

Abstract
Varicella-zoster virus (VZV) glycoprotein E (gE) is a multifunctional protein important for cell-cell spread, envelopment, and possibly entry. In contrast to other alphaherpesviruses, gE is essential for VZV replication. Interestingly, the N-terminal region of gE, comprised of amino acids 1 to 188, was shown not to be conserved in the other alphaherpesviruses by bioinformatics analysis. Mutational analysis was performed to investigate the functions associated with this unique gE N-terminal region. Linker insertions, serine-to-alanine mutations, and deletions were introduced in the gE N-terminal region in the VZV genome, and the effects of these mutations on virus replication and cell-cell spread, gE trafficking and localization, virion formation, and replication in vivo in the skin were analyzed. In summary, mutagenesis of the gE N-terminal region identified a new functional region in the VZV gE ectodomain essential for cell-cell spread and the pathogenesis of VZV skin tropism and demonstrated that different subdomains of the unique N-terminal region had specific roles in viral replication, cell-cell spread, and secondary envelopment.
Varicella-zoster virus (VZV) is a human alphaherpesvirus that causes two clinically different diseases: varicella (or chickenpox), during the primary infection, and zoster (or shingles), during virus reactivation from latency (3). The VZV genome is about 125 kb, the smallest among the human herpesviruses, and encodes at least 70 unique open reading frames (ORFs) (10, 17, 22). Although VZV and herpes simplex virus (HSV) types 1 and 2, the other two human alphaherpesviruses, show similarities in genome organization and share a number of homologous genes, VZV has unique mechanisms of pathogenesis that must be explained by its genetic differences from HSV. These mechanisms include T-cell tropism, which results in cell-associated viremia during primary VZV infection, and the characteristic formation of large polykaryocytes due to cell-cell fusion during skin infection (29).
The VZV genome encodes nine putative glycoproteins that are known or presumed to be involved in different steps during the viral replication cycle: attachment and entry into the target cell, envelopment of the viral particles, cell-cell spread, and egress. The glycoprotein E (gE) is a 623-amino-acid (aa) typical type I membrane glycoprotein encoded by ORF68. gE is the most abundant glycoprotein expressed on the plasma membrane and in the cytoplasm of infected cells, and it is present on the virion envelope (10, 20). gE is a multifunctional protein that has been shown to be involved in cell fusion and to localize to the trans-Golgi network (TGN), where the virus undergoes secondary envelopment (13, 33, 34, 51).
VZV gE moves from the endoplasmic reticulum to the Golgi membrane for its complete maturation (20). gE is recycled from the plasma membrane and targeted to the TGN through signal sequences contained in the C terminus (1, 63), which have been identified as the endocytosis motif YAGL (aa 582 to 585), and the TGN localization signal AYRV (aa 568 to 571) (48, 64). In addition, the C-terminal domain contains an “acid patch” (aa 588 to 601), with serines and threonines that are targets for phosphorylation (62); the differential phosphorylation of these residues by cellular casein kinase II and the viral ORF47 kinase regulates the trafficking of gE to the plasma membrane or to the TGN (26). When these gE C-terminal motifs were disrupted by mutagenesis in the context of the viral genome, the C-terminal domain and specifically the endocytosis motif were essential for VZV replication, whereas mutation of the TGN localization signal was compatible with replication but reduced VZV infection of skin and T cells in vivo (39). VZV gE also accelerated tight-junction formation between polarized epithelial cells, with or without concomitant expression of gI, and colocalized with the tight-junction protein ZO-1. Of interest, two regions of the gE ectodomain show similarities with the adhesion molecules E-cadherin and desmocollin (37).
Like its homolog in the other alphaherpesviruses, VZV gE forms noncovalent heterodimers with gI (ORF67), which facilitates gE endocytosis and accumulation in the TGN (27, 47, 59, 61). Although VZV gI is not essential for viral replication in vitro, impaired cell-cell spread and altered posttranslational modification and trafficking of gE were shown in cells infected with VZV gI deletion mutants (31, 36). In addition, deleting gI or its C terminus resulted in abnormal envelopment of cytoplasmic virions, suggesting that gE-gI heterodimers are required to produce mature, enveloped virions (59). Of interest, this process requires gD as well as gE/gI in HSV (19). Importantly, gI is essential for replication in skin and T-cell xenografts, indicating that gE trafficking and maturation, cell-cell spread, and virion assembly, as modulated by gI, are required in differentiated human cells in vivo (38).
Three facts render the function of gE in VZV of particular interest. First, in contrast to other alphaherpesviruses, VZV gE is essential for viral replication (31, 36). Second, gE and gI are the only glycoproteins encoded by the unique short region (US) in VZV (15, 16), which lacks the glycoprotein gD, the receptor-binding protein in HSV, pseudorabiesvirus (PrV), and bovine herpesvirus type 1 (BHV-1) (9, 54, 55). VZV gE has been considered likely to have gD-like functions (13). Third, although gE is conserved among the alphaherpesviruses, VZV gE has only 47% similarity and 27% identity to HSV-1 gE. The highest homology occurs between amino acids 328 and 500 of VZV gE, one of the two cysteine-rich domains of the glycoprotein (30), and a deletion in this region decreased gE/gI complex formation, which is consistent with the conservation of this domain to preserve the gE/gI interactions that are also observed in HSV-1 and the other alphaherpesviruses (61). Together, these facts strongly suggest that VZV gE, besides sharing the functions of gE in the other alphaherpesviruses, most of which involve interactions with gI, may have some other distinct and unique functions.
The purpose of these experiments was to extend our investigations of the functional domains of VZV gE by focusing on the large ectodomain of the protein. These experiments were based on the observation that the first 188 amino acids of the VZV gE N terminus did not appear to be conserved compared with gE homologues by bioinformatics methods. We took advantage of the cosmid system to carry out a mutational analysis of the functions of this unique gE N-terminal region in the context of the viral genome (12, 25, 31). Since deletion of this gE N-terminal region proved to be incompatible with VZV replication from cosmids, we further examined this segment of VZV gE using targeted deletions, linker insertion mutagenesis, and conservative mutations in which serines were mutated into alanines. We assessed the effects of these mutations on growth kinetics and syncytium formation, the intracellular trafficking and maturation of gE, interactions with gI, and virion formation in vitro and on VZV infection of skin xenografts in a SCIDhu model of VZV pathogenesis in vivo (7, 38-42).
MATERIALS AND METHODS
Cell lines.
Human melanoma cells, MeWo (21), were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, nonessential amino acids, and antibiotics.
Construction of ORF68 mutant cosmids.
The complete genome of the VZV parental Oka strain (P-Oka) is contained in four overlapping SuperCos 1 cosmid vectors (Stratagene, La Jolla, CA) designated pFsp73 (nucleotide [nt] 1 to 33128), pSpe14 (nt 21795 to 61868), pPme2 (nt 53755 to 96035), and pSpe23 (nt 94055 to 125124) (44). ORF68 (nt 115911 to 117782 in P-Oka; nt 115808 to 117679 in Dumas strain) is located in the unique short region of the VZV genome in the pSpe23 cosmid. A SacI fragment of about 6 kb (nt 112015 to 118091 in P-Oka) that contains the ORF68 sequence was cloned in pBluescript II SK+ plasmid (Stratagene, La Jolla, CA) in which the KpnI site was eliminated (pBSKΔKpnI). In this plasmid (named pBSKΔKpnI-SacI), the KpnI (nt 115230 in P-Oka) and BglII (nt 116390 in P-Oka) sites, located 680 bp upstream and 480 bp downstream to the ORF68 ATG, respectively, are unique sites, and they were used to insert the ORF68 mutations. The pBSKΔKpnI-SacI plasmid was modified to eliminate the EcoRI in the polylinker (pBSKΔKpnI-EcoRI-SacI); in this plasmid KpnI, BglII, and EcoRI (nt 117137 in P-Oka) sites are unique sites. pBSKΔKpnI-SacI plasmid was used as template for PCR-based mutagenesis.
gE mutagenesis in the N-terminal region of aa 1 to 188.
Eleven gE mutants in the region of amino acids 1 to 188 were constructed, consisting of two serine-to-alanine alterations, six linker insertions, and three deletions with linker insertion. PCR mutagenesis was performed with two steps of PCR to introduce the appropriate mutation in the KpnI-BglII fragment or in the BglII-EcoRI fragment.
(i) Serine-to-alanine mutation.
Alanine substitutions were made for serine in positions 31 and 49. The strategy used for the alanine substitution mutants was previously described (4). Briefly, in the first step two PCRs were made using a 5′-KpnI-outside forward primer (5′-GGTCCGGTACCTCCCTGTTT-3′) in combination with the internal mutagenic reverse primer and the 3′-BglII-outside reverse primer (5′-GGATTAAGATCTCCTTTAAACACG-3′) with the internal mutagenic forward primer (underlining indicates the restriction enzyme site). Thirty-nucleotide internal mutagenic primers were designed in which the mutation was inserted after the first 10 nt; in this way the two internal mutagenic primers overlap for 20 nt (4). In the second step, the two PCR products were combined and used as template for the second PCR. For this PCR, the two outside primers were used, and the 1,160 bp KpnI-BglII fragment, containing the mutation, was amplified. This fragment was cloned in the pCR4-TOPO vector (Invitrogen, Carlsbad, CA) and sequenced to verify the mutation.
(ii) Linker insertion mutagenesis.
For the linker insertion mutants, a 12-nt linker containing a NotI site (5′-TTGCGGCCGCAA-3′) was inserted after the amino acid in positions 16, 27, 51, 90, 146, and 187. This linker inserts a Leu-Arg-Pro-Gln peptide in frame with the gE sequence. The linker insertion mutants were also constructed in two PCR steps. In the first step, two amplification reactions were made using the 5′-KpnI-outside forward primer (see above) or the 5′-BglII-outside forward primer (5′-CATCCACGTTATCCCTACGTT-3′) in combination with the internal mutagenic reverse primer, as well as the 3′-BglII-outside reverse primer (see above) or the 3′-EcoRI-outside reverse primer (5′-GTACAACCGGAATTCATATGAGA-3′) in combination with the internal mutagenic forward primer (underlining indicates the restriction enzyme site). The internal primers contain the linker sequence 5′ of each mutagenic primer; the primers were designed to insert the linker sequence after the amino acids 16, 27, 51, 90, 146, and 187. The two PCR products were gel purified and restricted with NotI. The two fragments were combined and the ligase reaction was performed for 2 h at room temperature. The ligase product was used as the substrate for the second PCR. This PCR was performed with the outside primers to amplify the KpnI-BglII fragment or the BglII-EcoRI fragment with the linker sequence inserted in the appropriate position. These fragments were cloned in pCR4-TOPO vector and sequenced to verify the mutation.
(iii) Construction of deletion mutants with the NotI linker.
The linker insertion mutants 27, 51, and 187, cloned in pBSKΔKpnI-SacI or pBSKΔKpnI-EcoRI-SacI, were used to produce three deletions, ΔP27-Y51 (from amino acids 28 to 51), ΔP27-P187 (from amino acids 28 to 187), and ΔY51-P187 (from amino acids 52 to 187). These mutants were generated by restrictions using the NotI site located in the linker sequence, producing the plasmids pBSKΔKpnI-SacIΔP27-Y51, pBSKΔKpnI-EcoRI-SacIΔP27-P187, and pBSKΔKpnI-EcoRI-SacIΔY51-P187. These mutants contain the linker sequence inserted at the site of deletion between amino acids in positions 27 and 52, 27 and 188, and 51 and 188, respectively.
(iv) Cloning of the mutant gE in the pSpe23ΔAvrII cosmid.
The pCR4-TOPO plasmid containing the KpnI-BglII or BglII-EcoRI fragment with the alanine substitution and the linker insertion were digested with KpnI and BglII or BglII-EcoRI, and the fragment was inserted in pBSKΔKpnI-SacI or pBSKΔKpnI-EcoRI-SacI, substituting for the wild-type fragment. The pBSKΔKpnI-SacI and pBSKΔKpnI-EcoRI-SacI plasmids, containing the different gE mutations, were digested with AvrII and SgrAI and inserted in the pSpe23ΔAvrII cosmid, in which the AvrII site in position 112956 in the P-Oka genome (Dumas strain, nt 112854) is a unique site, substituting for the wild-type AvrII-SgrAI fragment (nt 112956 to 117355 in P-Oka).
(v) Cloning of the wild-type gE in the AvrII site in pSpe23ΔAvrII cosmid.
For the rescue of gE mutations which were incompatible with VZV growth, a fragment containing the intact ORF68 as well as 266 bp upstream and 204 bp downstream (nt 115645 to 117986 in P-Oka) was inserted in the AvrII restriction site (nt 112956 in P-Oka) in pSpe23ΔAvrII. The cosmids were sequenced to verify the rescue fragment inserted (Biotech Core, Inc., Sunnyvale, CA).
Construction of expression plasmids and transfection.
The wild-type gE sequence and the ΔP27-P187 mutant gE sequence were cloned in the expression plasmid pCDNA3.1+ (Invitrogen, Carlsbad, CA). The wild-type and mutated gE proteins were amplified using the gEp-fw (5′-CGCAAGCTTGCCACCATGGGACAGTTAATAAACCTG-3′) and gEp-rev primers (5′-CGCCTCGAGTCACCGGGTCTTATCTATATAC-3′) containing a HindIII and an XhoI site, respectively (underlined in the sequences above). The gEp-fw primer contains the Kozak sequence after the HindIII site. The PCR fragments were cloned in pCR4-TOPO plasmid and sequenced. The pCR4-TOPO plasmids containing the different PCR fragments were digested with HindIII and XhoI, and the fragment was inserted in pCDNA3.1+ in the HindIII and XhoI sites.
Melanoma cells were plated in 6 wells at a density of 1 × 106 cells per well. Twenty-four hours later, cells were transfected with 5 μg of plasmid with LipofectAMINE 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. Forty-eight hours posttransfection, the cells were fixed in 4% paraformaldehyde and processed for immunofluorescence.
Cosmid transfection and DNA isolation.
Cosmid DNA preparation and transfection procedures were done as previously described (31, 53). DNA was isolated from transfected cells using DNAzol (Gibco-BRL, Grand Island, N.Y.), and PCR and sequencing were performed to confirm the expected mutations.
Infectious focus assay.
Melanoma cells were seeded in a 6-well plate, infected with 1 × 103 PFU per well at day 0, and cultured for 6 days. Wells were trypsinized every day for 6 days, centrifuged, and resuspended in 1 ml of culture medium. The infected cells were serially diluted 10-fold, and 0.1 ml was added to melanoma cells in 24-well plates in triplicate. Cells were fixed in crystal violet in 20% ethanol, and plaques were counted in an inverted light microscope (magnification, ×4). For the rOka-ΔP27-Y51 and rOka-ΔY51-P187 mutants, cells were fixed in 4% paraformaldehyde and stained with polyclonal anti-VZV human immune serum (40) and secondary anti-human biotin (Vector Laboratories, Inc., Burlingame, CA). Cells were stained with the Fast Red substrate (Sigma). Statistical difference in growth kinetics was determined by the Student's t test.
Antibodies.
gE was detected with the anti-gE monoclonal antibody (MAb) purchased from Chemicon. Mutant rOka-ΔY51-P187 and ΔP27-P187 gE protein were detected with MAb 7G8 directed against the C terminus of the protein, a kind gift of Bagher Forghani, California Department of Health Services, Berkeley. The anti-gI polyclonal antibody was kindly provided by Saul Silverstein, Columbia University, NY. The trans-Golgi network was stained with the sheep anti-human TGN46 antibody (Serotec).
Confocal microscopy.
Melanoma cells infected with rOka and rOka-gE mutants viruses were fixed in 4% paraformaldehyde and incubated with 10% goat or donkey serum, 0.1% Triton X-100 in phosphate-buffered saline (PBS) for 1 h. The cells were incubated with the appropriate primary antibody in 1% goat serum, 0.1% Triton X-100 in PBS overnight at 4°C. The cells were washed three times in PBS and stained with the secondary antibodies Texas Red-coupled goat anti-mouse or donkey anti-mouse and fluorescein isothiocyanate-coupled goat anti-rabbit or donkey anti-sheep (Jackson ImmunoResearch, Inc., West Grove, PA) in 1% goat or donkey serum, 0.1% Triton X-100 in PBS. The cells were washed in PBS and mounted with Vectashield (Vector Laboratories, Inc., Burlingame, CA). Confocal analysis was performed at the Cell Sciences Imaging Facility (Stanford, CA) with a Zeiss LSM 510 confocal laser-scanning microscope (Carl Zeiss, Inc.).
Western blotting.
Melanoma cells were infected with rOka-gE mutant virus along with the rOka control, and cell lysates were collected in radioimmunoprecipitation assay buffer at 24 h and 72 h postinfection. Proteins were separated on a 10% polyacrylamide gel and transferred to a nitrocellulose membrane. Mouse anti-gE antibody 3B3 (51) was used at a dilution of 1:8,000. Rabbit anti-IE4 (a kind gift of Paul Kinchington, University of Pittsburgh, Pa.) was used at a dilution of 1:500, and mouse anti-α-tubulin monoclonal antibody (Sigma) was used at a 1:10,000 dilution. Antibodies were detected with horseradish peroxidase-conjugated sheep anti-mouse and sheep anti-rabbit antibody (Amersham) at dilutions of 1:2,000 to 1:8,000 and visualized by ECL (Amersham).
Electron microscopy.
Melanoma cell monolayers were prepared on carbon-coated and vacuum oven-baked coverslips and infected with rOka and the rOka-gE mutants for 72 h. The monolayers were rinsed three times with PBS, fixed in 1% glutaraldehyde in sodium cacodylate buffer for 2 h on ice, and processed as described before (7).
Skin xenografts infected with rOka and rOka gE mutants were recovered 21 days after inoculation. The tissue was cut and fixed with 4% paraformaldehyde for 30 min. Samples were processed as described elsewhere (7). The samples were viewed with a Hitachi S-4000 scanning electron microscope (Hitachi Instruments, Inc., Mito City, Japan). For transmission electron microscopy (TEM) experiments, monolayers were prepared in 12-well tissue culture plates, infected with each virus for 72 h, and processed as previously described (7). Skin xenografts were embedded in Epon and processed as described before (7). The samples were viewed with a JEM-1230 transmission electron microscope (JEOL-USA, Inc., Peabody, MA). Immunoscanning electron microscopy (SEM) and TEM were done by using antibody to label the protein of interest before EM was performed. Sections were placed on grids and blocked in 5% goat serum. Subsequently, the sections were incubated with anti-VZV antibody. Thereafter the grids were washed again and incubated with a secondary antibody conjugated with gold beads.
For TEM of rOka-ΔY51-P187-infected cells, primary human lung fibroblasts (MRC-5) were seeded on microscope glass coverslips and infected with 1 × 103 PFU of rOka and rOka-ΔY51-P187. After 48 h, the cells were fixed in 2% glutaraldehyde in PBS (pH 7.2) for 35 min at 4°C, rinsed in PBS, and postfixed in 1% osmium tetroxide for 1 h. Samples were washed in distilled water two times for 10 min and stained in 1% aqueous uranyl acetate for 15 min or 0.25% aqueous uranyl acetate overnight. Samples were dehydrated through a graded series of alcohol and infiltrated with 1:1 ethyl alcohol/epoxy for 30 min and 100% epoxy for 1 h. Gelatin capsules filled with embedding media (Epoxy) were placed on the cells, and samples were polymerized in the oven at 60°C for 12 h. Specimens were sectioned with Ultra microtome (Leica), and sections were collected on copper grids, stained with 2% uranyl acetate for 10 min and 1% lead citrate for 4 min, and examined with a Phillips CM-12 transmission electron microscope.
Infection of skin xenografts in SCIDhu mice.
Skin xenografts were made in homozygous CB-17scid/scid mice, using human fetal skin tissue obtained according to federal and state regulations (40, 41). Animal use was in accordance with the Animal Welfare Act and was approved by the Stanford University Administrative Panel on Laboratory Animal Care. rOka and rOka-gE mutant viruses were passed three times in primary human lung fibroblasts (MRC-5) and inoculated in the xenografts. Infectious virus titer was determined for each inoculum at the time of inoculation. Skin xenografts were harvested after 10 and 21 days, and virus recovered from each implant was analyzed by infectious focus assay. Xenografts infected with rOka-ΔY51-P187 mutant were collected after 10 and 22 days and analyzed by infectious focus assay and immunohistochemistry with polyclonal anti-VZV human immune serum (40). Virus recovered from the tissues was tested by PCR and sequencing to confirm the expected mutations.
RESULTS
Effects of mutagenesis of the gE N-terminal region on VZV replication.
To investigate the potential functions of the VZV gE ectodomain, we analyzed this 544-amino-acid domain for conserved domains using the “Conserved Domains Databases” (32). This analysis indicated that the first 188 amino acids had no significant relatedness to the gE proteins of the other alphaherpesviruses (Fig. 1). The alignment of VZV gE with these gE proteins started from amino acid 189 (Fig. 1B); similarity to gE from the closely related simian varicella virus (SVV; cercopithecine herpesvirus 9) was observed beginning with SVV gE amino acid 173 (Fig. 1B). In accordance with this conserved domain analysis, the N-terminal region of aa 1 to 188 analyzed with the “Basic Local Alignment Search Tool” (BLAST) (2) matched the VZV gE sequence but had no similarity with any other alphaherpesvirus gE proteins (data not shown). Analysis of the conserved motif was also performed with MEME (5) using the GCG software (Wisconsin Package version 11; Genetics Computer Group, Madison, WI). Conserved motifs were found comparing VZV gE and gE from HSV-1, SVV-9, and PrV-1; these motifs were located in VZV gE between aa 351 and 486 but not in the N terminus. To further confirm that the N-terminal region of VZV gE was not a conserved domain, VZV gE and gE from HSV-1, SVV-9, BHV-1, and PrV-1 were compared with CLUSTALW (56) and T-COFFEE (45) multiple-sequence alignments. This analysis did not show any significant alignment of the VZV N-terminal region of aa 1 to 188 with the other gE proteins (data not shown). These results indicated that the first 188 amino acids of the N terminus represented a domain that was specific for VZV gE, and it was not conserved in gE homologues of any other alphaherpesviruses.
FIG. 1.

Identification of the conserved domain in VZV gE. The 544-amino-acid ectodomain of VZV gE was analyzed for conserved domains with the “Conserved Domains Databases” using the Pfam database (32). (A) VZV gE ectodomain of amino acids 1 to 544 and the position of the conserved domain. (B) Alignment of VZV gE and gE in the other alphaherpesviruses. The first 80 amino acids of the alignment are shown. Numbers on the left indicate the amino acid from which the conserved domain starts; numbers at the far left are the accession numbers. BHV, bovine herpesvirus; VZV, varicella-zoster virus; SVV, simian varicella virus; EHV, equine herpesvirus; FeHV, feline herpesvirus; CaHV, canine herpesvirus; PRV, pseudorabiesvirus; GaHV, gallid herpesvirus; HSV, herpes simplex virus; CeHV, SA8.
To investigate the functions associated with this 188-amino-acid region of VZV gE, six linker insertions, two alanine substitutions, and two partial deletions were made by mutagenesis of ORF68 in the pOka cosmid pSpe23ΔAvrII (Fig. 2A and B). A dodecameric linker containing a NotI site and coding for the 4-amino-acid peptide Leu-Arg-Pro-Gln was inserted by PCR in frame after each of the gE amino acid residues 16, 27, 51, 90, 146, and 187. In addition, conservative alanine substitutions were made for serines in positions 31 and 49. Finally, deletions were created between gE amino acids P27-Y51 and Y51-P187; these mutations contained the linker sequence inserted at the site of each deletion. The mutated pSpe23ΔAvrII cosmid was transfected along with the cosmids pPme2, pSpe14, and pFsp73 (Fig. 2B). Separate transfections were done using two different clones of each mutated cosmid.
FIG. 2.

gE mutagenesis. (A) Schematic representation of the gE mutations. The gE glycoprotein and the positions of the mutations are represented. The position of the linker insertions (▿, line 1), serine-to-alanine alterations (thick bar, line 2), and deletions (Δ, line 3) are indicated. Amino acids are numbered from the N terminus to the C terminus of the gE protein. The black box represents the transmembrane domain. (B) Schema of the cosmid system. Line 1, schematic representation of the VZV genome; line 2, diagram of the overlapping cosmids containing the VZV genome (parental Oka); line 3, the SacI fragment from the pSpe23 cosmid containing the ORF68 gene; line 4, insertion of the gE rescue cassette in the AvrII site. pSpe23gE-R indicates the rescued gE mutants. (C) Summary of the results from gE mutagenesis. Recovery of recombinant virus, rescue of the mutation with lethal phenotype, and level of gE expression are indicated for each mutation. ND, not done; NL, normal. An asterisk indicates that the expression of mutant ΔY51-P187 and ΔP27-P187 gE was analyzed by immunofluorescence only (see Fig. 3, 4, and 6P to R). TRL, terminal repeat long; UL, unique long region; IRL, internal repeat long; IRS, inverted repeat short; US, unique short region; TRS, terminal repeat short.
Infectious virus was recovered using cosmids that introduced linker insertions into gE at P27, Y51, G90, I146, and P187 but not the G16 linker insertion (Fig. 2C). Infectious virus was also recovered from both of the alanine substitutions, S31A and S49A, and the two partial deletions, ΔP27-Y51 and ΔY51-P187 (Fig. 2C). Infectious virus was recovered in transfections done with only one of two independently derived mutated cosmids that had the linker insertion at I146 or the ΔY51-P187 deletion along with the other three intact cosmids. These recombinant viruses were designated rOka-P27, rOka-Y51, rOka-G90, rOka-I146, rOka-P187, rOka-S31A, rOka-S49A, rOka-ΔP27-Y51, and rOka-ΔY51-P187. The expected mutations were confirmed by PCR and sequencing (data not shown).
The linker insertion at G16 is located in the potential signal peptide (amino acids 1 to 24). Transfection of the G16 mutant cosmid with the other intact cosmids did not yield infectious VZV in four independent transfections with two separately derived cosmid clones. To demonstrate that the failure to recover infectious virus was likely due to disrupting the potential signal peptide, the intact ORF68, along with the flanking nucleotides from 266 bp upstream and 204 bp downstream of ORF68, was inserted in both orientations at the unique AvrII site in pSpe23-G16 to create repaired cosmids designated pSpe23-G16R (Fig. 2B) (36). Recombinant viruses were recovered from two independently derived pSpe23-G16R cosmid clones, and the presence of the G16 mutation as well as the rescue cassette was confirmed by sequencing. These experiments indicated that the first 16 amino acids of gE, which constitute most of the 24-residue signal peptide that is predicted by sequence analysis, were needed for VZV replication. This observation is consistent with our previous experiments showing that VZV gE is an essential protein (36).
The unique N-terminal region of gE is essential for viral replication.
Since the first 188 amino acids of VZV gE were not conserved in the alphaherpesvirus gE homologues (Fig. 1), we tested the effect of deleting this region on VZV replication. Based on the G16 linker insertion experiments, the region from amino acids 27 to 187 was deleted, leaving the potential signal peptide intact (Fig. 2A). The mutant cosmid, designated pSpe23-ΔP27-P187, contained the linker sequence at the site of the deletion. Transfection of two different pSpe23-ΔP27-P187 cosmids with the intact pPme2, pSpe14, and pFsp73 cosmids did not yield infectious virus in four independent transfections. Transfected cells were passed every 3 to 4 days for 4 weeks to detect delayed replication. These results suggested that the complete deletion of the N-terminal region of gE, leaving the signal peptide intact, was not compatible with VZV replication.
To provide evidence that the failure to recover virus was accounted for by removing the region of the ectodomain that is unique to VZV gE, we inserted the intact ORF68 rescue cassette, with flanking nucleotides, into the AvrII site in both orientations in pSpe23-ΔP27-P187 (Fig. 2B). Recombinant virus was recovered in transfection with two independently derived pSpe23-ΔP27-P187R cosmid clones. In addition, transfection of the pSpe23 cosmid in which a copy of the ΔP27-P187-mutated gE with flanking sequences was inserted into the AvrII site in both orientations was also compatible with viral replication.
The failure to recover VZV from cosmids carrying the ΔP27-P187 mutation could be explained in two ways. The N-terminal deletion might cause misfolding of gE, which is then degraded rapidly, or deletion of this N-terminal region might abolish an essential function. To demonstrate that the lethal phenotype was not related to instability or failure to produce the mutant protein, the ORF68 mutation encoding ΔP27-P187 was cloned in the expression plasmid pcDNA3.1+, and melanoma cells were transfected with this plasmid along with a plasmid expressing VZV gI. Intact gE, when expressed with gI, was localized both at the plasma membrane and in the perinuclear region in the TGN (Fig. 3A to C and G to I); the TGN localization was confirmed by gE colocalization with the TGN marker TGN46 (Fig. 4A to C). The mutant ΔP27-P187 gE was expressed in the cytoplasm, although in a more diffuse pattern than that of intact gE, and showed some colocalization with gI and with the TGN marker (Fig. 3D to F and 4D to F). However, most importantly, ΔP27-P187 gE did not reach the plasma membrane and did not colocalize with gI at this site (Fig. 3J to L). These data suggested that the failure to recover infectious VZV with pSpe23-ΔP27-P187 mutant cosmids was likely related to impaired trafficking and localization of the mutant ΔP27-P187 gE protein. Thus, this unique region of the VZV gE ectodomain, which is distinct from the domains in gE homologues that are required for gE/gI heterodimer formation and from the trafficking motifs in the VZV gE C terminus, appears to be necessary for VZV gE expression on the plasma membrane.
FIG. 3.

Localization of mutant gE ΔP27-P187 in melanoma cells. Melanoma cells were cotransfected with wild-type gE (A to C and G to I) or mutant ΔP27-P187 gE (D to F and J to L) and gI, and they were analyzed 48 h posttransfection. Transfected cells were fixed and permeabilized (A to F) or fixed without permeabilization to detect the staining at the plasma membrane (G to L). gE (red) was detected with MAb 7G8, which recognizes the C terminus of the protein; gI (green) was detected with an anti-rabbit antibody against gI; nuclei were stained with 4′,6′-diamidino-2-phenylindole (blue). Magnification, ×63. wt, wild type.
FIG. 4.

Localization of mutant gE ΔP27-P187 and the trans-Golgi network (TGN) in melanoma cells. Melanoma cells were cotransfected with wild-type gE (A to C) or mutant ΔP27-P187 (D to F) and gI, and they were analyzed 48 h posttransfection. gE (red) was detected with MAb 7G8; the TGN was stained with the sheep anti-human TGN46 antibody; nuclei were stained with 4′,6′-diamidino-2-phenylindole (blue). Magnification, ×63. wt, wild type.
Growth kinetics and plaque morphologies of rOka-gE N-terminal mutant viruses in vitro.
The growth kinetics of the alanine substitution mutant rOka-S49A were the same as those for rOka, with a peak titer at day 5 (Fig. 5A). In contrast, rOka-S31A, the second alanine substitution mutant, showed lower titers than rOka between days 3 and 6, and the peak titer, which occurred at day 4, was significantly lower than the peak rOka titer at day 5 (P = 0.029) (Fig. 5A). Plaque sizes of these mutants were not different from rOka. The growth kinetics of the linker insertion mutants rOka-Y51, rOka-G90, rOka-I146, and rOka-P187 were the same as those for rOka (Fig. 5A and B), and there were no significant differences in plaque sizes. However, the rOka-P27 mutant showed delayed growth compared to rOka during the first 3 days, although titers were similar to those of rOka at days 4 to 6 (Fig. 5A). rOka-P27 showed a minor reduction in mean plaque size, which was 0.87 mm ± 0.17 mm SD (standard deviation), versus 0.98 mm ± 0.20 mm SD for rOka (P = 0.011). The titers produced by the deletion mutant rOka-ΔP27-Y51 were the same as those of rOka at all time points (Fig. 5C), but the plaque size was reduced; the mean was 0.69 mm ± 0.23 mm SD for the rOka-ΔP27-Y51 mutant and 1.01 mm ± 0.18 mm SD for rOka (P < 0.05). Replication of rOka-ΔY51-P187 was reduced (Fig. 5C), and plaque size was also significantly smaller, with a mean of 0.65 mm ± 0.16 mm SD for rOka-ΔY51-P187 and 1.01 mm ± 0.18 mm SD for rOka (P < 0.05). Thus, the linker insertion at P27, the alanine substitution at S31, and deleting the unique ectodomain residues beginning at P27 and extending through Y51 altered VZV replication in vitro, but the linker insertion at Y51 did not. While linker insertions at G90, I146, and P187 within the Y51-P187 segment did not affect VZV replication, deleting the region that had been targeted for linker insertion mutagenesis caused diminished infectious virus yields and cell-cell spread in vitro, indicating that important functional domains exist within this portion of the unique VZV gE N terminus.
FIG. 5.
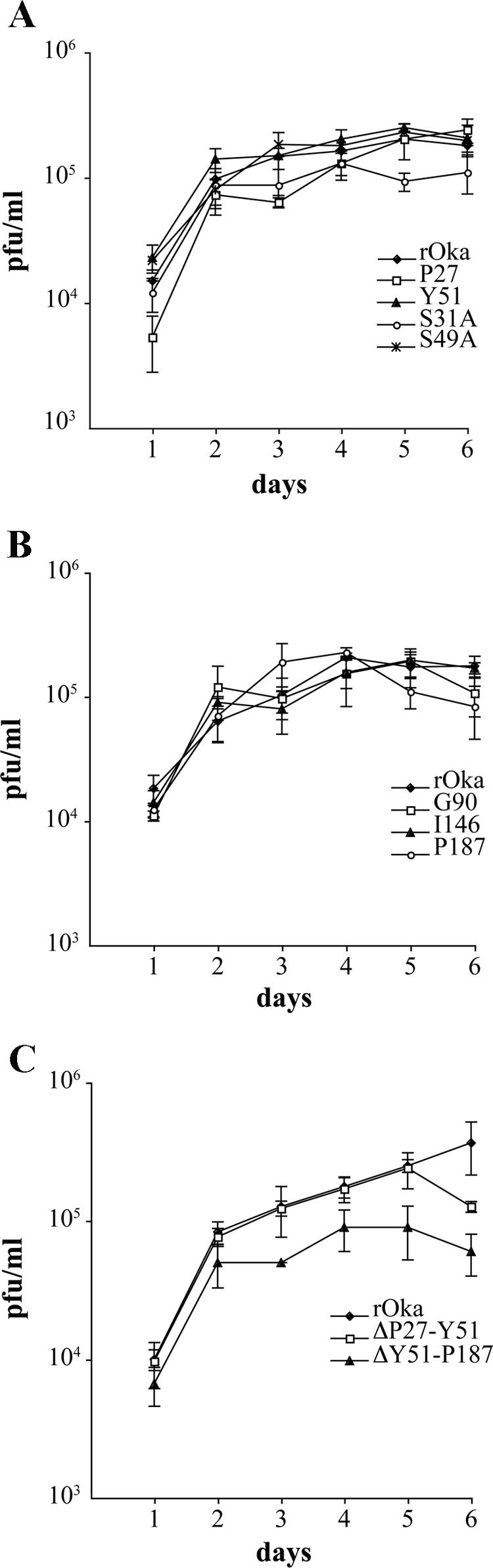
Replication of the gE N-terminal mutants in vitro. Melanoma cells were inoculated on day 0 with 1 × 103 PFU of rOka, rOka-P27, rOka-Y51, rOka-S31A, and rOka-S49A (A); rOka, rOka-G90, rOka-I146, and rOka-P187 (B); or rOka-ΔP27-Y51 and ΔY51-P187 (C). Aliquots were harvested from day 1 to day 6. Each point represents the mean of three wells. The error bars represent the standard deviations. Significance between the rOka and the rOka mutants was determined by Student's t tests.
Cellular localization, expression, and maturation of gE in cells infected with gE N-terminal mutant viruses.
By confocal microscopy, at 24 h postinfection gE expression in cells infected with rOka was localized predominantly to the perinuclear region (TGN) but had started to accumulate at the plasma membrane, and most gE was colocalized with gI (Fig. 6A to C). By 72 h postinfection, gE was abundantly expressed at the plasma membrane (data not shown). VZV gE trafficking and colocalization with gI were not altered in cells infected with the four mutants that showed alterations of the growth kinetics or plaque size, including rOka-P27, rOka-S31A, rOka-ΔP27-Y51, and rOka-ΔY51-P187 at 24 h (Fig. 6G to R) or 72 h postinfection (data not shown), compared to rOka (Fig. 6 A to C) or the mutant rOka-Y51, which had the same characteristics as rOka in the infectious focus assay (Fig. 6D to F). However, a qualitative assessment of confocal images suggested that more single infected cells were observed at 24 h in cells infected with rOka-ΔY51-P187 compared to rOka, consistent with impaired cell-cell spread (Fig. 6P to R). No differences were observed when gE expression and colocalization with gI at 24 and 72 h postinfection was examined for the other mutants, rOka-S49A, rOka-G90, rOka-I146, and rOka-P187, that had exhibited no changes in growth or plaque size compared to rOka (data not shown). These results indicated that the alterations in growth kinetics and plaque morphology of some of the rOka gE mutants with nonlethal changes in the gE N terminus were not related to an abnormal localization and trafficking of gE or failure to colocalize with gI.
FIG. 6.

Localization of gE and gI in cells infected with gE N-terminal mutants at 24 h postinfection. Melanoma cells were infected with rOka (A to C) or the rOka gE mutants rOka-Y51 (D to F), rOka-P27 (G to I), rOka-S31A (J to L), rOka-ΔP27-Y51 (M to O), and rOka-ΔY51-P187 (P to R), and they were then fixed and permeabilized after 24 h. Cells were labeled with anti-rabbit antibody against gI (in green) and with monoclonal antibody against gE (Chemicon) or monoclonal antibody 7G8 (P to R) (in red); nuclei were stained with 4′,6′-diamidino-2-phenylindole (blue). Right column, colocalization of gE and gI signal and 4′,6′-diamidino-2-phenylindole staining. Magnification, ×40.
Expression and maturation of gE in cells infected with the mutant viruses was compared to rOka-infected cells by immunoblot at 24 and 72 h postinfection, using IE4 as a loading control for viral proteins (Fig. 7, central panel). Mature gE is an O-linked and N-linked glycoprotein with a molecular mass of about 94 kDa, which represents the predominant product at 72 h postinfection; two immature forms of about 81 kDa and 88 kDa, which represent the high-mannose and the O-glycosylated forms, respectively, are also detectable by immunoblot at earlier time points (20, 43). The expression of gE by rOka-P27 was reduced at 24 h postinfection, while rOka-S31A showed only a minor reduction (Fig. 7A, top panel, lanes 3 and 5); gE accumulated at 72 h postinfection in cells infected with rOka-P27 and rOka-S31A mutants, although the level of gE expression remained quite low in rOka-P27-infected cells (Fig. 7B, top panel, lane 3). The rOka-Y51, rOka-S49A, rOka-G90, rOka-I146, and rOka-P187 mutants showed no modification in the level of gE expression, assessed relative to IE4, or maturation compared to gE in cells infected with rOka either at 24 h and 72 h postinfection (Fig. 7, top panel, lanes 4 and 6 to 9). In contrast, gE expression was decreased in rOka-ΔP27-Y51-infected cells at both time points (Fig. 7, top panel, lanes 10). Finally, in cells infected with rOka-P27 and rOka-ΔP27-Y51 mutants, the mature form of gE was not the predominant form at 72 h postinfection, suggesting a delay or possible alteration in the maturation process (Fig. 7B, lanes 3 and 10). The maturation of gE in cells infected with rOka-ΔY51-P187 mutant could not be assessed, since the epitope of the anti-gE MAb 3B3, located between amino acids 149 and 161 (51), was deleted, and anti-gE polyclonal antibody did not detect this mutant gE by immunoblot. However, analysis of melanoma cells infected with rOka-ΔY51-P187 by flow cytometry with the anti-gE MAb 7G8 and the polyclonal anti-VZV human immune serum (40) showed mutant gE on plasma membranes (data not shown).
FIG. 7.

Analysis of gE expression and maturation. Melanoma cells were inoculated with the gE mutant viruses; the cells were harvested at 24 h and 72 h postinfection and analyzed by Western blotting with mouse anti-gE MAb 3B3 (top panel), anti-rabbit polyclonal antibody to IE4 (central panel), and MAb anti-α-tubulin (bottom panel). Cell lysates from uninfected melanoma cells (lane 1) or inoculated with rOka (lane 2), rOka-P27 (lane 3), rOka-Y51 (lane 4), rOka-S31A (lane 5), rOka-S49A (lane 6), rOka-G90 (lane 7), rOka-I146 (lane 8), rOka-P187 (lane 9), and rOka-ΔP27-Y51 (lane 10) at 24 h postinfection (A) and 72 h postinfection (B) are shown. The molecular markers are indicated on the left. The arrow indicates the IE4-specific band of about 52 kDa.
Effect of mutations of the gE N-terminal region on VZV replication in skin in vivo.
Three of the four mutants that showed reduced replication in vitro, including rOka-P27, rOka-S31A, and rOka-ΔY51-P187, and two mutants that showed no difference from rOka, including rOka-Y51 and rOka-S49A, were evaluated in vivo by inoculating skin xenografts with these viruses. The inoculum titers were the following: rOka, 2.9 × 105 PFU/ml; rOka-P27, 3.1 × 105 PFU/ml; and rOka-S49A, 3.5 × 105 PFU/ml. In the second experiment, inoculum titers were the following: rOka, 5 × 105 PFU/ml; rOka-Y51, 3.7 × 105 PFU/ml; and rOka-S31A, 4 × 105 PFU/ml. The growth kinetics of rOka-S49A and rOka-Y51 were similar to those of rOka at both day 10 and day 21 after inoculation (Fig. 8A and B). In contrast, rOka-P27 replicated at a level comparable to that of rOka at day 10 but was significantly lower at day 21 (P = 0.017) (Fig. 8A). The rOka-S31A mutant showed almost no growth at day 10 (infectious virus was recovered from only one of five xenografts) and a very poor level of replication after 21 days (Fig. 8B). Sequencing of isolates recovered after 21 days showed persistence of the expected mutations (data not shown). When rOka-ΔY51-P187 was compared with rOka, the inoculum titers were 5 × 105 PFU/ml for rOka and 7.7 × 104 PFU/ml for the mutant virus. rOka-ΔY51-P187 failed to replicate at 10 and 22 days postinfection (Fig. 8C). Thus, the P27 insertion had some effect, although limited, whereas the S31 residue appeared to be an important virulence determinant, and the gE region between residues 51 and 187 was essential for the pathogenesis of VZV infection in skin.
FIG. 8.

Effect of the gE N-terminal mutations on VZV replication in skin xenografts. Skin xenografts were inoculated with MRC-5 cells infected with rOka, rOka-P27, and rOka-S49A (A); rOka, rOka-Y51, and rOka-S31A (B); and rOka and rOka-ΔY51-P187 (C). The infected xenografts were collected at days 10 and 21 or days 10 and 22 for the rOka-ΔY51-P187 mutant. The number of samples from which infectious virus was recovered per total number of xenografts inoculated is indicated on the horizontal axis. Each bar represents the mean titer, and the error bar indicates the standard error. The asterisk indicates significance (P < 0.05).
Virion formation in melanoma cells and skin xenografts infected with rOka-gE N-terminal mutant viruses.
To investigate possible alterations in viral egress and virion morphogenesis that might be associated with the altered pathogenicity of rOka-P27, rOka-S31A, and rOka-ΔY51-P187 compared to that of rOka-S49A and rOka-Y51, we further analyzed these mutants by electron microscopy in melanoma cells and in infected skin xenografts. VZV infection of melanoma cells is characterized by the formation of “viral highways,” which represent sites on cell surfaces where viral particles accumulate and facilitate cell-cell spread (23). When patterns of egress of rOka, rOka-P27, rOka-Y51, rOka-S31A, and rOka-S49A mutant virions were analyzed by SEM in melanoma cells, the distribution of viral particles on surfaces of rOka-infected cells was consistent with the formation of the expected viral highways (Fig. 9A). In cells infected with rOka-P27, many viral particles were observed on the cell surface, but the viral highway pattern was almost lost (Fig. 9B). The pattern of rOka-Y51 egress showed that the viral highways were still detectable but less distinctive than that of rOka (Fig. 9C). In rOka-S31A-infected cells, viral particles were distributed in a less distinctive viral highway pattern (Fig. 9D), while rOka-S49A was not different from rOka (data not shown).
FIG. 9.

Egress of VZV particles from infected melanoma cells. Melanoma cells infected with rOka (A), rOka-P27 (B), rOka-Y51 (C), and rOka-S31A (D) were analyzed by SEM to investigate the pattern of egress of the viral particles. The pattern of viral highways was most evident in rOka infection (dashed lines).
To investigate whether the mutant form of gE was incorporated into virions, melanoma cells infected with rOka-P27, rOka-S31A, and rOka-Y51 were examined by SEM using gold-labeled anti-gE MAb 3B3. Analysis of rOka-P27- and rOka-S31A-infected cells showed that gE was present on most viral particles, although envelope gE was detected on fewer virions than in the rOka-infected cells, where almost every viral particle was labeled (Fig. 10A to D and G to H). Surprisingly, virions in rOka-Y51-infected cells showed the least gE on surface particles (Fig. 10E to F). Similar results were obtained when infected melanoma cells were analyzed by TEM with immunogold gE labeling. In rOka-P27- and rOka-S31A-infected cells, most viral particles had gE incorporation with a pattern similar to that of rOka (Fig. 10I to J and L), while the rOka-Y51 viral particles showed only limited gE (Fig. 10K). In addition, two capsids enclosed by one envelope were common in rOka-Y51-infected cells, suggesting an effect of the Y51 mutation on the late stage of final envelopment (Fig. 10K, open arrowhead). These differences between rOka-Y51 and rOka were unexpected, since rOka-Y51 did not show any alteration in viral replication in vitro and in vivo or any modifications in gE trafficking, expression, or maturation. Since the 3B3 MAb detected the Y51 mutant gE in Western blot assays and the 3B3 MAb epitope is between amino acids 149 to 161, it is unlikely that failure of the monoclonal antibody to bind to gE in the immunogold experiments could explain the results.
FIG. 10.

Incorporation of gE in virion envelopes. Melanoma cells were infected with rOka (A, B, and I), rOka-P27 (C, D, and J), rOka-Y51 (E, F, and K), and rOka-S31A (G, H, and L), and they were analyzed by SEM (A to H) and TEM (I to L) after immunostaining with ultrasmall gold beads. Backscatter electron signal, shown in panels B, D, F, and H, was performed to document the specificity of the anti-gE immunolabeling; the labeling is highlighted by white circles. White arrows on panels C, F, and G indicate virions without detectable gE labeling. The black arrows in panels I to L indicate the gold beads on the envelopes of viral particles. Two capsids in the same envelope were observed in rOka-Y51 particles (K, open arrowhead).
SEM and TEM analysis were performed on the skin xenografts collected at 21 days postinoculation with rOka-S31A and rOka-Y51. Fewer rOka-S31A virions were observed in infected epidermal cells by SEM than for rOka, while no significant difference was observed between rOka-Y51 and the control (data not shown). TEM analysis of the rOka-Y51-infected skin cells showed many nucleocapsids in the nucleus (Fig. 11B) and virions in cytoplasmic vacuoles (Fig. 11C); no differences from rOka were observed.
FIG. 11.

TEM analysis of infected skin xenografts. Extracytoplasmic viral particles were evident in the rOka-infected samples (A); many nucleocapsids in the nucleus and viral particles in cytoplasmic vacuoles were observed in rOka-Y51-infected xenografts (B and C). Magnification for panel A, ×5,000; magnification for panels B and C, ×2,500. Cyt, cytoplasm; Nuc, nucleus.
Since the rOka-ΔY51-P187 mutant was completely impaired for growth in the skin in vivo (Fig. 8C), we analyzed virion formation in vitro in infected MRC-5 cells. The cells were infected with either rOka or rOka-ΔY51-P187 and analyzed by TEM 48 h postinfection (Fig. 12). Nucleocapsids were observed in the nucleus of cells infected with rOka (Fig. 12A and B) and rOka-ΔY51-P187 (Fig. 12C and D). Viral particles emerging from the cells infected with rOka were easily detected (Fig. 12E and F), while only rare intra- and extracellular viral particles were observed in cells infected with rOka-ΔY51-P187 (Fig. 12G and H). Intracytoplasmic aggregates were observed in rOka-ΔY51-P187-infected cells that were absent in the cells infected with rOka (Fig. 12I and J).
FIG. 12.

TEM analysis of MRC-5 cells infected with rOka (A, B, E, F, and I) and rOka-ΔY51-P187 (D, C, G, H, and J). Nucleocapsids in the nucleus of infected cells (black arrows) were observed in rOka (A and B) and rOka-ΔY51-P187 (C and D). Intra- and extracytoplasmic viral particles (open arrows) were present in the rOka-infected cells (E and F), while only rare extracytoplasmic virions in rOka-ΔY51-P187-infected cells are indicated in panels G and H (open arrow). Lysosome bodies (white arrow) were evident in the cytoplasm of rOka-infected cells (I), and intracytoplasmic aggregates (white arrow) were also present in the rOka-ΔY51-P187-infected cells (J). Cyt, cytoplasm; Nuc, nucleus. Magnifications are indicated.
DISCUSSION
All of the alphaherpesviruses have genes that encode variants of the glycoproteins gE and gI. However, in contrast to the other alphaherpesviruses, VZV gE is required for viral replication (31, 36). Our bioinformatics analyses indicated that VZV gE resembles the gE homologues in regions known to mediate gE-gI interactions, but VZV gE also has a large N-terminal region that is not conserved, even in the closely related virus HSV-1. Our mutational analysis of this unique 188-amino-acid gE N terminus has demonstrated that it mediates functions required for VZV replication that are distinct from residues involved in gE-gI interactions, and that these functions are necessary even when the endocytosis motif in the C terminus, which is also an essential domain (39), is intact.
Complete deletion of the unique gE N terminus was not compatible with VZV replication even when the sequence of the putative signal peptide, amino acids 1 to 16, was present. The mutant gE protein was expressed and transported to the TGN in transfected cells, suggesting that the failure to recover virus was not likely to be due to lack of synthesis of the protein. However, gE without the 188-amino-acid N terminus did not traffic to plasma membranes despite coexpression with gI and evidence of gE and gI colocalization in the cytoplasm. This result indicates that trafficking and accumulation of gE at the plasma membrane is essential for VZV replication. When gI is deleted in VZV, gE remains detectable on plasma membrane, albeit in an abnormal punctate distribution, and some cell-cell fusion with polykaryocyte formation is preserved (11, 31). Therefore, the failure of gE lacking the unique N terminus to reach or be retained on plasma membranes suggests that this region of gE may be needed for stable expression at this vital site for VZV spread from cell to cell and that this function is not mediated only by gE-gI interactions, as it appears to be in other alphaherpesviruses (14, 18, 24, 35). It is possible that this nonconserved, unusually long N-terminal region of VZV gE is important for bridging to adjacent uninfected cells. This concept is consistent with the hypothesis that gE has some gD-like functions, given recent evidence that gD participates in events at cell junctions in HSV-1-infected cells, along with HSV-1 gE and gI (28). Since VZV replication is completely dependent on cell-cell spread in vitro in fibroblasts and melanoma cells, the loss of such a function would be predicted to be lethal for VZV, whereas gD could compensate for the deletion of HSV-1 gE. Since gE retrieval from plasma membranes appears to be the route by which it is localized properly to sites of virion envelopment, this defect would also be expected to interfere with late stages of virion assembly.
Partial deletions of the unique gE N-terminal domain were tolerated, although cell fusion was decreased in cells infected with the rOka-ΔP27-Y51 and rOka-ΔY51-P187 mutants, and infectious virus yields of rOka-ΔY51-P187 were diminished. Whereas gE trafficking to plasma membrane and colocalization with gI was normal, the decrease in cell-cell spread could be explained by lower levels of gE, reflecting less efficient expression or more rapid degradation of the mutant protein. In addition to decreased syncytium formation, rOka-ΔY51-P187 exhibited a marked alteration of secondary envelopment of virions and accumulation of cytoplasmic aggregates in vitro and was incompatible with VZV infection of skin in vivo. VZV skin tropism requires polykaryocyte formation, and our experiments in the SCIDhu skin model demonstrate that VZV mutants retain some infectivity, as long as cell fusion is preserved, even when virion formation and secondary envelopment are severely compromised (7, 29). Cell-cell spread of human cytomegalovirus in the absence of enveloped particles and extracellular infectious virus has also been reported (52). Therefore, the consequences of deleting gE residues Y51-P187 suggest a role for this domain in fusion of epidermal cells in vivo.
Experiments with the gE N-terminal linker insertion mutants indicated that the N-terminal region does not contain domains important for binding to gI and are consistent with evidence that gE/gI heterodimer formation is mediated by the cysteine-rich domain of VZV gE, which is in the most conserved region of the gE alphaherpesvirus homologues (61). The fact that linker insertions throughout the unique gE N terminus were well tolerated also suggests that functions of this region are not highly dependent upon the conformational state of the gE protein. The linker insertion at P27 had minor effects on replication in vitro, which was associated with diminished gE due either to decreased synthesis or to reduced stability of the mutant gE. Of interest, this mutation had a substantial impact on the egress of VZ virions along “viral highways,” which is highly characteristic of VZV-infected cells (23). Nevertheless, the P27 linker insertion was compatible with incorporation of gE into virions. Virion morphogenesis was normal, and this phenotypic difference in vitro was associated with only a minor impairment in pathogenic potential in skin in vivo.
The disruption of the gE N terminus by a linker insertion at Y51 resulted in a mutant, rOka-Y51, in which gE incorporation into virions was reduced, and the detection of two nucleocapsids in the same intracellular vesicle suggested a possible alteration of secondary envelopment. Egress along “viral highways” was also somewhat diminished. Nevertheless, rOka-Y51 exhibited normal infectious virus yields, plaque sizes, and gE trafficking in infected cells in vitro, and replication in skin xenografts in vivo was not altered. Thus, typical extensive incorporation of gE into VZ virion envelopes does not appear to be required for cell-cell spread and polykaryocyte formation as long as gE expression on plasma membrane is preserved.
Interestingly, a gE N-terminal mutation in which the serine at position 31 was replaced with an alanine residue had substantial effects on the capacity of VZV to infect differentiated human epidermal cells within the intact skin microenvironment in vivo. The diminished pathogenicity of rOka-S31A in skin was not associated with any changes in vitro except a slight decrease in gE expression levels and maximum virus yields. The rOka-S31A experiments suggest that S31 residue may be important for interaction with other viral and cellular proteins during VZV infection of epidermal cells in vivo.
The involvement of this unique gE N-terminal region in VZV cell-cell spread and cell fusion is further suggested by the phenotype of the spontaneously occurring VZV mutant VZV-MSP, in which the single mutation D150N accelerates cell-cell spread in vitro and in skin xenografts (51). We speculate that this N-terminal region may contain a binding domain for cellular proteins that acts as a receptor for cell-cell spread, resembling nectin-1 interactions with HSV-1 gD (28). Further, these N-terminal gE functions are distinct from the functions of the alphaherpesvirus gE homologs that occur through gE/gI heterodimer formation and support the evidence that these are mediated by conserved domains like the cysteine-rich region at amino acids 328 to 500, which is downstream from the unique VZV gE N terminus (6, 30, 50, 58). As in HSV, this conserved region of VZV gE may support efficient cell-cell spread by directing virions to the cell junctions through binding to cellular components at this site (14, 18, 24, 35, 49, 60). In this regard, VZV gE was shown to enhance cell-cell contact in polarized epithelial cells with or without gI and colocalized with the tight-junction protein ZO-1 (37). However, our evidence that mutations in the unique VZV gE domain affect VZV cell-to-cell spread suggests that this region provides additional functions required for cell fusion.
VZV gE also has the capacity to form a complex with gH in the virion (34). The interactions between these two VZV glycoproteins may also be important for secondary envelopment, in addition to the known contributions of the conserved gE N-terminal and C-terminal regions of the alphaherpesvirus gE proteins to this process (8, 19, 35, 36, 39, 57, 60). It is possible that the defect in secondary envelopment observed in cells infected with rOka-ΔY51-P187 reflects disruption of an important gE-gH interaction, which is required for VZV to replicate in skin. Finally, the ability of gE to form dimers with phosphorylation patterns that differ from the monomeric form and that are detectable on the cell membrane suggested that gE has properties shared by known cell surface receptors (46). Mutations in the unique VZV gE ectodomain may interfere with functions that require gE dimer formation.
In summary, we have identified a new functional region in the VZV gE ectodomain that is not conserved in gE in the other alphaherpesviruses and that contains subdomains which are determinants of virulence in skin, which is an essential stage of VZV pathogenesis. These experiments provide further evidence that gE is a multifunctional protein that plays a unique role in VZV replication.
Acknowledgments
This work was supported by grants from the National Institute of Allergy and Infectious Diseases, AI20459 and AI053846, and from the National Cancer Institute, CA49605 (A.M.A.) and AI 22795 (C.G.).
We thank Nafisa Ghori at the Department of Microbiology and Immunology, Stanford University, for assistance with transmission electron microscopy and Lee Kozar of the Bioinformatic Resource at Stanford Medical Center for helpful discussions and assistance with sequence analysis. We thank Anne Schaap for technical assistance.
REFERENCES
- 1.Alconada, A., U. Bauer, and B. Hoflack. 1996. A tyrosine-based motif and a casein kinase II phosphorylation site regulate the intracellular trafficking of the varicella-zoster virus glycoprotein I, a protein localized in the trans-Golgi network. EMBO J. 15:6096-6110. [PMC free article] [PubMed] [Google Scholar]
- 2.Altschul, S. F., T. L. Madden, A. A. Schaffer, J. Zhang, Z. Zhang, W. Miller, and D. J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arvin, A. M. 2001. Varicella-zoster virus, p. 2731-2768. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 4th ed., vol. 2. Lippincott Williams & Wilkins, Philadelphia, Pa. [Google Scholar]
- 4.Baiker, A., C. Bagowski, H. Ito, M. Sommer, L. Zerboni, K. Fabel, J. Hay, W. Ruyechan, and A. M. Arvin. 2004. The immediate-early 63 protein of varicella-zoster virus: analysis of functional domains required for replication in vitro and for T-cell and skin tropism in the SCIDhu model in vivo. J. Virol. 78:1181-1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bailey, T. L., and C. Elkan. 1994. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 2:28-36. [PubMed] [Google Scholar]
- 6.Basu, S., G. Dubin, M. Basu, V. Nguyen, and H. M. Friedman. 1995. Characterization of regions of herpes simplex virus type 1 glycoprotein E involved in binding the Fc domain of monomeric IgG and in forming a complex with glycoprotein I. J. Immunol. 154:260-267. [PubMed] [Google Scholar]
- 7.Besser, J., M. Ikoma, K. Fabel, M. H. Sommer, L. Zerboni, C. Grose, and A. M. Arvin. 2004. Differential requirement for cell fusion and virion formation in the pathogenesis of varicella-zoster virus infection in skin and T cells. J. Virol. 78:13293-13305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brack, A. R., B. G. Klupp, H. Granzow, R. Tirabassi, L. W. Enquist, and T. C. Mettenleiter. 2000. Role of the cytoplasmic tail of pseudorabies virus glycoprotein E in virion formation. J. Virol. 74:4004-4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Campadelli-Fiume, G., F. Cocchi, L. Menotti, and M. Lopez. 2000. The novel receptors that mediate the entry of herpes simplex viruses and animal alphaherpesviruses into cells. Rev. Med. Virol. 10:305-319. [DOI] [PubMed] [Google Scholar]
- 10.Cohen, J. I., and S. E. Straus. 2001. Varicella-zoster virus and its replication, p. 2707-2730. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 4th ed., vol. 2. Lippincott Williams & Wilkins, Philadelphia, Pa. [Google Scholar]
- 11.Cohen, J. I., and H. Nguyen. 1997. Varicella-zoster virus glycoprotein I is essential for growth of virus in Vero cells. J. Virol. 71:6913-6920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cohen, J. I., and K. E. Seidel. 1993. Generation of varicella-zoster virus (VZV) and viral mutants from cosmid DNAs: VZV thymidylate synthetase is not essential for replication in vitro. Proc. Natl. Acad. Sci. USA 90:7376-7380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cole, N. L., and C. Grose. 2003. Membrane fusion mediated by herpesvirus glycoproteins: the paradigm of varicella-zoster virus. Rev. Med. Virol. 13:207-222. [DOI] [PubMed] [Google Scholar]
- 14.Collins, W. J., and D. C. Johnson. 2003. Herpes simplex virus gE/gI expressed in epithelial cells interferes with cell-to-cell spread. J. Virol. 77:2686-2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davison, A. J. 1983. DNA sequence of the US component of the varicella-zoster virus genome. EMBO J. 2:2203-2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davison, A. J., and D. J. McGeoch. 1986. Evolutionary comparisons of the S segments in the genomes of herpes simplex virus type 1 and varicella-zoster virus. J. Gen. Virol. 67:597-611. [DOI] [PubMed] [Google Scholar]
- 17.Davison, A. J., and J. E. Scott. 1986. The complete DNA sequence of varicella-zoster virus. J. Gen. Virol. 67:1759-1816. [DOI] [PubMed] [Google Scholar]
- 18.Dingwell, K. S., and D. C. Johnson. 1998. The herpes simplex virus gE-gI complex facilitates cell-to-cell spread and binds to components of cell junctions. J. Virol. 72:8933-8942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Farnsworth, A., K. Goldsmith, and D. C. Johnson. 2003. Herpes simplex virus glycoproteins gD and gE/gI serve essential but redundant functions during acquisition of the virion envelope in the cytoplasm. J. Virol. 77:8481-8494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grose, C. 2002. The predominant varicella-zoster virus gE and gI glycoprotein complex, p. 195-223. In A. Holzenburg and E. Bogner (ed.), Structure-function relationships of human pathogenic viruses. Kluwer Academic/Plenum Publishers, New York, N.Y.
- 21.Grose, C., D. M. Perrotta, P. A. Brunell, and G. C. Smith. 1979. Cell-free varicella-zoster virus in cultured human melanoma cells. J. Gen. Virol. 43:15-27. [DOI] [PubMed] [Google Scholar]
- 22.Grose, C., S. Tyler, G. Peters, J. Hiebert, G. M. Stephens, W. T. Ruyechan, W. Jackson, J. Storlie, and G. A. Tipples. 2004. Complete DNA sequence analyses of the first two varicella-zoster virus glycoprotein E (D150N) mutant viruses found in North America: evolution of genotypes with an accelerated cell spread phenotype. J. Virol. 78:6799-6807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harson, R., and C. Grose. 1995. Egress of varicella-zoster virus from the melanoma cell: a tropism for the melanocyte. J. Virol. 69:4994-5010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson, D. C., M. Webb, T. W. Wisner, and C. Brunetti. 2001. Herpes simplex virus gE/gI sorts nascent virions to epithelial cell junctions, promoting virus spread. J. Virol. 75:821-833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kemble, G. W., P. Annunziato, O. Lungu, R. E. Winter, T. A. Cha, S. J. Silverstein, and R. R. Spaete. 2000. Open reading frame S/L of varicella-zoster virus encodes a cytoplasmic protein expressed in infected cells. J. Virol. 74:11311-11321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kenyon, T. K., J. I. Cohen, and C. Grose. 2002. Phosphorylation by the varicella-zoster virus ORF47 protein serine kinase determines whether endocytosed viral gE traffics to the trans-Golgi network or recycles to the cell membrane. J. Virol. 76:10980-10993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kimura, H., S. E. Straus, and R. K. Williams. 1997. Varicella-zoster virus glycoproteins E and I expressed in insect cells form a heterodimer that requires the N-terminal domain of glycoprotein I. Virology 233:382-391. [DOI] [PubMed] [Google Scholar]
- 28.Krummenacher, C., I. Baribaud, R. J. Eisenberg, and G. H. Cohen. 2003. Cellular localization of nectin-1 and glycoprotein D during herpes simplex virus infection. J. Virol. 77:8985-8999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ku, C. C., J. Besser, A. Abendroth, C. Grose, and A. M. Arvin. 2005. Varicella-zoster virus pathogenesis and immunobiology: new concepts emerging from investigations with the SCIDhu mouse model. J. Virol. 79:2651-2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Litwin, V., W. Jackson, and C. Grose. 1992. Receptor properties of two varicella-zoster virus glycoproteins, gpI and gpIV, homologous to herpes simplex virus gE and gI. J. Virol. 66:3643-3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mallory, S., M. Sommer, and A. M. Arvin. 1997. Mutational analysis of the role of glycoprotein I in varicella-zoster virus replication and its effects on glycoprotein E conformation and trafficking. J. Virol. 71:8279-8288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marchler-Bauer, A., and S. H. Bryant. 2004. CD-Search: protein domain annotations on the fly. Nucleic Acids Res. 32:W327-W331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maresova, L., T. J. Pasieka, and C. Grose. 2001. Varicella-zoster virus gB and gE coexpression, but not gB or gE alone, leads to abundant fusion and syncytium formation equivalent to those from gH and gL coexpression. J. Virol. 75:9483-9492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maresova, L., T. J. Pasieka, E. Homan, E. Gerday, and C. Grose. 2005. Incorporation of three endocytosed varicella-zoster virus glycoproteins, gE, gH, and gB, into the virion envelope. J. Virol. 79:997-1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McMillan, T. N., and D. C. Johnson. 2001. Cytoplasmic domain of herpes simplex virus gE causes accumulation in the trans-Golgi network, a site of virus envelopment and sorting of virions to cell junctions. J. Virol. 75:1928-1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mo, C., J. Lee, M. Sommer, C. Grose, and A. M. Arvin. 2002. The requirement of varicella zoster virus glycoprotein E (gE) for viral replication and effects of glycoprotein I on gE in melanoma cells. Virology 304:176-186. [DOI] [PubMed] [Google Scholar]
- 37.Mo, C., E. E. Schneeberger, and A. M. Arvin. 2000. Glycoprotein E of varicella-zoster virus enhances cell-cell contact in polarized epithelial cells. J. Virol. 74:11377-11387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moffat, J., H. Ito, M. Sommer, S. Taylor, and A. M. Arvin. 2002. Glycoprotein I of varicella-zoster virus is required for viral replication in skin and T cells. J. Virol. 76:8468-8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moffat, J., C. Mo, J. J. Cheng, M. Sommer, L. Zerboni, S. Stamatis, and A. M. Arvin. 2004. Functions of the C-terminal domain of varicella-zoster virus glycoprotein E in viral replication in vitro and skin and T-cell tropism in vivo. J. Virol. 78:12406-12415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moffat, J. F., M. D. Stein, H. Kaneshima, and A. M. Arvin. 1995. Tropism of varicella-zoster virus for human CD4+ and CD8+ T lymphocytes and epidermal cells in SCID-hu mice. J. Virol. 69:5236-5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moffat, J. F., L. Zerboni, P. R. Kinchington, C. Grose, H. Kaneshima, and A. M. Arvin. 1998. Attenuation of the vaccine Oka strain of varicella-zoster virus and role of glycoprotein C in alphaherpesvirus virulence demonstrated in the SCID-hu mouse. J. Virol. 72:965-974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moffat, J. F., L. Zerboni, M. H. Sommer, T. C. Heineman, J. I. Cohen, H. Kaneshima, and A. M. Arvin. 1998. The ORF47 and ORF66 putative protein kinases of varicella-zoster virus determine tropism for human T cells and skin in the SCID-hu mouse. Proc. Natl. Acad. Sci. USA 95:11969-11974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Montalvo, E. A., R. T. Parmley, and C. Grose. 1985. Structural analysis of the varicella-zoster virus gp98-gp62 complex: posttranslational addition of N-linked and O-linked oligosaccharide moieties. J. Virol. 53:761-770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Niizuma, T., L. Zerboni, M. H. Sommer, H. Ito, S. Hinchliffe, and A. M. Arvin. 2003. Construction of varicella-zoster virus recombinants from parent Oka cosmids and demonstration that ORF65 protein is dispensable for infection of human skin and T cells in the SCID-hu mouse model. J. Virol. 77:6062-6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Notredame, C., D. G. Higgins, and J. Heringa. 2000. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302:205-217. [DOI] [PubMed] [Google Scholar]
- 46.Olson, J. K., G. A. Bishop, and C. Grose. 1997. Varicella-zoster virus Fc receptor gE glycoprotein: serine/threonine and tyrosine phosphorylation of monomeric and dimeric forms. J. Virol. 71:110-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Olson, J. K., and C. Grose. 1998. Complex formation facilitates endocytosis of the varicella-zoster virus gE:gI Fc receptor. J. Virol. 72:1542-1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Olson, J. K., and C. Grose. 1997. Endocytosis and recycling of varicella-zoster virus Fc receptor glycoprotein gE: internalization mediated by a YXXL motif in the cytoplasmic tail. J. Virol. 71:4042-4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Polcicova, K., K. Goldsmith, B. L. Rainish, T. W. Wisner, and D. C. Johnson. 2005. The extracellular domain of herpes simplex virus gE is indispensable for efficient cell-to-cell spread: evidence for gE/gI receptors. J. Virol. 79:11990-12001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rizvi, S. M., and M. Raghavan. 2001. An N-terminal domain of herpes simplex virus type I gE is capable of forming stable complexes with gI. J. Virol. 75:11897-11901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Santos, R. A., C. C. Hatfield, N. L. Cole, J. A. Padilla, J. F. Moffat, A. M. Arvin, W. T. Ruyechan, J. Hay, and C. Grose. 2000. Varicella-zoster virus gE escape mutant VZV-MSP exhibits an accelerated cell-to-cell spread phenotype in both infected cell cultures and SCID-hu mice. Virology 275:306-317. [DOI] [PubMed] [Google Scholar]
- 52.Silva, M. C., J. Schroer, and T. Shenk. 2005. Human cytomegalovirus cell-to-cell spread in the absence of an essential assembly protein. Proc. Natl. Acad. Sci. USA 102:2081-2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sommer, M. H., E. Zagha, O. K. Serrano, C. C. Ku, L. Zerboni, A. Baiker, R. Santos, M. Spengler, J. Lynch, C. Grose, W. Ruyechan, J. Hay, and A. M. Arvin. 2001. Mutational analysis of the repeated open reading frames, ORFs 63 and 70 and ORFs 64 and 69, of varicella-zoster virus. J. Virol. 75:8224-8239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Spear, P. G., R. J. Eisenberg, and G. H. Cohen. 2000. Three classes of cell surface receptors for alphaherpesvirus entry. Virology 275:1-8. [DOI] [PubMed] [Google Scholar]
- 55.Spear, P. G., and R. Longnecker. 2003. Herpesvirus entry: an update. J. Virol. 77:10179-10185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tirabassi, R. S., and L. W. Enquist. 1999. Mutation of the YXXL endocytosis motif in the cytoplasmic tail of pseudorabies virus gE. J. Virol. 73:2717-2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tyborowska, J., K. Bienkowska-Szewczyk, M. Rychlowski, J. T. Van Oirschot, and F. A. Rijsewijk. 2000. The extracellular part of glycoprotein E of bovine herpesvirus 1 is sufficient for complex formation with glycoprotein I but not for cell-to-cell spread. Arch. Virol. 145:333-351. [DOI] [PubMed] [Google Scholar]
- 59.Wang, Z. H., M. D. Gershon, O. Lungu, Z. Zhu, S. Mallory, A. M. Arvin, and A. A. Gershon. 2001. Essential role played by the C-terminal domain of glycoprotein I in envelopment of varicella-zoster virus in the trans-Golgi network: interactions of glycoproteins with tegument. J. Virol. 75:323-340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wisner, T., C. Brunetti, K. Dingwell, and D. C. Johnson. 2000. The extracellular domain of herpes simplex virus gE is sufficient for accumulation at cell junctions but not for cell-to-cell spread. J. Virol. 74:2278-2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yao, Z., W. Jackson, B. Forghani, and C. Grose. 1993. Varicella-zoster virus glycoprotein gpI/gpIV receptor: expression, complex formation, and antigenicity within the vaccinia virus-T7 RNA polymerase transfection system. J. Virol. 67:305-314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yao, Z., W. Jackson, and C. Grose. 1993. Identification of the phosphorylation sequence in the cytoplasmic tail of the varicella-zoster virus Fc receptor glycoprotein gpI. J. Virol. 67:4464-4473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhu, Z., M. D. Gershon, Y. Hao, R. T. Ambron, C. A. Gabel, and A. A. Gershon. 1995. Envelopment of varicella-zoster virus: targeting of viral glycoproteins to the trans-Golgi network. J. Virol. 69:7951-7959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhu, Z., Y. Hao, M. D. Gershon, R. T. Ambron, and A. A. Gershon. 1996. Targeting of glycoprotein I (gE) of varicella-zoster virus to the trans-Golgi network by an AYRV sequence and an acidic amino acid-rich patch in the cytosolic domain of the molecule. J. Virol. 70:6563-6575. [DOI] [PMC free article] [PubMed] [Google Scholar]