressing RAX(S18A) were approximately 81% viable (P = .04, n = 5; Figure 1C). In contrast, cells overexpressing RAX were only approximately 50% viable (P = .003, n = 5; Figure 1C). These data indicate that RAX expression and activation by phosphorylation correlates with reduced cell survival during serum depletion.
Figure 1.

Expression of siRNA to RAX promotes cell survival in medium with low serum. (A) Western blots using cell lysates from MEFs expressing the indicated siRNAs as well as from cell lines that express exogenous RAX or RAX(S18A). (B) Growth rate of MEF cell lines under serum-replete (10%) conditions. (C) Viability measured by TUNEL assay in medium with low (0.1%) serum of cell lines with reduced RAX levels caused by siRNA expression or expressing either exogenous RAX or RAX(S18A).
RAX expression is required for IFNγ/TNFα-induced apoptosis
To investigate the role of RAX during apoptosis initiated by cell stress other than IL-3 deprivation,16 we measured the effect of IFNγ/TNFα. Importantly, even in cells with reduced RAX, PKR was up-regulated by IFNγ and TNFα treatments, as expected,3 while RAX expression remained unchanged (Figure 2A). Expression of exogenous RAX, but not the nonphosphorylatable RAX(S18A) mutant, sensitized cells to IFNγ/TNFα-induced apoptosis. Vector-only control cells treated with IFNγ and low concentrations of TNFα (10 ng/mL) for 24 hours remained approximately 98% viable, while cells expressing exogenous RAX were only approximately 65% viable (P = .002, n = 9; Figure 2B). Furthermore, after treatment with IFNγ for 24 hours and increasing concentrations of TNFα for an additional 24 hours, apoptosis was markedly repressed in RAX(S18A)- and RAX siRNA–expressing cells (Figure 2C). Thus, the si164 cells remained approximately 69% viable, while control cells were only approximately 28% viable (P < .001, n = 9) in high concentrations of TNFα (80 ng/mL; Figure 2C). Similarly, cells expressing the dominant-negative RAX(S18A) mutant remained approximately 58% viable, while vector-only control cells were approximately 32% viable (80 ng/mL TNFα, P = .01, n = 3). In contrast, exogenous expression of RAX promoted cell death such that only approximately 26% of cells were viable compared with vector-only control cells that were approximately 63% viable after 20 ng/mL TNFα treatment (P = .016, n = 3; Figure 2C). Since the levels of TNFα and IFNγ surface receptors were approximately equal for all of these cells, the difference in cytotoxic effect observed for these cytokines can be attributed to RAX, RAX(S18A), or RAX siRNA expression.
Figure 2.

Reduced levels of RAX prevent IFNγ/TNFα-induced apoptosis. (A) IFNγ up-regulates PKR but not RAX in MEF cell lines. Western blots using antibodies to PKR or RAX with lysates from cells treated with 10 ng/mL IFNγ and TNFα. (B) Viability of MEF cells expressing either exogenous RAX or RAX(S18A) compared with vector control cells after treatment with 10 ng/mL IFNγ and/or 10 ng/mL TNFα for 24 hours. (C) Viability of MEFs expressing RAX, RAX(S18A), or reduced RAX that were pretreated with 20 ng/mL IFNγ for 24 hours followed by increasing concentrations of TNFα for 24 hours measured by TUNEL assay. (D) Relative PKR activity was assessed by autophosphorylation assay. Results represent the averaging of 3 separate experiments that quantify activated PKR and total PKR by autoradiography and Western blotting. Cells were treated with 10 ng/mL IFNγ for 24 hours followed by addition of 10 ng/mL TNFα for the indicated times. (E) Relative eIF2α phosphorylation determined by quantifying Western blots from 3 separate experiments using antibody to eIF2α and phosphoserine-51 eIF2α following treatment with 10 ng/mL IFNγ for 24 hours and with 10 ng/mL TNFα for the indicated times.
Since RAX is the only identified cellular activator of PKR to date, we examined PKR activation by measuring the change in PKR autophosphorylation activity relative to the total PKR protein level after IFNγ/TNFα cotreatment. While an increase in PKR activity was not detected in the si164-expressing cells following 24 hours of IFNγ treatment, control siRNA– and vector-only–expressing cells displayed an approximately 2-fold increase in activated PKR (P = .03, n = 3; Figure 2D). Significantly, cells stably expressing exogenous RAX had increased PKR activity compared with vector control cells (approximately 3-fold vs approximately 2-fold, P = .02, n = 3), while expression of the nonphosphorylatable RAX(S18A) mutant caused a small inhibition of PKR activation (approximately 1.4-fold activation vs approximately 2-fold, P = .001, n = 3) following IFNγ-only treatment (Figure 2D). Furthermore, after an additional 24 hours of TNFα treatment, cells expressing exogenous RAX exhibited significantly increased PKR activity compared with vector-only control cells (approximately 2.7-fold vs approximately 1.7-fold, P = .01, n = 3), but PKR was not activated to same extent in cells expressing the RAX(S18A) mutant as control cells (ie, approximately 1.2-fold vs approximately 1.7-fold, P = .05, n = 3; Figure 2D). Significantly, the si164-expressing cells not only failed to activate PKR during IFNγ/TNFα cotreatment, but also endogenous PKR activity was reduced 4-fold compared with control siRNA-expressing cells (P < .001, n = 3; Figure 2D).
Since eIF2α is a primary substrate for PKR, we measured eIf2α phosphorylation at serine 51 to confirm PKR activation.11 After 24 hours with IFNγ, control siRNA– and vector-only–expressing cells exhibited an approximately 2.7-fold increase in eIF2α phosphorylation relative to total protein level (P = .007, n = 3), and subsequent cotreatment of these cells with TNFα for up to 24 hours did not further increase eIF2α phosphorylation compared with un-treated cells (Figure 2E). Significantly, RAX overexpressing cells displayed an initial 3-fold increase in relative eIF2α phosphorylation following IFNγ treatment (P = .02, n = 3), which rapidly escalated following TNFα cotreatment to an approximately 7-fold increase compared with untreated cells (P = .03, n = 3; Figure 2E). In contrast, cells expressing the nonphosphorylatable RAX(S18A) mutant displayed markedly reduced eIF2α phosphorylation after either IFNγ or IFNγ/TNFα cotreatment compared with vector-only control cells (ie, approximately 1.9-fold vs approximately 2.7-fold after 24 hours of IFNγ, P = .07, n = 3; Figure 2E). Importantly, the si164-expressing cells displayed no significant increase in eIF2α phosphorylation after either IFNγ or IFNγ/TNFα treatment (Figure 2E). Thus, failure of IFNγ/TNFα cotreatment to promote apoptosis is likely caused, at least in part, by the failure of RAX to properly activate PKR.
Reduced RAX expression is associated with aberrant transcription factor activation by IFNγ/TNFα
Several reports indicate that PKR-mediated signal transduction enhances TNFα-mediated apoptosis by activating regulators of gene expression.13,21,25,32-34 For example, cotreatment with IFNγ/ TNFα activates PKR, which in turn initiates IκB degradation and STAT1 transcription that mediates IRF1 up-regulation. Since reducing the expression of cellular RAX blocked PKR activation following IFNγ/TNFα cotreatment, we examined whether RAX may also affect these 3 regulators of gene expression. After IFNγ/TNFα cotreatment, the si164-expressing cells exhibited significantly reduced IκBα degradation and IRF-1 failed to be up-regulated compared with control cells (Figure 3A). Significantly, cells stably expressing exogenous RAX displayed enhanced IκB degradation and rapidly up-regulated IRF1 following IFNγ treatment compared with control cells, which up-regulated IRF1 only after prolonged exposure (> 2 hours) to IFNγ/TNFα (Figure 3A). Thus, the rate of IRF1 up-regulation was proportional to the level of RAX expression.
Figure 3.

Regulators of transcription and survival are affected by RAX expression during IFNγ/TNFα treatment. (A) Western blotting was used to monitor IκBα degradation and IRF-1 production after cells had been treated with 10 ng/mL IFNγ followed by treatment with 10 ng/mL TNFα for the indicated times. (B) The level of STAT1 and phosphoserine 727 STAT1 were monitored after IFNγ/TNFα treatment by Western blotting.
STAT1 expression and activation as monitored by phosphorylation at serine 727 were also determined after IFNγ and TNFα treatment. Irrespective of RAX expression, STAT1 was up-regulated following IFNγ-only treatment and remained so after TNFα addition (Figure 3B). However, the si164-expressing cells showed a marked decrease in STAT1 phosphorylation at serine 727 during IFNγ/TNFα treatment (Figure 3B). These data support a role for RAX affecting regulators of gene expression that mediate IFNγ/TNFα signaling.13,21,24
Expression of the nonphosphorylatable RAX(S18A) mutant suppresses Fancc-null cells' hypersensitivity to IFNγ/TNFα
The Fanconi anemia complementation group C (Fancc) gene product may protect cells from PKR-mediated cytotoxicity by cooperating with Hsp70 to inhibit PKR activity.29 MEFs derived from Fancc–/– mice have constitutively activated PKR, which may contribute to their hypersensitivity to IFNγ/TNFα.26 Since RAX-dependent PKR activation appears to be necessary for IFNγ/TNFα-induced apoptosis, we examined how expression of RAX or the nonphosphorylatable RAX(S18A) mutant affected these cells' hypersensitive phenotype to IFNγ/TNFα.
We tested Fancc–/– MEFs that stably express similar levels of either FLAG-tagged RAX or the nonphosphorylatable RAX(S18A) mutant (Figure 4A). Importantly, overexpression of RAX or RAX(S18A) did not alter TNF-R1, IFNγ-Rα, or PKR expression, indicating that differences observed in these cells are likely specific to RAX (Figure 4A). The Fancc–/– cells with increased RAX expression were more sensitive than vector-only control cells to treatment with either IFNγ (approximately 75% vs approximately 85% viable, P = .04, n = 4) or TNFα alone (approximately 60% vs approximately 88%, P < .001, n = 4), and also exhibited enhanced cell death following IFNγ/TNFα cotreatment (approximately 34% vs approximately 60%, P < .001, n = 4; Figure 4B). Significantly, expression of the nonphosphorylatable RAX(S18A) mutant, at least in part, suppressed IFNγ/TNFα apoptosis compared with vector-only control cells (approximately 69% vs approximately 60% viable, P = .08, n = 4; Figure 4B).
Figure 4.

Hypersensitivity of Fancc–/– cells to IFNγ/TNFα or mitomycin C can be suppressed by expression of the dominant negative RAX(S18A) mutant. (A) Western blots with lysates from Fancc–/– MEF cells expressing either RAX or RAX(S18A). (B) Survival as determined using TUNEL assay after treatment with 10 ng/mL IFNγ and/or 10 ng/mL TNFα of Fancc–/– cell lines. (C) Autoradiographs and Western blots from 3 separate PKR autophosphorylation assays were quantified and used to graph the increase in active PKR relative to total PKR protein level after treatment with 10 ng/mL IFNγ for 24 hours followed by treatment with 10 ng/mL TNFα for the indicated times. (D) Survival of normal MEF and Fancc–/– MEF cell lines expressing either exogenous RAX or RAX(S18A) after 24 hours of treatment with the indicated concentrations of mitomycin C. Viability was measured using TUNEL assay.
Next, we examined PKR activation in Fancc–/– cells expressing exogenous RAX or RAX(S18A) following IFNγ/TNFα treatment. After 24 hours of IFNγ, PKR activity was up-regulated approximately 1.5-fold in vector-only control Fancc–/– cells compared with untreated cells (P = .002, n = 3), while cells expressing exogenous RAX displayed an approximately 2-fold increase in PKR activity (P = .07, n = 3; Figure 4C). In contrast, cells expressing the nonphosphorylatable RAX(S18A) mutant failed to activate PKR even as the level of PKR expression increased following IFNγ treatment, and thus displayed a small reduction in relative PKR activity (approximately 20%, P = .10, n = 3) following IFNγ-only treatment (Figure 4C). Furthermore, PKR activation after IFNγ/TNFα cotreatment was partially blocked by RAX(S18A) compared with vector-only control cells (approximately 1.4-fold vs approximately 2.2-fold, P = .03, n = 3; Figure 4C). Importantly, Fancc–/– cells expressing exogenous RAX had an approximately 3.3-fold increase in PKR activity (compared with approximately 2.2-fold for vector-only control cells, P = .04, n = 3) following IFNγ/TNFα cotreatment (Figure 4C). Thus, increasing RAX expression in Fancc–/– MEF cells significantly increased PKR activation that may be responsible for the decreased viability observed.
Since a molecular hallmark of FA cells is sensitivity to DNA damage induced by crosslinking agents such as mitomycin C (MMC), we tested whether RAX expression in Fancc–/– MEF cells altered these cells' hypersensitivity to MMC. Similar to findings by others, Fancc–/– vector-only control cells were much less viable than normal MEF vector-only control cells after treatment with increasing concentrations of MMC for 24 hours (Figure 4D). At the highest dose tested (400 ng/mL), Fancc–/– vector-only control cells were approximately 44% viable compared with normal MEFs that were approximately 72% viable (P = .03, n = 4; Figure 4D). MMC did not alter endogenous RAX or PKR expression (data not shown). However, exogenous RAX expression greatly sensitized (approximately 26% vs approximately 44% viable, P = 0.01, n = 4), while expression of the nonphosphorylatable RAX(S18A) mutant protected Fancc–/– cells (approximately 56% vs approximately 44% viable, P = .05, n = 4) from MMC compared with vector-only control cells (Figure 4D). These findings may indicate that the RAX-PKR signaling pathway contributes to hypersensitivity to inflammatory cytokines and DNA damage observed in Fancc–/– cells.
RAX is required for host antiviral defense to VSV infection
PKR may be activated by dsRNA produced during viral infection including with VSV.35 To investigate whether the PKR activator RAX is also necessary for host antiviral defense or whether PKR functions independently following infection, MEFs with reduced RAX were tested for sensitivity to VSV. After 24 hours of infection with an MOI of 1 or 5, si164-expressing cells were only approximately 15% or approximately 10% viable, respectively, compared with control siRNA cells that remained approximately 78% viable (P = .03 and .01, respectively, n = 3; Figure 5A). Furthermore, exogenous expression of RAX, but not the nonphosphorylatable RAX(S18A) mutant, increased cell death following viral infection such that only approximately 41% compared with approximately 88% for vector-only control cells were viable (MOI 1, P = .002, n = 3; Figure 5A).
Figure 5.

RAX deficiency causes failure of host antiviral defense to VSV infection. (A) Viability of MEF cell lines, as measured by trypan blue dye exclusion, 24 hours after infection with VSV at the indicated multiplicity of infection (MOI). (B) The increase in PKR autophosphorylation activity relative to total PKR level after the indicated time of infection with VSV at an MOI of 1 was determined by densitometry analysis of autoradiographs and Western blots from 3 separate experiments as described and graphed. (C) Densitometry analysis of Western blots from 3 separate experiments using antibodies specific for eIF2α and phosphoserine-51 eIF2α was used to determine and graph the fold increase in phospho-eIF2α relative to total eIF2α at the indicated times after VSV infection at an MOI of 1. (D) Western blotting with antibody R1 was used to monitor viral protein production of N, P, and M proteins at the indicated times after infection. (E) Virus titers of supernatants from infected cells 24 hours after infection were measured by plaque assay using BHK-21 cells. pfu indicates plaque-forming unit.
The si164-expressing cells failed to activate PKR compared with an approximately 2.5-fold activation in control siRNA cells following 4 hours of VSV infection (MOI 1, P = .001, n = 3; Figure 5B). In addition, PKR activity was reduced 50% by 24 hours of infection in the si164-expressing cells (P = .02, n = 3; Figure 5B). Significantly, in cells expressing exogenous RAX, relative PKR activity was approximately 2.7-fold higher than uninfected cells by 4 hours of infection, which was maintained throughout the infection (P = .01, n = 3; Figure 5B). Furthermore, after 24 hours of infection, the relative PKR activity in RAX-expressing cells was twice that of vector-only control cells (ie, approximately 3-fold vs approximately 1.5-fold, P = .001, n = 3; Figure 5B). In contrast, cells expressing the nonphosphorylatable RAX(S18A) mutant displayed a significant inhibition of PKR activity after 4 hours compared with vector-only control cells (ie, approximately 1.8-fold vs approximately 2.6-fold, respectively, P = .03, n = 3; Figure 5B). Thus, unlike the si164-expressing cells, where RAX is reduced by more than 80% and PKR fails to be activated, PKR is still activated to some extent in the RAX(S18A)-expressing cells but not to the same level as observed in vector-only control cells.
MEF si164-expressing cells with reduced RAX also fail to phosphorylate and inactivate eIF2α to the same degree as control cells during VSV infection (Figure 5C). Thus, after 4 hours, eIF2α phosphorylation in si164-expressing cells rises to approximately 3.5-fold versus uninfected cells, compared with the approximately 10-fold increase observed in control cells (P = .02, n = 3; Figure 5C). Importantly, exogenous expression of RAX promoted almost twice the increase in eIF2α phosphorylation by 2 hours of infection compared with vector-only cells (ie, approximately 15-fold vs approximately 8-fold, P = .01, n = 3; Figure 5C). This increase in eIF2α phosphorylation is maintained through 24 hours after infection, a time by which the RAX-expressing cells exhibit an approximately 7-fold increase compared with only an approximately 2-fold increase in vector-only control cells (P = .03, n = 3; Figure 5C).
Most likely as a direct consequence of the inability to activate PKR, phosphorylate eIF2α, and block new protein synthesis during VSV infection, the viral N, P, and M proteins were produced more rapidly and to a higher level in si164-expressing cells compared with that in control cells (4 hours vs 24 hours; Figure 5D). Significantly, VSV titer from si164-expressing cells was over 1000-fold higher than that from control siRNA-expressing cells (1 × 108 vs 6 × 104, P < .001, n = 3; Figure 5E). In contrast, cells expressing exogenous RAX displayed both a decrease in viral protein production and an approximately 100-fold reduced virus titer (P = .05, n = 3) compared with vector control cells (Figure 5D-E).
Knock-down of RAX expression or forced expression of RAX(S18A) promotes anchorage-independent colony formation in soft agar
Since RAX can activate PKR, and PKR has been associated with inhibition of cell growth and viability, we assessed the oncogenic potential of RAX by assaying anchorage-independent growth capacity in soft agar of MEF cells with reduced RAX expression, exogenous RAX, or the nonphosphorylatable RAX(S18A) mutant. Interestingly, while vector-only control MEF cells form colonies (defined as groups of 50 or more cells) in soft agar with an efficiency of approximately 4.4%, cells expressing exogenous RAX display about a 7-fold reduction in transformation efficiency (0.6% vs 4.4%, respectively, P = .001, n = 4; Figure 6). In contrast, expression of the nonphosphorylatable RAX(S18A) mutant more than tripled the anchorage-independent growth capability of these cells compared with vector-only MEF cells (14.9% vs 4.4%, P = .001, n = 4, respectively; Figure 6). In addition, si164 cells, with reduced RAX, displayed about twice the level of colony formation in soft agar as that of control cells (7.4% vs 4.4%, P = .004, n = 4; Figure 6). Taken together, these results suggest that RAX may contribute to growth inhibition.
Figure 6.
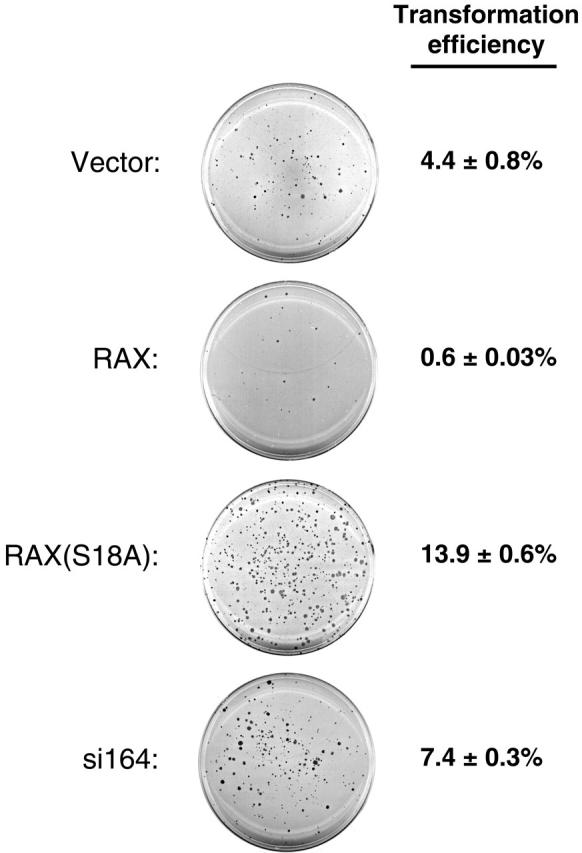
Reduced levels of RAX or expression of RAX(S18A) promotes anchorage-independent colony formation. MEF cells with reduced RAX or expressing exogenous RAX or RAX(S18A) were assayed for colony formation after 14 days of growth in soft agar. A representative crystal violet–stained plate for each cell line is shown. The transformation efficiency for each cell line was determined by averaging the number of colonies formed (groups of approximately 50 or more cells) from 4 separate plates divided by the total number of cells plated (1 × 104) ± SD.
Discussion
Our results demonstrate that RAX is required to effectively activate PKR and initiate apoptosis during a variety of cytotoxic cell-stress applications, including serum depletion, cytotoxic drug or cytokine treatment, and viral infection. Cells with reduced RAX expression display a significant defect in the stress response as evidenced by failure to fully activate PKR and phosphorylate eIF2α, degrade IκB, induce IRF-1, and phosphorylate STAT1 following IFNγ/TNFα treatment. In contrast, increased RAX expression in MEFs promoted the opposite phenotype of more rapid PKR activation, which increased eIF2α phosphorylation, IκB degradation, IRF1 expression, and STAT1 phosphorylation during IFNγ/TNFα treatment, leading to more rapid apoptosis. Thus, understanding the mechanism by which RAX regulates the molecular components of stress signaling and gene expression will be an important area of future research.
Our results also suggest that RAX is required for the host antiviral response to VSV infection, since viral protein production and titer are drastically reduced in cells expressing exogenous RAX, while alternatively, an increase in viral productivity occurs in RAX “knock-down” cells. Increased RAX expression promotes enhanced PKR activity and apoptosis following VSV infection, while cells with reduced RAX expression fail to activate PKR and display increased cell death due to rampant viral production. Although the exact mechanism by which RAX regulates the response to VSV infection remains to be determined, RAX is clearly required for PKR activation. Furthermore, since RAX, like PKR, is a dsRNA-binding protein, it seems likely that some dsRNA species produced during infection may promote RAX activation of PKR. Alternatively, it is possible that some other cellular signal activated during VSV infection initiates the RAX-dependent antiviral mechanism.
Interestingly, and consistent with findings using cells that express either a dominant-negative PKR mutant, a nonphosphorylatable eIF2α mutant, or TRBP, reduced RAX expression or expression of the nonphosphorylatable RAX(S18A) mutant promotes cellular transformation, characterized by anchorage-independent colony growth.36-39 These results suggest a role for the RAX-PKR–signaling axis in negative growth regulation such that blocking RAX expression or activation can induce cell transformation.40 However, while RAX activation of PKR may contribute to suppression of cell transformation in immortalized MEF cells, additional studies are required to determine whether RAX has a tumor suppressor activity in vivo.
Previously, we reported that expression of the nonphosphorylatable RAX(S18A) mutant led to prolonged survival (a dominant-negative phenotype) following IL-3 withdrawal from factor-dependent myeloid cells.16 Results here extend those findings to include a role for RAX phosphorylation in the cellular response to diverse stress applications and suggest that these stresses may promote RAX phosphorylation and activation. Significantly, forced expression of the nonphosphorylatable RAX(S18A) mutant in normal and Fancc–/– MEFs was found to significantly block cell hypersensitivity to IFNγ/TNFα or MMC. These findings support the notion that defects in extracellular signaling pathways and the RAX-PKR stress-response signaling axis may contribute to the DNA repair defect and inflammatory cytokine hypersensitivity phenotype reported for FA cells.29,30 These findings also suggest that blocking RAX-dependent PKR activation in FA-C patient cells may have potential therapeutic value by enhancing survival during stress. However, this possibility will now need to be tested in primary cells from FA-C patients.
In summary, results indicate that cells with decreased RAX expression not only fail to activate PKR but also display prolonged survival in response to various cytotoxic stresses, including serum starvation, treatment with cytotoxic cytokines or chemotherapy, or viral infection. We therefore propose that under various diverse stress situations RAX functions as the upstream activator of PKR in a stress → RAX → PKR signaling axis that leads to cell death. In addition, RAX expression/activation also appears to function as a negative regulator of growth since blocking expression or introducing the nonphosphorylatable RAX(S18A) mutant promotes anchorage-independent growth of MEF cells. Thus, inhibiting RAX activation by phosphorylation may not only facilitate cell survival following a stress application but also may promote cell transformation. Conversely, activating RAX may prove to be a novel antineoplastic strategy.
Acknowledgments
We would like to thank S. Smallwood for providing technical assistance with the VSV work.
Prepublished online as Blood First Edition Paper, April 11, 2006; DOI 10.1182/blood-2005-11-006817.
Supported by National Institutes of Health grant 5R01HL054083-8. W.L.B. was funded by National Research Service Award (NRSA) CA09126-27.
R.L.B. and W.L.B. contributed equally to this work.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Ito T, Jagus R, May WS. Interleukin 3 stimulates protein synthesis by regulating double-stranded RNA-dependent protein kinase. Proc Natl Acad Sci U S A. 1994;91: 7455-7459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mundschau LJ, Faller DV. Platelet-derived growth factor signal transduction through the interferon-inducible kinase PKR: immediate early gene induction. J Biol Chem. 1995;270: 3100-3106. [DOI] [PubMed] [Google Scholar]
- 3.Yeung MC, Liu J, Lau AS. An essential role for the interferon-inducible, double-stranded RNA-activated protein kinase PKR in the tumor necrosis factor-induced apoptosis in U937 cells. Proc Natl Acad Sci U S A. 1996;93: 12451-12455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gil J, Garcia MA, Gomez-Puertas P, et al. TRAF family proteins link PKR with NF-kappa B activation. Mol Cell Biol. 2004;24: 4502-4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hsu LC, Park JM, Zhang K, et al. The protein kinase PKR is required for macrophage apoptosis after activation of Toll-like receptor 4. Nature. 2004;428: 341-345. [DOI] [PubMed] [Google Scholar]
- 6.Silva AM, Whitmore M, Xu Z, Jiang Z, Li X, Williams BR. Protein kinase R (PKR) interacts with and activates mitogen-activated protein kinase kinase 6 (MKK6) in response to double-stranded RNA stimulation. J Biol Chem. 2004;279: 37670-37676. [DOI] [PubMed] [Google Scholar]
- 7.Gilbert SJ, Duance VC, Mason DJ. Does protein kinase R mediate TNF-alpha- and ceramide-induced increases in expression and activation of matrix metalloproteinases in articular cartilage by a novel mechanism? Arthritis Res Ther. 2004;6: R46-R55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peel AL, Bredesen DE. Activation of the cell stress kinase PKR in Alzheimer's disease and human amyloid precursor protein transgenic mice. Neurobiol Dis. 2003;14: 52-62. [DOI] [PubMed] [Google Scholar]
- 9.Jiang Z, Zamanian-Daryoush M, Nie H, Silva AM, Williams BR, Li X. Poly(I-C)-induced Toll-like receptor 3 (TLR3)-mediated activation of NFkappa B and MAP kinase is through an interleukin-1 receptor-associated kinase (IRAK)-independent pathway employing the signaling components TLR3-TRAF6-TAK1-TAB2-PKR. J Biol Chem. 2003;278: 16713-16719. [DOI] [PubMed] [Google Scholar]
- 10.Chang RC, Wong AK, Ng HK, Hugon J. Phosphorylation of eukaryotic initiation factor-2alpha (eIF2alpha) is associated with neuronal degeneration in Alzheimer's disease. Neuroreport. 2002;13: 2429-2432. [DOI] [PubMed] [Google Scholar]
- 11.Williams BR. Signal integration via PKR. Sci STKE. 2001;2001: RE2. [DOI] [PubMed] [Google Scholar]
- 12.Williams BR. PKR: a sentinel kinase for cellular stress. Oncogene. 1999;18: 6112-6120. [DOI] [PubMed] [Google Scholar]
- 13.Deb A, Haque SJ, Mogensen T, Silverman RH, Williams BR. RNA-dependent protein kinase PKR is required for activation of NF-kappa B by IFN-gamma in a STAT1-independent pathway. J Immunol. 2001;166: 6170-6180. [DOI] [PubMed] [Google Scholar]
- 14.Donze O, Deng J, Curran J, Sladek R, Picard D, Sonenberg N. The protein kinase PKR: a molecular clock that sequentially activates survival and death programs. EMBO J. 2004;23: 564-571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ito T, Yang M, May WS. RAX, a cellular activator for double-stranded RNA-dependent protein kinase during stress signaling. J Biol Chem. 1999;274: 15427-15432. [DOI] [PubMed] [Google Scholar]
- 16.Bennett RL, Blalock WL, May WS. Serine 18 phosphorylation of RAX, the PKR activator, is required for PKR activation and consequent translation inhibition. J Biol Chem. 2004;279: 42687-42693. [DOI] [PubMed] [Google Scholar]
- 17.Patel RC, Sen GC. PACT, a protein activator of the interferon-induced protein kinase, PKR. EMBO J. 1998;17: 4379-4390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Patel CV, Handy I, Goldsmith T, Patel RC. PACT, a stress-modulated cellular activator of interferon-induced double-stranded RNA-activated protein kinase, PKR. J Biol Chem. 2000;275: 37993-37998. [DOI] [PubMed] [Google Scholar]
- 19.Huang X, Hutchins B, Patel RC. The C-terminal, third conserved motif of the protein activator PACT plays an essential role in the activation of double-stranded-RNA-dependent protein kinase (PKR). Biochem J. 2002;366: 175-186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peters GA, Hartmann R, Qin J, Sen GC. Modular structure of PACT: distinct domains for binding and activating PKR. Mol Cell Biol. 2001;21: 1908-1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zamanian-Daryoush M, Mogensen TH, DiDonato JA, Williams BR. NF-kappaB activation by double-stranded-RNA-activated protein kinase (PKR) is mediated through NF-kappaB-inducing kinase and IkappaB kinase. Mol Cell Biol. 2000;20: 1278-1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang YL, Reis LF, Pavlovic J, et al. Deficient signaling in mice devoid of double-stranded RNA-dependent protein kinase. EMBO J. 1995;14: 6095-6106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar A, Yang YL, Flati V, et al. Deficient cytokine signaling in mouse embryo fibroblasts with a targeted deletion in the PKR gene: role of IRF-1 and NF-kappaB. EMBO J. 1997;16: 406-416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ramana CV, Grammatikakis N, Chernov M, et al. Regulation of c-myc expression by IFN-gamma through Stat1-dependent and -independent pathways. EMBO J. 2000;19: 263-272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheshire JL, Williams BR, Baldwin AS Jr. Involvement of double-stranded RNA-activated protein kinase in the synergistic activation of nuclear factor-kappaB by tumor necrosis factor-alpha and gamma-interferon in preneuronal cells. J Biol Chem. 1999;274: 4801-4806. [DOI] [PubMed] [Google Scholar]
- 26.Pang Q, Keeble W, Diaz J, et al. Role of double-stranded RNA-dependent protein kinase in mediating hypersensitivity of Fanconi anemia complementation group C cells to interferon gamma, tumor necrosis factor-alpha, and double-stranded RNA. Blood. 2001;97: 1644-1652. [DOI] [PubMed] [Google Scholar]
- 27.D'Andrea AD, Grompe M. The Fanconi anaemia/BRCA pathway. Nat Rev Cancer. 2003;3: 23-34. [DOI] [PubMed] [Google Scholar]
- 28.Zhang X, Li J, Sejas DP, Rathbun KR, Bagby GC, Pang Q. The Fanconi anemia proteins functionally interact with the protein kinase regulated by RNA (PKR). J Biol Chem. 2004;279: 43910-43919. [DOI] [PubMed] [Google Scholar]
- 29.Pang Q, Christianson TA, Keeble W, Koretsky T, Bagby GC. The anti-apoptotic function of Hsp70 in the interferon-inducible double-stranded RNA-dependent protein kinase-mediated death signaling pathway requires the Fanconi anemia protein, FANCC. J Biol Chem. 2002;277: 49638-49643. [DOI] [PubMed] [Google Scholar]
- 30.Pang Q, Christianson TA, Koretsky T, et al. Nucleophosmin interacts with and inhibits the catalytic function of eukaryotic initiation factor 2 kinase PKR. J Biol Chem. 2003;278: 41709-41717. [DOI] [PubMed] [Google Scholar]
- 31.Pang Q, Keeble W, Christianson TA, Faulkner GR, Bagby GC. FANCC interacts with Hsp70 to protect hematopoietic cells from IFN-gamma/TNF-alpha-mediated cytotoxicity. EMBO J. 2001;20: 4478-4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Der SD, Yang YL, Weissmann C, Williams BR. A double-stranded RNA-activated protein kinase-dependent pathway mediating stress-induced apoptosis. Proc Natl Acad Sci U S A. 1997;94: 3279-3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wong AH, Tam NW, Yang YL, et al. Physical association between STAT1 and the interferon-inducible protein kinase PKR and implications for interferon and double-stranded RNA signaling pathways. EMBO J. 1997;16: 1291-1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kirchhoff S, Koromilas AE, Schaper F, Grashoff M, Sonenberg N, Hauser H. IRF-1 induced cell growth inhibition and interferon induction requires the activity of the protein kinase PKR. Oncogene. 1995;11: 439-445. [PubMed] [Google Scholar]
- 35.Kibler KV, Shors T, Perkins KB, et al. Double-stranded RNA is a trigger for apoptosis in vaccinia virus-infected cells. J Virol. 1997;71: 1992-2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meurs EF, Galabru J, Barber GN, Katze MG, Hovanessian AG. Tumor suppressor function of the interferon-induced double-stranded RNA-activated protein kinase. Proc Natl Acad Sci U S A. 1993;90: 232-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Benkirane M, Neuveut C, Chun RF, et al. Oncogenic potential of TAR RNA binding protein TRBP and its regulatory interaction with RNA-dependent protein kinase PKR. EMBO J. 1997;16: 611-624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Donze O, Jagus R, Koromilas AE, Hershey JW, Sonenberg N. Abrogation of translation initiation factor eIF-2 phosphorylation causes malignant transformation of NIH 3T3 cells. EMBO J. 1995;14: 3828-3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perkins DJ, Barber GN. Defects in translational regulation mediated by the alpha subunit of eukaryotic initiation factor 2 inhibit antiviral activity and facilitate the malignant transformation of human fibroblasts. Mol Cell Biol. 2004;24: 2025-2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koromilas AE, Roy S, Barber GN, Katze MG, Sonenberg N. Malignant transformation by a mutant of the IFN-inducible dsRNA-dependent protein kinase. Science. 1992;257: 1685-1689. [DOI] [PubMed] [Google Scholar]