

Abstract
Background
Termination of translation in eukaryotes requires two release factors, eRF1, which recognizes all three nonsense codons and facilitates release of the nascent polypeptide chain, and eRF3 stimulating translation termination in a GTP-depended manner. eRF3 from different organisms possess a highly conservative C region (eRF3C), which is responsible for the function in translation termination, and almost always contain the N-terminal extension, which is inessential and vary both in structure and length. In the yeast Saccharomyces cerevisiae the N-terminal region of eRF3 is responsible for conversion of this protein into the aggregated and functionally inactive prion form.
Results
Here, we examined functional importance of the N-terminal region of a non-prion form of yeast eRF3. The screen for mutations which are lethal in combination with the SUP35-C allele encoding eRF3C revealed the sup45 mutations which alter the N-terminal domain of eRF1 and increase nonsense codon readthrough. However, further analysis showed that synthetic lethality was not caused by the increased levels of nonsense codon readthrough. Dominant mutations in SUP35-C were obtained and characterized, which remove its synthetic lethality with the identified sup45 mutations, thus indicating that synthetic lethality was not due to a disruption of interaction with proteins that bind to this eRF3 region.
Conclusion
These and other data demonstrate that the N-terminal region of eRF3 is involved both in modulation of the efficiency of translation termination and functioning of the eRF1/eRF3 complex outside of translation termination.
Background
Termination of protein synthesis takes place in ribosomes in a response to stop codons in the "decoding" site. The proteins that control this process are called polypeptide chain release factors (RFs). In eubacteria there are two class 1 polypeptide chain release factors, RF1 and RF2, recognizing UAA/UAG and UAA/UGA stop codons, respectively, whereas eukaryotes employ only one such factor, eRF1, which is able to decode all three nonsense codons [1]. In addition, translation termination in eubacteria and eukaryotes is stimulated by class 2 release factors, RF3 and eRF3, respectively. Both class 2 factors are GTPases enhancing the termination efficiency by stimulating activity of class 1 release factors in a GTP-dependent manner [2-4]. RF3 is able to remove RF1 or RF2 from the ribosome after hydrolysis of the ester bond in peptidyl tRNA, i.e. to stimulate recycling of class 1 factors [5]. In contrast to eubacterial RF3, eukaryotic eRF3 functions in termination by applying its GTPase activity to assist eRF1 with the stop codon recognition and/or ensures rapid and efficient hydrolysis of peptidyl tRNA [6]. Importantly, this was shown upon the excess of eRF1 when quantitative peptide release did not require recycling of eRF1. In the yeast Saccharomyces cerevisiae eRF1 and eRF3 are encoded by the essential SUP45 and SUP35 genes, and often designated as Sup45p and Sup35p, respectively [3].
Members of the eRF1 family have been identified in a number of eukaryotes and they all show a high degree of sequence similarity. Determination of the crystal structure of human eRF1 has shown that it is composed of three domains and resembles by overall shape and dimensions a tRNA molecule with the N-terminal and middle domains corresponding to the tRNA's anticodon stem and aminoacyl acceptor stem, respectively [7]. eRF3 also has a complex structure and can be divided into at least two regions: a non-conserved N-terminal region and a conserved C-terminal region, showing a considerable sequence similarity to the translation elongation factor eEF1A, which brings aminoacyl-tRNAs to the ribosomal A site. The C-terminal part (domain C) of the S. cerevisiae eRF3 is responsible for the function of the protein in translation termination. The N-terminal region of yeast eRF3 is non-essential for the translation termination and viability and may be further subdivided into the N-terminal (N) domain, which was shown to be required for maintenance of the cytoplasmically-inherited prion determinant [PSI+] [8,9] and the middle (M) domain, which also is important for [PSI+] propagation [10,11]. The nonsense suppressor phenotype of [PSI+] reflects eRF3 aggregation and functional inactivation [12,13]. Thus, the N domain of eRF3 may inhibit translation termination in S. cerevisiae in a [PSI+] prion-dependent manner. However, other data have shown that this domain also affects function of a non-prion form of yeast eRF3, since deletion of the eRF3 N-terminal extension (N and M domains) decreased nonsense codon readthrough caused by depletion of eRF1 [14]. This agrees with the results showing that N-terminal extension of the Schizosaccharomyces pombe eRF3 may inhibit translation termination by blocking eRF1 binding to eRF3 [15]. However, the N-terminal region of eRF3 from the filamentous fungus Podospora anserina confers quite opposite effect on translation termination, since deletion of this protein region caused suppression of nonsense mutations [16]. The effects of N-terminal truncation of eRF3 have a species-specific character probably because of the lack of sequence similarity between the N-terminal regions of eRF3 homologs from different organisms. Nevertheless, despite of structural dissimilarity, the N-terminal regions of eRF3 from yeast and mammals are able to interact with poly(A)+ binding protein PABP, which probably serves for coupling termination and initiation steps of translation [17,18] and for regulation of mRNA deadenylation and decay [19]. The N-terminal region of S. cerevisiae eRF3 also is able to bind other proteins, such as Itt1p, a protein with unknown function, and Sla1p assisting in the cytoskeleton assembly [20,21]. These interactions may modulate translation termination efficiency and link translation with other cellular processes.
Biochemical studies of S. cerevisiae eRF3 are hampered by its propensity to aggregate [22,23]. This makes genetic approach preferable to investigate function of this release factor. Here, we identified mutations which alter eRF1 and make the N-terminal region of eRF3 indispensable. The study of these mutations and their lethal interaction with the SUP35 deletion allele, encoding the eRF3 C-terminal domain, indicate the role of N-terminal region of this factor in non-translational function of the eRF1/eRF3 termination complex in yeast.
Results
Identification and characterization of mutants in which deletion of the eRF3 NM region is lethal
The SUP35-C strain YJV159-rS35C carrying the pHT-SUP35 (SUP35 ADE3) centromeric plasmid was mutagenized with UV irradiation and over 150,000 of grown colonies were screened using a yeast colony color sectoring assay. Among them, 23 were of Sect- phenotype. Such clones could arise due to mutations in the SUP35-C gene, since it is essential [8], or due to mutations in other genes. To discriminate between these possibilities, the centromeric pRS315-SUP35C (SUP35-C LEU2) plasmid was introduced into the YJV159-rS35C strain carrying pHT-SUP35. In 22 cases this restored Sect+ phenotype indicating that mutations were within SUP35-C and corresponding mutants were discarded. One mutant, which segregated white sectors very poorly, preserved its leaky Sect- phenotype in the presence of the pRS315-SUP35C plasmid. The mutant, designated as YJV-rS35C-SL23, could not grow at 37°C on YPD plate. This mutant carrying pHT-SUP35 was transformed with a genomic library based on the LEU2 centromeric plasmid p366, and two transformants able to grow at non-permissive temperature were selected. The obtained temperature resistant transformants were characterized by the frequent loss of pHT-SUP35 at permissive temperature, thus being Sect+. Plasmid from one transformant was isolated and partial sequencing of the cloned DNA fragment has shown that it contained the SUP45 gene. Subsequent delimitation of the functional region of the isolated DNA fragment indicated that SUP45 was responsible for both temperature resistance and Sect+ phenotype. Transformation of this mutant with various SUP45-carrying plasmids caused the same effects. Thus, mutation that made the NM region of eRF3 indispensable arose in the SUP45 gene. Transformation of the mutant YJV-rS35C-SL23 carrying pHT-SUP35 with the multicopy plasmid YEplac181-SUP35NM encoding the eRF3 NM region did not restore the Sect+ phenotype. This indicated that expression of the entire eRF3 protein is necessary for viability of this mutant.
The obtained sup45 mutation, designated as sup45-sl23ts, caused growth inhibition in the strain YJV-rS35C-SL23, but was lethal in other sup45-sl23ts strains with the SUP35-C allele. For example, the URA3 SUP35 plasmid pRS316-SUP35 of the SUP35-disrupted strain 33G-D373-rSL23-ΔS35 carrying sup45-sl23ts could not be replaced with the LEU2 SUP35-C plasmids upon incubation on 5FOA medium (Fig. 1). It is also important that in this strain the plasmid pRS316-SUP35 also could not be replaced with the centromeric pRS315-SUP35MC plasmid, encoding eRF3 which lacks only the N domain (Fig. 1). Thus, just the absence of the eRF3 prion-forming domain was responsible for synthetic lethality.
Figure 1.

The sup45-sl23ts mutation is lethal in the strain 33G-D373-rSL23-ΔS35, carrying the SUP35-C or SUP35-MC alleles. The LEU2 plasmids with SUP35-C or SUP35-MC can not replace the resident URA3 plasmid pRS316-SUP35 carrying wild type SUP35 in the strain 33G-D373-rSL23-ΔS35. Transformants were incubated on 5FOA-containing medium for 3 days. Plasmids: SUP35-C, pRS315-SUP35C; SUP35-MC, pRS315-SUP35MC; SUP35, pRS315-SUP35; empty vector, pRS315.
Synthetic lethality was also reproduced in the S1-R-H8 strain. This strain, carrying SUP35-C, was disrupted for SUP45 and contained the URA3 SUP45 centromeric plasmid pRG415-SUP45, which could not be replaced with the plasmid pRS315-sup45-sl23 carrying the mutant sup45-sl23ts allele. In contrast, in the control strain S1-H8 with chromosomal wild type SUP35, pRS315-sup45-sl23 could replace the resident pRG415-SUP45 plasmid. The lethal effect of combination with SUP35-C was also found for another sup45 mutation, sup45-36ts [24]: the pRS315-sup45-36 plasmid did not replace pRG415-SUP45 in the S1-R-H8 strain, but replaced this plasmid in the SUP45 disruptant S1-H8 bearing the wild type SUP35 gene.
The possible reason of lethality manifested by the sup45 mutations in combination with SUP35-C is the decreased level of eRF1. However, the amount of eRF1 was not affected in the sup45-36ts [25] and sup45-sl23ts mutants (data not shown). S. cerevisiae eRF1 and eRF3 interact with each other through the binding sites located at their C-termini [26,27], though the eRF3 N-terminal region also may be involved in this interaction [28]. Mutations in eRF1 breaking its interaction with eRF3 decrease cell viability [27]. The observed disparity in genetic interaction of the sup45-sl23ts and sup45-36ts mutations with SUP35 and SUP35-C suggests that these mutations influence binding of eRF1 to eRF3, and this effect is more pronounced when eRF3 lacks its N-terminal region. However, the study of the ability of Escherichia coli-expressed His6-eRF1 and His6-eRF1-sl23 immobilized on TALON beads to precipitate eRF3 and eRF3C from yeast cell lysates [27] did not show a noticeable effect of sup45-sl23ts mutation on the interaction of eRF1 with both eRF3 and eRF3C (data not shown).
The sup45-sl23ts and sup45-36ts mutations altered the eRF1 N domain (Ser-30 to Phe and Leu-34 to Ser substitutions, respectively), increased nonsense codon readthrough and caused temperature sensitivity [25]. At elevated temperature corresponding mutations did not cause any noticeable increase in the levels of nonsense codon readthrough (Fig. 2 and data not shown), but affected cell morphology. The sup45-36ts mutation caused defect of cytokinesis and resulted in appearance of multinucleated cells with elongated buds. The defect of cytokinesis in this mutant is related to mislocalization of the myosin light chain Mlc1p and its temperature sensitivity could be suppressed by Mlc1p overproduction [25]. In contrast, temperature sensitivity of the sup45-sl23ts mutant could not be alleviated by excess of Mlc1p. Cells of the sup45-sl23ts mutant were increased in size (Fig. 3) and contained single nuclei (data not shown). Temperature sensitivity of the sup45-sl23ts mutant can not be explained by a defect of translation termination, since increase of the number of sup45-sl23ts copies allowed mutant to grow at 37°C, but did not decrease the levels of nonsense codon readthrough either at permissive or non-permissive temperature (Fig. 2). It is also noteworthy that extra copies of the sup45-sl23ts allele did not restore viability of the strain YJV-rS35C-SL23 carrying chromosomal sup45-sl23ts and SUP35-C, indicating that temperature sensitivity and synthetic lethality were caused by the impairment of different cellular processes.
Figure 2.

Extra copies of sup45-sl23ts improve growth of the strain 33G-D373-rSL23 carrying the chromosomal sup45-sl23ts mutation at 37°C (A), but do not influence nonsense codon readthrough (B). Empty vector, YEplac181; multi-sup45-sl23, YEplac181-sup45-sl23. Photos were taken after incubation of transformants on leucine omission medium for 4 days. The strain 33G-D373-rSL23 carrying the plasmids indicated above, was transformed with either one of the pUKC815, 817, 818, 819 plasmids and grown in medium selective for plasmids to mid log phase. Then appropriate aliquots of yeast culture were taken and β-galactosidase activity was assayed. Suppression efficiency (average from three independent transformants, each in three parallels) was estimated as described [44].
Figure 3.

Cell morphology defects of the sup45 mutants incubated at 37°C. The strain 33G-D373-ΔS45 disrupted for SUP45 and bearing the plasmids pRS315-sup45-sl23 (sup45-sl23), pRS315-sup45-36 (sup45-36) or pRS315-SUP45 (SUP45), was grown in liquid YPD medium for 15 h at 37°C. Bar = 6 μm.
Mutations in SUP35-C abolishing its synthetic lethality with sup45-sl23ts or sup45-36ts
Mutations in SUP35-C which make it compatible with sup45-sl23ts were obtained by passing the corresponding plasmid in the E. coli "mutator" strain. Five independent pools of the mutagenized plasmid were obtained and used to transform the strain YJV-rS35C-SL23 which carried the chromosomal sup45-sl23ts and SUP35-C alleles and the centromeric SUP35 ADE3 pHT-SUP35 plasmid. Over 100,000 of transformed cells were incubated on leucine omission medium at 37°C and 116 Leu+ temperature resistant clones were selected. Streaking of these transformants on leucine omission medium showed that 75 of them produced colonies with Sect+ phenotype. The plasmid DNA from 8 Sect+ transformants was isolated and used for repeated transformation of the strain YJV-rS35C-SL23 carrying pHT-SUP35. All Leu+ transformants bearing the isolated plasmids were temperature resistant and able to lose the resident pHT-SUP35 (SUP35 ADE3 URA3) plasmid on medium supplemented with uracil, suggesting that they carried mutant SUP35-C. It is noteworthy that, since these mutations were dominant, they restored the normal growth rate and temperature resistance of the sup45-sl23ts mutant YJV-rS35C-SL23 carrying the chromosomal SUP35-C allele. These SUP35-C mutant alleles supported viability of the sup45-sl23ts mutant 33G-D373-rSL23-ΔS35 disrupted for SUP35 and allowed its growth at non-permissive temperature, indicating that suppression of temperature sensitivity did not depend on genetic background.
Sequencing of the mutant SUP35-C alleles revealed that they had nucleotide substitutions that caused either Glu-539 to Lys (five cases, isolated from four mutagenized plasmid pools), Ala-596 to Val (two cases, both derived from the same plasmid pool) or Ala-597 to Val (one case) replacements. It is noteworthy that these amino acid substitutions occurred in the positions located downstream to the eRF3 regions implicated in GTP hydrolysis [3,8]. The corresponding alleles were respectively designated as SUP35-C14, SUP35-C11 and SUP35-C42.
The isolated compensatory SUP35-C mutations were studied for the ability to alleviate temperature sensitivity caused by the sup45-36ts mutant allele as well as to suppress its synthetic lethality with SUP35-C. Introduction of either one of the plasmids with the SUP35-C14, SUP35-C11 or SUP35-C42 alleles into the sup45-36ts mutant 33G-D373-r36-ΔS35, which was disrupted for SUP35 and carried the pRG415-SUP35 (URA3 SUP35) plasmid, allowed the loss of the resident plasmid on 5-FOA-containing medium at permissive temperature (Fig. 4), but did not cause resistance to 37°C. Thus, the compensatory mutations suppressed lethality caused by the combination of either sup45-sl23ts or sup45-36ts with SUP35-C, but did not suppress temperature sensitivity of sup45-36ts.
Figure 4.
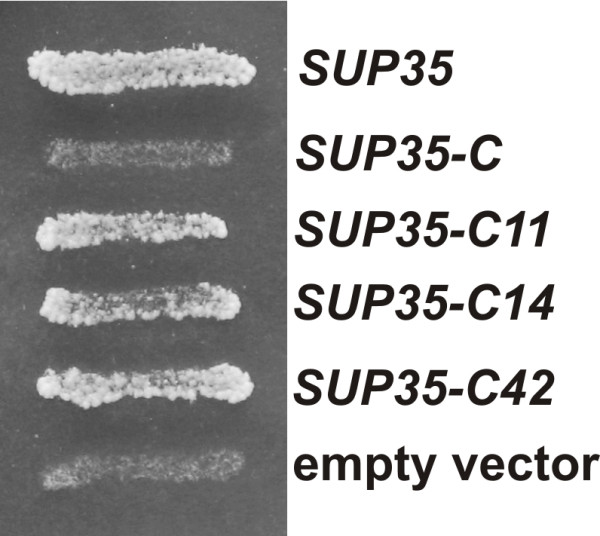
Compensatory mutations in plasmid SUP35-C restore viability of the strain carrying the sup45-36ts and SUP35-C alleles. The sup45-36ts mutant 33G-D373-r36-ΔS35 disrupted for SUP35 and carrying the SUP35 URA3 pRS316-SUP35 plasmid was transformed with either one of the LEU2 plasmids pRS315-SUP35 (SUP35), pRS315-SUP35C (SUP35-C), pRS315-SUP35-C11 (SUP35-C11), pRS315-SUP35-C14 (SUP35-C14), pRS315-SUP35-C42 (SUP35-C42) or pRS315 (empty vector). Transformants were incubated on 5FOA-containing medium for 3 days.
Synthetic lethality between sup45-sl23ts or sup45-36ts and SUP35-C is not caused by the increased levels of nonsense codon readthrough
We also tested, whether the SUP35-C compensatory mutations influenced the nonsense codon readthrough caused by the sup45-sl23ts and sup45-36ts mutations. For this, the strain 33G-D373-rSL23 carrying the chromosomal sup45-sl23ts mutation and wild type SUP35, was transformed with centromeric plasmids containing either the SUP35-C allele (control) or alleles with the SUP35-C14, SUP35-C11 or SUP35-C42 compensatory mutations along with either one of the pUKC815, 817, 818, 819 plasmids. Quantitative assay demonstrated that the nonsense codon readthrough was noticeably decreased only in the presence of SUP35-C11. This compensatory mutation also decreased the levels of nonsense readthrough in the sup45-36ts mutant 33G-D373-r36 carrying wild type SUP35 but to a lesser extent than in the isogenic sup45-sl23ts mutant 33G-D373-rSL23 (Fig. 5). Thus, though the effect of SUP35-C compensatory mutations on the levels of nonsense readthrough was not significant in most cases, they all abolished synthetic lethality, making unlikely that the increased levels of readthrough was the reason of lethal interaction between the sup45ts mutations and SUP35-C allele.
Figure 5.

Influence of the compensatory mutations in SUP35-C on the levels of nonsense codon readthrough in the sup45-sl23ts 33G-D373-rSL23 (A) and sup45-36ts 33G-D373-r36 (B) mutants. Plasmids: SUP35-C, pRS315-SUP35C; SUP35-C11, pRS315-SUP35C11; SUP35-C14, pRS315-SUP35C14; SUP35-C42, pRS315-SUP35C42. Suppression efficiency (average from three independent transformants, each in three parallels) was estimated with the use of the pUKC815, 817, 818, 819 plasmids.
To confirm this we have constructed the strains 33G-D373-rSL23-r35C and 33G-D373-r36-r35C, which carried, respectively, the sup45-sl23ts and sup45-36ts mutations and the SUP35-C allele, as well as the centromeric pCM183-SUP45 plasmid with the TRP1 selectable marker and SUP45 under the control of regulatable tetO2 promoter. The synthetic lethality of sup45ts and SUP35-C combination in these strains was manifested only upon the repression of wild type SUP45 by doxycycline. As a control we used the isogenic strains 33G-D373-rSL23 and 33G-D373-r36, which had the wild type SUP35 gene instead of the SUP35-C allele. To monitor the levels of nonsense codon readthrough upon the repression of wild type SUP45, all strains were transformed with either one of the pUKC815, 817, 818, 819 plasmids.
Since chromosomally-encoded mutant eRF1 and its wild type counterpart encoded by the plasmid were electrophoretically indistinguishable, we could not estimate a decline in the amount of eRF1 upon the repression of the plasmid SUP45 in the studied strains. However, in another strain with the same tetO2-SUP45 plasmid, 17 h repression decreased the amount of eRF1 to approximately 5% of its initial levels. Importantly, this drop in wild type eRF1 amount was accompanied with enrichment of cultures with dead cells up to 20% for the 33G-D373-rSL23-r35C and 33G-D373-r36-r35C strains, while the number of such cells in the 33G-D373-rSL23 and 33G-D373-r36 cultures was less than 10%. The in vivo quantification of UAA, UAG, and UGA nonsense readthrough has shown that its levels in the strains with synthetic lethal mutations was not higher than in the control strains (Fig. 6). This disagrees with the assumption that the cause of synthetic lethality between sup45 mutations and SUP35-C allele is too high levels of nonsense codon readthrough. Incubation of the strain 33G-D373-rSL23-r35C carrying the tetO2-SUP45 plasmid in medium containing doxycycline did not cause any change in the polyribosome content or distribution (data not shown), indicating absence of the sharp effect on translation.
Figure 6.

Interaction between the sup45ts and SUP35-C alleles does not result in increase of the efficiency of nonsense codon readthrough. Data for the 33G-D373-rSL23 (sup45-sl23ts SUP35) and 33G-D373-rSL23-r35C (sup45-sl23ts SUP35-C) strains (A), as well as for 33G-D373-r36 (sup45-36ts SUP35) and 33G-D373-r36-r35C (sup45-36ts SUP35-C) strains (B) are presented. All strains carrying the pCM183-SUP45 plasmid (tetO2-SUP45) were transformed with either one of the pUKC815, 817, 818, 819 plasmids and grown in tryptophane omission medium to mid log phase. Then the cells were collected, resuspended in analogous medium containing 10 μg/ml doxycycline. After 17 h of incubation in this medium, appropriate aliquots of yeast culture were taken and β-galactosidase activity was assayed. All data represent an average of at least three independent experiments.
Discussion
Here, we have demonstrated that the sup45-sl23ts mutation did not decrease the levels of eRF1, and sup45-sl23ts extra copies neither suppressed synthetic lethality, nor decreased nonsense codon readthrough. This indicates that both defect of translation termination and lethal interaction between sup45-sl23ts and SUP35-C were not due to a shortage of eRF1, making improbable suggestion that these effects were due to inefficient recycling of mutant eRF1 by eRF3 lacking the N-terminal region. Furthermore, such suggestion contradicts to our previous data indicating that the NM region of eRF3 may negatively affect its ability to recycle eRF1. It was shown that shortage of eRF1 in the strain expressing full length eRF3 resulted in higher levels of nonsense codon readthrough than in the strain expressing eRF3C [14], while deletion of the eRF3 NM region did not affect nonsense readthrough at the normal levels of eRF1 [examined in the presence of the tRNA suppressor SUP16 (SUQ5) to enable detection of a decrease in nonsense readthrough levels; our unpublished data]. This suggests that the presence of eRF3 NM region has a significant negative effect on translation termination only at the decreased eRF1 levels. Deficiency in eRF1 should make its recycling important for translation termination and, therefore, these data agreed with the hypothesis that eRF3 is involved in the recycling of eRF1 and indicated that NM region of eRF3 interferes with this process. Thus, deletion of NM region should enhance the eRF3 activity, making improbable that the reason of synthetic lethal interaction between SUP35-C and sup45-sl23ts or sup45-36ts lies in inefficient translation termination. Furthermore, the results of this work suggest that combination of SUP35-C with either sup45-sl23ts or sup45-36ts is lethal not due to an increase of nonsense codon readthrough.
The obtained SUP35-C mutations compensate for the lack of the eRF3 N-terminal region, thus ruling out the possibility that synthetic lethality was due to a disruption of interaction with proteins that bind to this eRF3 region, including interaction with the poly(A)+-binding protein PABP [18,19]. This makes unlikely that synthetic lethality was caused by a disturbance in regulation of mRNA decay or by alteration of translation initiation by its uncoupling from termination. The latter is also supported by the observation that synthetic lethality was not accompanied by noticeable alteration of the polyribosomal profile.
Both identified sup45 mutations, sup45-sl23ts and sup45-36ts, that make the N-terminal region of eRF3 indispensable, altered the N domain of eRF1 and, furthermore, they caused substitutions of amino acids located in proximity to each other. Both sup45-sl23ts and sup45-36ts increased the levels of nonsense codon readthrough and, therefore, affected translation termination. In addition, they did not allow yeast cells to grow at 37°C. Earlier it was found that temperature sensitivity of the sup45-36ts mutant was not due to a defect of translation termination. Inability to grow at non-permissive temperature was caused by the impairment of cytokinesis resulting from mislocalization of the eRF1-binding protein, myosin light chain Mlc1p [25]. Though cytokinesis defect was not shown for the sup45-sl23ts mutant, its temperature sensitivity also was not caused by a defect in translation termination, suggesting that yeast eRF1 may have different functions outside of translation. It is also important that the compensatory SUP35-C mutations alleviated both effects of the sup45-sl23ts mutation, temperature sensitivity and increased nonsense readthrough, indicating that interaction with eRF3 probably is important for eRF1 not only for promoting translation termination, but also for its uncharacterized function. In contrast, interaction with eRF3 is not required for function of eRF1 in cytokinesis, since eRF1 in a complex with Mlc1p can not bind eRF3 [25].
Conclusion
The data presented here show that the effect of synthetic lethality can not be deduced from a defect of translation termination. The study of the SUP35-C compensatory mutations has shown that they are able to suppress both temperature sensitivity of sup45-sl23ts and its lethal interaction with SUP35-C. However, temperature sensitivity of sup45-sl23ts and its synthetic lethality with SUP35-C are caused by different reasons, since increasing the copy-number of sup45-sl23ts alleviated temperature sensitivity but did not abolish synthetic lethality. Therefore, these data show that the eRF1/eRF3 complex may be involved in different processes outside of translation termination.
In many yeast species the N-terminal part of the eRF3 release factors is responsible for their prion properties [29-33]. Such conservation suggests that the prion properties of yeast eRF3 have biological significance. In this work we have shown that the prion-forming domain of eRF3 from the yeast S. cerevisiae is important for its functioning outside of translation termination. However, this does not mean that the N-terminal extension of eRF3 factors from other organisms plays a similar role. Indeed, point mutations altering S. cerevisiae eRF3C can compensate for the lack of its NM region indicating that the C domain of eRF3 from other organisms may contain all information required for this function.
Methods
Media and strains
Yeast strains were grown on standard organic (YPD) and synthetic (SD) media [34]. The modified YPD media (YPDm) [2% peptone, 0.5% yeast extract (Oxoid), 3% glucose and 2% agar] was used in colony color sectoring assay experiments, because it favored accumulation of red pigment in the ade2 mutants. The 5-fluoroorotic acid (5FOA) medium was prepared as described [35]. The expression of the tetO2-controlled SUP45 was repressed by incubation of corresponding strains on medium which contained 10 μg/ml doxycycline and was selective for the pCM183-SUP45 plasmid. LB and 2 × YT media were used for bacteria [36]. Appropriate amounts of antibiotics, amino acids, and bases were added when necessary. Yeast cells were grown at 30°C, if not indicated otherwise, and bacteria at 37°C. DNA transformation of lithium acetate-treated yeast cells was performed as described previously [37]. E. coli cells were transformed by the method described in [38].
The E. coli mutator strain XL1-Red [endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac mutD5 mutS mutT Tn10 (Tetr)] (Stratagene) was used for obtaining mutations in the plasmid SUP35-C gene. For this, the plasmid pRS315-SUP35C was introduced into this E. coli strain. After incubation for 48 h, over 500 transformant colonies were suspended in 10 ml of 2 × YT supplemented with ampicillin (100 μg ml-1). Cell culture was additionally incubated for 8 h with shaking, plasmid DNA was isolated and used to transform yeast. The E. coli strain DH5α [supE44 Dlac U169 (f80 lacZDM15) hsdR17 recA1 endA1 gyrA96 thi-1 relA1] [36] was used in cloning experiments.
The yeast strains used are listed along with their genotypes in Table 1. Construction of strains obtained in this study is described below. The strain YJV159-rS35C, which contains the 5'-deletion SUP35-C allele (encoding the eRF3 fragment lacking amino acids 1-253, eRF3C) in place of the wild-type SUP35 allele, was obtained as follows. The pFL44-SUP35C integrative plasmid [8] bearing the SUP35-C allele was linearized by SalI site internal to the SUP35 coding sequence and integrated into the SUP35 gene of the strain YJV159 by selecting transformants on uracil omission medium. The Ura+ integrants expressing both eRF3 and eRF3C were obtained and placed onto 5FOA-containing medium to select for plasmid excision. The obtained Ura- clones were screened by Western blotting to select strains expressing eRF3C only. Replacement of SUP35 with SUP35-C was further confirmed by Southern analysis. The same integration/excision method was used to obtain the temperature-sensitive sup45 mutants 33G-D373-rSL23 (sup45-sl23ts) and 33G-D373-r36 (sup45-36ts). The integrative plasmids pJJ244-sup45-sl23 and pJJ244-sup45-36 bearing the sup45-sl23ts and sup45-36ts alleles, respectively, were linearized by MscI and integrated into the chromosomal SUP45 gene of the strain 33G-D373. The obtained Ura+ transformants were placed on 5FOA-containing medium for plasmid excision and Ura- clones with the suppressor (growth on tryptophane and histidine omission media) and temperature sensitive (inability to grow on YPD at 37°C) phenotypes were selected. Then, the obtained strains 33G-D373-rSL23 and 33G-D373-r36 were disrupted for SUP35 to obtain respectively the strains 33G-D373-rSL23-ΔS35 and 33G-D373-r36-ΔS35. To perform this, the XhoI-Ppu10I DNA fragment of the plasmid pSUP35::TRP1 was used to transform the strains 33G-D373-rSL23 and 33G-D373-r36 harboring the pRS316-SUP35 (SUP35 URA3) plasmid. The clones unable to grow on 5FOA plates (without pRS316-SUP35) were selected. Disruption of SUP35 in these strains was confirmed by PCR analysis. The strains 33G-D373-rSL23-rS35C (sup45-sl23tsSUP35-C) and 33G-D373-r36-rS35C (sup45-36tsSUP35-C) were constructed in the same way as YJV159-rS35C. However, the pRS315-SUP45 (SUP45 LEU2) plasmid was introduced into the 33G-D373-rSL23 and 33G-D373-r36 strains before the integration/excision procedure to prevent synthetic lethality of the sup45ts and SUP35-C alleles. The pRS315-SUP45 plasmid was then replaced with the pCM183-SUP45 (tetO2-SUP45 TRP1) plasmid.
Table 1.
S cerevisiae strains used in this study
| Strain | Genotype | Source/reference |
| YJV159 | MATa ade2 ade3 his3 leu2-3,112 trp1 ura3 | [46] |
| YJV159-rS35C | Same as in YJV159, but with SUP35-C instead of SUP35 | This study |
| YJV159-rS35C-SL23 | Same as in YJV159-rS35C, but with sup45-sl23ts instead of SUP45 | This study |
| 33G-D373 | MATa ura3-52 leu2-3 112 trp1-289 his7-1 lys9-A21 pheA10 ade2-144, 717 | [47] |
| 33G-D373-rSL23 | Same as in 33G-D373, but with sup45-sl23ts instead of SUP45 | This study |
| 33G-D373-rSL23-rS35C | Same as 33G-D373-rSL23 but with SUP35-C instead of SUP35 | This study |
| 33G-D373-r36 | Same as in 33G-D373, but with sup45-36ts instead of SUP45 | This study |
| 33G-D373-r36-rS35C | Same as 33G-D373-r36 but with SUP35-C instead of SUP35 | This study |
| 33G-D373-rSL23-ΔS35 | Same as in 33G-D373-rSL23, but with sup35::TRP1 | This study |
| 33G-D373-r36-ΔS35 | Same as in 33G-D373-r36, but with sup35::TRP1 | This study |
| 33G-D373-ΔS45 | Same as in 33G-D373, but with sup45::ADE2 | [25] |
| S1-H8 | MATa leu2-3,112 his3-Δ1 ura3-52 trp1-289 sup45::TRP1 | [14] |
| S1-R-H8 | Same as in s1-H8, but with SUP35-C instead of SUP35 | [14] |
his7-1 and lys9-A21 and ade2-1, ochre mutations, trp1-289, amber mutation.
Plasmids
The plasmids used are listed along with their essential characteristics in Table 2. The pRS315-SUP35, pRS316-SUP35, pHT-SUP35 and pSUP35::TRP1 plasmids were constructed as follows. The SUP35-containing XhoI-XbaI DNA fragment of the plasmid pEMBLyex4-SUP35 [8] was inserted into the XbaI-SalI sites of pRS315, pRS316, pET-30a(+) (Novagen) and pBluescript II KS(+) (Stratagene) vectors giving the plasmids pRS315-SUP35, pRS316-SUP35, pET-S35 and pBS-SUP35, respectively. Next, the XbaI-NotI DNA fragment of pET-S35 containing SUP35 was cloned into the same sites of the centromeric ADE3 URA3 vector pHT4467 [39] resulting in pHT-SUP35. The TRP1-containing Acc65I-BglII DNA fragment of the pJJ281 plasmid [40] was cloned into the BsrGI and BclI sites of pBS-SUP35. HpaI-HpaI DNA fragment of the resulting plasmid was deleted, and thus, the plasmid pSUP35::TRP1 used for disruption of the chromosomal SUP35 gene was constructed. The MluI/Klenow-XbaI DNA fragment of the plasmid pEMBLyex4-SUP35C containing the 5'-deletion SUP35-C allele [8] was inserted between the SmaI and XbaI sites of pRS315 and as a result the plasmid pRS315-SUP35C was obtained. The plasmid pRS315-SUP35MC was constructed by the insertion of PvuII-XbaI DNA fragment of pEMBLyex4-SUP35MC [8] which contained the mutant SUP35-MC allele encoding eRF3 without the N domain into the SmaI and XbaI sites of pRS315. Mutant alleles SUP35-C11, SUP35-C14 and SUP35-C42, cloned into the shuttle vector pRS315, were sequenced using a set of primers specific for SUP35.
Table 2.
Plasmids used in this study
| Plasmid | Characteristics | Source/reference |
| YEplac181 | Multicopy LEU2 vector | [48] |
| pRS315 | Centromeric LEU2 vector | [49] |
| pRS316 | Centromeric URA3 vector | [49] |
| pRG415-SUP45 | Centromeric URA3 vector with SUP45 | [14] |
| pRS315-SUP45 | Same as pRS315, but with SUP45 | [25] |
| pRS315-sup45-36 | Same as pRS315, but with the sup45-36ts mutant allele | [25] |
| pRS315-sup45-sl23 | Same as pRS315, but with the sup45-sl23ts mutant allele | [25] |
| YEplac181-sup45-sl23 | Same as YEplac181, but the sup45-sl23ts mutant allele | This study |
| pRS315-SUP35 | Same as pRS315, but with SUP35 | This study |
| pRS315-SUP35-C11 | Same as pRS315, but with SUP35-C11 | This study |
| pRS315-SUP35-C14 | Same as pRS315, but with SUP35-C14 | This study |
| pRS315-SUP35-C42 | Same as pRS315, but with SUP35-C42 | This study |
| pRS316-SUP35 | Same as pRS316, but with SUP35 | This study |
| pFL44-SUP35C | Integrative URA3 vector with SUP35-C | [8] |
| pSUP35::TRP1 | Integrative vector with the sup35::TRP1 disruption allele | This study |
| pHT-SUP35 | Centromeric ADE3 URA3 vector containing SUP35 | This study |
| pRS315-SUP35C | Same as pRS315, but with SUP35-C | This study |
| pRS315-SUP35MC | Same as pRS315, but with SUP35-MC | This study |
| pJJ244-sup45-sl23 | Integrative URA3 vector with the sup45-sl23ts allele | This study |
| pJJ244-sup45-36 | Integrative URA3 vector with the sup45-36ts allele | This study |
| pCM183-SUP45 | Centromeric TRP1 vector containing SUP45 under the control of regulatable tetO2 promoter | This study |
The SUP45 gene was cloned using a yeast genomic library based on the plasmid p366, which was identical to YCp50 [41], but had LEU2 instead of URA3 (gift of P. Hieter). Isolation and sequencing of the sup45-sl23ts allele was described earlier [25]. The plasmid YEplac181-sup45-sl23 was obtained by insertion of the XbaI-XhoI DNA fragment of pRS315-sup45-sl23 containing the mutant sup45-sl23ts allele into the XbaI and SalI sites of YEplac181. The XhoI-SacI DNA fragments carrying the sup45 mutant alleles of the plasmids pRS315-sup45-36 and pRS315-sup45-sl23 were cloned between the SalI and SacI sites of the integrative URA3 vector pJJ244 [40] to obtain the plasmids pJJ244-sup45-36 and pJJ244-sup45-sl23, respectively. The pCM183-SUP45 plasmid bearing the SUP45 gene under the control of regulatable tetO2 promoter was constructed as follows. Using the plasmid pRS315-SUP45 as template, PCR was applied to place the NcoI site (CCATGG) at the SUP45 translation initiation codon. The obtained PCR fragment was digested with NcoI and XhoI and cloned into the same sites of the E. coli plasmid pET-30a(+) (Novagen) to generate the plasmid pL51-3. The KpnI-XhoI fragment of pL51-3, containing the coding sequence of SUP45, was inserted into the KpnI and SalI sites of YEplac112 resulting in the YEplac112-atgSUP45 plasmid. Then, the EcoICRI-PstI fragment of YEplac112-atgSUP45 was cloned into the HpaI and PstI sites of pCM183 [42].
β-galactosidase activity in cell lysates was quantified as described [43]. The levels of UAA, UAG and UGA readthrough were determined using the pUKC815/817/818/819 series vectors, as described previously [44].
Colony color sectoring assay
The assay is based on the facts that ade2 yeast cells accumulate a red pigment and form red colonies but ade2 ade3 cells do not accumulate this pigment and, consequently, form white colonies because the ade3 mutation blocks the pathway at a point prior to the accumulation of the pigment [45]. Thus, a strain that is ade2 ade3, but carries ADE3 on an autonomously replicating plasmid, forms on medium non-selective for this plasmid red colonies (cells with the plasmid) that contain white sectors (cells that have lost the plasmid). We refer this phenotype as Sect+ (ability to lose the ADE3 plasmid), and, consequently, the phenotype of red clones without white sectors or with a negligible number of such sectors is Sect- (inability to lose the ADE3 plasmid). Multiple suspensions of the SUP35-C ade2 ade3 strain YJV159-rS35C carrying the centromeric plasmid pHT-SUP35 (SUP35 ADE3) were prepared in liquid uracil omission medium; each suspension deriving from a separate colony grown on uracil omission medium was diluted, and plated on ten YPDm plates at approximately 5,000 cells per plate. These cells were mutagenized with UV irradiation to approximately 10% survival and then incubated for 4 or 5 days. As expected, most clones were Sect+. Cells from Sect- colonies were streaked on YPDm and after 2 days, individual colonies were restreaked on YPDm. Only strains that continued to give red colonies or red colonies with rare white sectors were studied further.
Authors' contributions
VU, performed a screen for the synthetic lethal mutations and participated in molecular genetic studies, IV carried out the molecular genetic studies and drafted the manuscript, NK-P, AP, AV and OG carried out the molecular genetic studies, VS participated in planning of experiments and drafted the manuscript, MT-A designed the study and wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgments
Acknowledgements
We are grateful to P. Hieter for providing the yeast genomic library and G. Fominov for critical reading of the manuscript. This work was supported by grants from the Howard Hughes Medical Institute (grant 55000337) and Russian Foundation for Basic Research.
Contributor Information
Valery N Urakov, Email: urakov@cardio.ru.
Igor A Valouev, Email: igva@cardio.ru.
Natalia V Kochneva-Pervukhova, Email: Nataly@cardio.ru.
Anna N Packeiser, Email: molgen@cardio.ru.
Alexander Yu Vishnevsky, Email: auvish@mail.ru.
Oleg O Glebov, Email: og@mrc-lmb.cam.ac.uk.
Vladimir N Smirnov, Email: v.n.smirnov@mtu-net.ru.
Michael D Ter-Avanesyan, Email: mdter@cardio.ru.
References
- Frolova L, Le Goff X, Rasmussen HH, Cheperegin S, Drugeon G, Kress M, Arman I, Haenni AL, Celis JE, Philippe M, Justesen J, Kisselev LL. A highly conserved eukaryotic protein family possessing properties of polypeptide chain release factor. Nature. 1994;372:701–703. doi: 10.1038/372701a0. [DOI] [PubMed] [Google Scholar]
- Zhouravleva G, Frolova L, Le Goff X, Le Guellec R, Inge-Vechtomov SG, Kisselev LL, Philippe M. Termination of translation in eukaryotes is governed by two interacting polypeptide chain release factors, eRF1 and eRF3. EMBO J. 1995;14:4065–4072. doi: 10.1002/j.1460-2075.1995.tb00078.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stansfield I, Jones KM, Kushnirov VV, Dagkesamanskaya AR, Poznyakovski AI, Paushkin SV, Nierras CR, Cox BS, Ter-Avanesyan MD, Tuite MF. The products of the SUP45 (eRF1) and SUP35 genes interact to mediate translation termination in Saccharomyces cerevisiae. EMBO J. 1995;14:4365–4373. doi: 10.1002/j.1460-2075.1995.tb00111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frolova L, Le Goff X, Zhouravleva G, Davydova E, Philippe M, Kisselev L. Eukaryotic polypeptide chain release factor eRF3 is an eRF1- and ribosome-dependent guanosine triphosphatase. RNA. 1996;2:334–341. [PMC free article] [PubMed] [Google Scholar]
- Zavialov AV, Buckingham RH, Ehrenberg M. A posttermination ribosomal complex is the guanidine nucleotide exchange factor for peptide release factor RF3. Cell. 2001;107:115–124. doi: 10.1016/S0092-8674(01)00508-6. [DOI] [PubMed] [Google Scholar]
- Alkalaeva EZ, Pisarev AV, Frolova LY, Kisselev LL, Pestova TV. In vitro reconstitution of eukaryotic translation reveals cooperativity between release factors eRF1 and eRF3. Cell. 2006;125:1125–1136. doi: 10.1016/j.cell.2006.04.035. [DOI] [PubMed] [Google Scholar]
- Song H, Mugnier P, Das AK, Webb HM, Evans DR, Tuite MF, Hemmings BA, Barford D. The crystal structure of human eukaryotic release factor eRF1 – mechanism of stop codon recognition and peptidyl-tRNA hydrolysis. Cell. 2000;100:311–321. doi: 10.1016/S0092-8674(00)80667-4. [DOI] [PubMed] [Google Scholar]
- Ter-Avanesyan MD, Kushnirov VV, Dagkesamanskaya AR, Didichenko SA, Chernoff YO, Inge-Vechtomov SG, Smirnov VN. Deletion analysis of the SUP35 gene of the yeast Saccharomyces cerevisiae reveals two non-overlapping functional regions in the encoded protein. Mol Microbiol. 1993;7:683–692. doi: 10.1111/j.1365-2958.1993.tb01159.x. [DOI] [PubMed] [Google Scholar]
- Ter-Avanesyan MD, Dagkesamanskaya AR, Kushnirov VV, Smirnov VN. The SUP35 omnipotent suppressor gene is involved in the maintenance of the non-Mendelian determinant [psi+] in the yeast Saccharomyces cerevisiae. Genetics. 1994;137:671–676. doi: 10.1093/genetics/137.3.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JJ, Sondheimer N, Lindquist SL. Changes in the middle region of Sup35 profoundly alter the nature of epigenetic inheritance for the yeast prion [PSI+] Proc Natl Acad Sci USA. 2002;99:16446–16453. doi: 10.1073/pnas.252652099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley ME, Liebman SW. The Sup35 domains required for maintenance of weak, strong or undifferentiated yeast [PSI+] prions. Mol Microbiol. 2004;51:1649–1659. doi: 10.1111/j.1365-2958.2003.03955.x. [DOI] [PubMed] [Google Scholar]
- Paushkin SV, Kushnirov VV, Smirnov VN, Ter-Avanesyan MD. Propagation of the yeast prion-like [PSI+] determinant is mediated by oligomerization of the SUP35 -encoded polypeptide chain release factor. EMBO J. 1996;15:3127–3134. [PMC free article] [PubMed] [Google Scholar]
- Patino MM, Liu JJ, Glover JR, Lindquist SL. Support for the prion hypothesis for inheritance of a phenotypic trait in yeast. Science. 1996;273:622–626. doi: 10.1126/science.273.5275.622. [DOI] [PubMed] [Google Scholar]
- Valouev IA, Kushnirov VV, Ter-Avanesyan MD. Yeast polypeptide chain release factors eRF1 and eRF3 are involved in cytoskeleton organization and cell cycle regulation. Cell Motil Cytoskeleton. 2002;52:161–173. doi: 10.1002/cm.10040. [DOI] [PubMed] [Google Scholar]
- Kong C, Ito K, Walsh MA, Wada M, Liu Y, Kumar S, Barford D, Nakamura Y, Song H. Crystal structure and functional analysis of the eukaryotic class II release factor eRF3 form S. pombe. Mol Cell. 2004;14:233–245. doi: 10.1016/S1097-2765(04)00206-0. [DOI] [PubMed] [Google Scholar]
- Gagny B, Silar P. Identification of the genes encoding the cytosolic translation release factors from Podospora anserina and analysis of their role during the life cycle. Genetics. 1988;149:1763–1775. doi: 10.1093/genetics/149.4.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino S, Imai M, Kobayashi T, Uchida N, Katada T. The eukaryotic polypeptide chain releasing factor (eRF3/GSPT) carrying the translation termination signal to the 3'-poly(A) tail of mRNA. Direct association of eRF3/GSPT with polyadenylate-binding protein. J Biol Chem. 1999;274:16677–16680. doi: 10.1074/jbc.274.24.16677. [DOI] [PubMed] [Google Scholar]
- Cosson B, Couturier A, Chabelskaya S, Kiktev D, Inge-Vechtomov S, Philippe M, Zhouravleva G. Poly(A)-binding protein acts in translation termination via eukaryotic release factor interaction and does not influence [PSI+] propagation. Mol Cell Biol. 2002;22:3301–3315. doi: 10.1128/MCB.22.10.3301-3315.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoda N, Kobayashi T, Uchida N, Funakoshi Y, Kikuchi Y, Hoshino S, Katada T. Translation termination factor eRF3 mediates mRNA decay through the regulation of deanylation. J Biol Chem. 2003;278:28287–38291. doi: 10.1074/jbc.C300300200. [DOI] [PubMed] [Google Scholar]
- Urakov VN, Valouev IA, Lewitin EI, Paushkin SV, Kosorukov VS, Kushnirov VV, Smirnov VN, Ter-Avanesyan MD. Itt1p, a novel protein inhibiting translation termination in Saccharomyces cerevisiae. BMC Mol Biol. 2001;2:9–18. doi: 10.1186/1471-2199-2-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailleul PA, Newnam GP, Steenbergen JN, Chernoff YO. Genetic study of interactions between the cytoskeletal assembly protein Sla1 and prion-forming domain of the release factor Sup35 (eRF3) in Saccharomyces cerevisiae. Genetics. 1999;153:81–94. doi: 10.1093/genetics/153.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover JR, Kowal AS, Schirmer EC, Patino MM, Liu JJ, Lindquist S. Self-seeded fibers formed by Sup35 the protein determinant of [PSI+], a heritable prion-like factor of S. cerevisiae. Cell. 1997;89:811–819. doi: 10.1016/S0092-8674(00)80264-0. [DOI] [PubMed] [Google Scholar]
- King C-Y, Tittmann P, Gross H, Gebert R, Aebi M, Wuthrich K. Prion-inducing domain 2-114 of yeast Sup35 protein transforms in vitro into amyloid-like filaments. Proc Natl Acad Sci USA. 1997;94:6618–6622. doi: 10.1073/pnas.94.13.6618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breining P, Piepersberg W. Yeast omnipotent suppressor SUP1 (SUP45): nucleotide sequence of the wild type and a mutant gene. Nucleic Acids Res. 1986;14:5187–5197. doi: 10.1093/nar/14.13.5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valouev IA, Urakov VN, Kochneva-Pervukhova NV, Ter-Avanesyan MD. Translation termination factors function outside of translation: yeast eRF1 interacts with myosin light chain, Mlc1p, to effect cytokinesis. Mol Microbiol. 2004;53:687–696. doi: 10.1111/j.1365-2958.2004.04157.x. [DOI] [PubMed] [Google Scholar]
- Ebihara K, Nakamura Y. C-terminal interaction of translational release factors eRF1 and eRF3 of fission yeast: G-domain uncoupled binding and the role of conserved amino acids. RNA. 1999;5:739–750. doi: 10.1017/S135583829998216X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eurwilaichitr L, Graves FM, Stansfield I, Tuite MF. The C-terminus of eRF1 defines a functionally important domain for translation termination in Saccharomyces cerevisiae. Mol Microbiol. 1999;32:485–496. doi: 10.1046/j.1365-2958.1999.01346.x. [DOI] [PubMed] [Google Scholar]
- Paushkin SV, Kushnirov VV, Smirnov VN, Ter-Avanesyan MD. Interaction between yeast Sup45p (eRF1) and Sup35p (eRF3) polypeptide chain release factors: implications for prion-dependent regulation. Mol Cell Biol. 1997;17:2798–2805. doi: 10.1128/mcb.17.5.2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoso A, Chien P, Osherovich LZ, Weissman JS. Molecular basis of a yeast prion species barrier. Cell. 2000;100:277–288. doi: 10.1016/S0092-8674(00)81565-2. [DOI] [PubMed] [Google Scholar]
- Kushnirov VV, Kochneva-Pervukhova NV, Chechenova MB, Frolova NS, Ter-Avanesyan MD. Prion properties of the Sup35 protein of yeast Pichia methanolica. EMBO J. 2000;19:324–331. doi: 10.1093/emboj/19.3.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernoff YO, Galkin AP, Lewitin E, Chernova TA, Newnam GP, Belenkiy SM. Evolutionary conservation of prion-forming abilities of the yeast Sup35 protein. Mol Microbiol. 2000;35:865–876. doi: 10.1046/j.1365-2958.2000.01761.x. [DOI] [PubMed] [Google Scholar]
- Nakayashiki T, Ebihara K, Bannai H, Nakamura Y. Yeast [PSI+] "prions" that are crosstransmissible and susceptible beyond a species barrier through a quasi-prion state. Mol Cell. 2001;7:1121–1130. doi: 10.1016/S1097-2765(01)00259-3. [DOI] [PubMed] [Google Scholar]
- Hara H, Nakayashiki T, Crist CG, Nakamura Y. Prion domain interaction responsible for species discrimination in yeast [PSI+] transmission. Genes to Cells. 2004;8:925–939. doi: 10.1111/j.1365-2443.2003.00694.x. [DOI] [PubMed] [Google Scholar]
- Sherman F, Fink GR, Hicks JB. Methods in yeast genetics. Cold Spring Harbor Laboratory Press; 1986. [Google Scholar]
- Boeke JD, LaCroute F, Fink GR. A positive selection for mutants lacking orotidine-5'-phosphate decarboxylase activity in yeast: 5-fluoro-orotic acid resistance. Mol Gen Genet. 1984;197:345–346. doi: 10.1007/BF00330984. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EE, Maniatis T. Molecular Cloning: A Laboratory Manual. 2. Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Gietz RD, Woods RA. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 2002;350:87–96. doi: 10.1016/s0076-6879(02)50957-5. [DOI] [PubMed] [Google Scholar]
- Inoue H, Nojima H, Okayama H. High efficiency transformation of Escherichia coli with plasmids. Gene. 1990;96:23–28. doi: 10.1016/0378-1119(90)90336-P. [DOI] [PubMed] [Google Scholar]
- Lafontaine DLJ, Preiss T, Tollervey D. Yeast 18S rRNA dimethylase Dim1p: a quality control mechanism in ribosome synthesis? Mol Cell Biol. 1998;18:2360–2370. doi: 10.1128/mcb.18.4.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones JS, Prakash L. Yeast Saccharomyces cerevisiae selectable markers in pUC18 polylinkers. Yeast. 1990;6:363–366. doi: 10.1002/yea.320060502. [DOI] [PubMed] [Google Scholar]
- Rose MD, Novick P, Thomas JH, Botstein D, Fink GR. A Saccharomyces cerevisiae genomic bank based on a centromere-containing shuttle vector. Gene. 1987;60:237–243. doi: 10.1016/0378-1119(87)90232-0. [DOI] [PubMed] [Google Scholar]
- Gari E, Piedrafita L, Aldea M, Herrero E. A set of vectors with tetracycline-regulatable promoter system for modulated gene expression in Saccharomyces cerevisiae. Yeast. 1997;13:837–848. doi: 10.1002/(SICI)1097-0061(199707)13:9<837::AID-YEA145>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Miller JH. Experiments in Molecular Genetics. Cold Spring Harbor Laboratory Press; 1972. [Google Scholar]
- Stansfield I, Akhmaloka , Tuite MF. A mutant allele of the SUP45 (SAL4) gene of Saccharomyces cerevisiae shows temperature-dependent allosuppressor and omnipotent suppressor phenotypes. Curr Genet. 1995;27:417–426. doi: 10.1007/BF00311210. [DOI] [PubMed] [Google Scholar]
- Roman H. Studies of gene mutation in Saccharomyces. Cold Spring Harbor Symp Quant Biol. 1956;21:175–185. doi: 10.1101/sqb.1956.021.01.015. [DOI] [PubMed] [Google Scholar]
- Fortes P, Kufel J, Fornerod M, Policarpou-Schwarz M, Lafontaine D, Tollervey D, Mattaj IW. Genetic and physical interactions involving the yeast nuclear cap-binding complex. Mol Cell Biol. 1999;19:6543–6553. doi: 10.1128/mcb.19.10.6543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernoff YO, Inge-Vechtomov SG, Derkach IL, Ptyushkina MV, Tarunina OV, Dagkesamanskaya AR, Ter-Avanesyan MD. Dosage-dependent translational suppression in yeast Saccharomyces cerevisiae. Yeast. 1992;8:489–499. doi: 10.1002/yea.320080702. [DOI] [PubMed] [Google Scholar]
- Gietz RD, Sugino A. New yeast- Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene. 1988;74:527–534. doi: 10.1016/0378-1119(88)90185-0. [DOI] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]