

Abstract
All sequenced poxviruses encode orthologs of the vaccinia virus L1 and F9 proteins, which are structurally similar and share about 20% amino acid identity. We found that F9 further resembles L1 as both proteins are membrane components of the mature virion with similar topologies and induce neutralizing antibodies. In addition, a recombinant vaccinia virus that inducibly expresses F9, like a previously described L1 mutant, had a conditional-lethal phenotype: plaque formation and replication of infectious virus were dependent on added inducer. However, only immature virus particles are made when L1 is repressed, whereas normal-looking intracellular and extracellular virions formed in the absence of F9. Except for the lack of F9, the polypeptide components of such virions were indistinguishable from those of wild-type virus. These F9-deficient virions bound to cells, but their cores did not penetrate into the cytoplasm. Furthermore, cells infected with F9-negative virions did not fuse after a brief low-pH treatment, as did cells infected with virus made in the presence of inducer. In these respects, the phenotype associated with F9 deficiency was identical to that produced by the lack of individual components of a previously described poxvirus entry/fusion complex. Moreover, F9 interacted with proteins of that complex, supporting a related role. Thus, despite the structural relationships of L1 and F9, the two proteins have distinct functions in assembly and entry, respectively.
Vaccinia virus (VACV) is a member of the Poxviridae, a family of large enveloped double-stranded DNA viruses that replicate entirely in the cytoplasm (19). The nearly 200 genes of VACV encode enzymes and factors for transcription (5) and replication (21) of the genome, assembly of virus particles (8), cell entry (20), and modulation of the immune response (1). Assembly begins, in areas of the cytoplasm that are partially cleared of cellular organelles, with the creation of crescent membranes that enclose electron-dense material known as viroplasm. Membrane curvature, imposed by an external protein lattice, results in the formation of the spherical immature virion (IV) containing structural proteins, enzymes, and the viral genome. Further development, including the disassembly of the scaffold, proteolytic cleavages, and core condensation, results in the infectious barrel-shaped mature virion (MV). Most MVs remain in the cytoplasm until cell lysis enables their spread. A subpopulation of MVs undergoes wrapping by a double membrane derived from modified trans-Golgi or endosomal cisternae, allowing them to transit along microtubules to the periphery of the cell. There, the outer membrane fuses with the plasmalemma, resulting in exocytosis of the extracellular virion (EV), which is essentially an MV with an additional membrane. Prior to infecting another cell, the EV covering membrane is discarded (17), allowing the MV membrane to fuse with the cell membrane (4, 6, 7, 15, 39).
Sequence-based predictions suggest that the Western Reserve (WR) strain of VACV encodes 44 transmembrane proteins (www.poxvirus.org), of which some are still uncharacterized. Those proteins associated with the MV membrane can be placed into at least four functional groups involved in membrane crescent formation, e.g., A14 and A17 (25, 27, 38, 41); virion maturation, e.g., A9 and L1 (24, 43); virus attachment to glycosaminoglycans, e.g., H3 and D8 (14, 18); and virus entry, e.g., A16, A21, A28, G9, H2, and L5 (22, 23, 30, 32, 36). When expression of any one of the aforementioned entry proteins is repressed, typical-looking MVs and EVs form but they are defective in entry and induction of syncytia after brief low-pH treatment. These proteins as well as two more that have not yet been characterized, J5 and G3, are associated in a multiprotein complex (31). It seems likely that all of these proteins are essential components of the fusion apparatus. Roles for the A27 and L1 MV membrane proteins in virus entry have been considered previously because they are targets of neutralizing antibody. However, an A27 deletion mutant can enter cells and induce fusion and the primary role of the protein is related to wrapping of the MV (26, 28, 30, 40). The situation with L1 is ambiguous; a conditional-lethal null mutant is blocked in virus morphogenesis and MVs are not formed (24), thus far precluding the assessment of an additional role in entry. Another VACV protein, F9, is related to L1: the two proteins have a 20% sequence identity throughout their length, six invariant cysteines, and a C-terminal transmembrane domain (29). Both L1 and F9 are conserved in all poxviruses, suggesting that they have nonredundant functions. The atomic structure of the L1 protein has been determined (35), whereas the F9 protein has not been characterized except to demonstrate that it has intramolecular disulfide bonds formed by the poxvirus-encoded redox system (34). Here, we show that the F9 protein is located at the surface of the MV membrane and that F9, unlike L1, is not required for morphogenesis but has an essential role in virus entry and cell-cell fusion.
MATERIALS AND METHODS
Cells and viruses.
VACV strain WR was propagated in HeLa S3 cells in Eagle's minimal essential medium (Quality Biologicals) containing 2.5% fetal bovine serum (12). Recombinant vF9Li stocks were prepared in medium containing 100 μM isopropyl-β-d-thiogalactopyranoside (IPTG). WR-NPsiinfeklEGFP, a recombinant VACV encoding an enhanced green fluorescent fusion protein (EGFP) under the control of a VACV early promoter, was used in a flow cytometric virus neutralization assay (11). MVs were purified by sedimentation through two consecutive 36% sucrose cushions and a 25 to 40% continuous sucrose gradient as described previously (13). The numbers of purified virus particles and infectious units were determined by optical density at 260 nm and plaque assay, respectively, and used to calculate the specific infectivity or particle-to-PFU ratio.
Generation of recombinant VACV.
Recombinant viruses were derived from the WR strain of VACV. vF9Li was constructed from vT7LacOI (3), which encodes a constitutively expressed Escherichia coli lac repressor and the bacteriophage T7 RNA polymerase under the transcriptional control of the lac operator. The F9L locus of vT7LacOI was altered by homologous recombination with a PCR product containing (i) the F9L gene transposed from left to right and under the transcriptional control of the lac operator-regulated T7 promoter, (ii) the EGFP gene regulated by the viral late P11 promoter, and (iii) F9L flanking sequences. Recombinant virus expressing the EGFP reporter was clonally purified by three rounds of plaque isolation in the presence of 100 μM IPTG. The modifications at the F9L locus were confirmed by DNA sequencing.
vF9-V5 expresses the F9 protein, with a V5 epitope tag at its C terminus. The construction strategy employed was similar to that used for vE10-hemagglutinin (HA) except that EGFP served as the reporter (33).
Antibodies.
G. Cohen and R. Eisenberg (University of Pennsylvania) provided R192 and R180 rabbit antibodies raised against secreted baculovirus-expressed F9 and L1 recombinant proteins, respectively. Rabbit polyclonal antibodies against the following VACV peptides or proteins were used: A10 (R. Doms and B. Moss, unpublished), A4 (10), A21 (37), L5 (36), A16 (23), and A28 (G. Nelson and B. Moss, unpublished). The anti-L1 monoclonal antibody 7D11 (42) was prepared from a hybridoma kindly provided by A. Schmalljohn (United States Army Medical Research Institute for Infectious Diseases). Anti-HA and anti-V5 antibodies conjugated to horseradish peroxidase were from Roche Applied Science and Invitrogen, respectively.
SDS-PAGE.
Cells were lysed in 0.2% NP-40 (10 mM Tris, pH 7.4, 10 mM CaCl2, 10 mM NaCl) containing micrococcal nuclease and complete protease inhibitor cocktail (Roche Applied Science) at 4°C for 10 min. After addition of lithium dodecyl sulfate sample buffer and reducing agent (Invitrogen), cell lysates were heated to 70°C for 10 min. Equal volumes of lysate were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) on either a 12% Bis-Tris or a 4 to 12% gradient polyacrylamide gel in 2-(N-morpholino)ethanesulfonic acid (MES)-SDS running buffer (Invitrogen).
Western blot analysis.
Proteins separated by SDS-PAGE were electrophoretically transferred to nitrocellulose and blocked in pH 7.5 phosphate-buffered saline (PBS) containing 5% dry milk. The membranes were incubated for 1 h with a 1:1,000 dilution of primary antibody, washed three times with 0.05% Tween 20, and subsequently incubated with a 1:2,000 dilution of horseradish peroxidase-conjugated antibody to rabbit immunoglobulin G (IgG). Bound antibody was detected with SuperSignal West Pico chemiluminescent substrate (Pierce Biotechnology Inc.).
Biotinylation of purified virions.
Purified MVs were incubated with 1 mg/ml of EZ-Link sulfo-NHS-SS-biotin ([sulfosuccinimidyl 2-(biotinamido)-ethyl-1,3-dithiopropionate]; Pierce Biotechnology Inc.) for 30 min at 4°C on a rocking platform. Excess biotin was quenched according to the manufacturer's protocol. Virus was pelleted, lysed with SDS-PAGE sample buffer, and then incubated with NutrAvidin beads (Pierce Biotechnology Inc.) for 1 h at room temperature. Unbound proteins were recovered, and the column was washed three times; biotinylated proteins were eluted with SDS-PAGE sample buffer containing 50 mM dithiothreitol (DTT).
Cell binding and penetration assay.
HeLa cells on glass coverslips were infected with 10 PFU per cell of purified F9+ virions or the equivalent number of F9− virions in the presence of 300 μg of cycloheximide per ml. After 1 h at 4°C, the cells were washed three times and then shifted to 37°C for 2 h. The cells were washed three times, fixed with 4% paraformaldehyde, and made permeable with 0.05% Triton X-100. After being blocked with 1% bovine serum albumin, the cells were incubated with a 1:1,000 dilution of anti-L1 mouse monoclonal antibody 7D11 and anti-A4 rabbit polyclonal antibody. The cells were washed three times and then incubated for 1 h with a mixture of goat anti-mouse IgG coupled to Alexa Fluor 488 at a 1:200 dilution, goat anti-rabbit IgG coupled to Alexa Fluor 568 at a 1:200 dilution, and diamidino-2-phenylindole dihydrochloride (DAPI) from Molecular Probes at a 1:3,000 dilution. Images were collected with a Leica TCS-NT laser scanning confocal microscope.
Northern blot analysis.
BS-C-1 cells were treated with 40 μg/ml of cytosine arabinoside (AraC) for 1 h prior to infection with 5 PFU per cell of purified F9+ virions or the same number of F9− virions. After 3 h of incubation in the presence of AraC, total RNA was extracted using the RNeasy minikit (QIAGEN) and a Northern blot assay was performed using the NorthernMax-Gly kit (Ambion) with an [α-32P]dCMP-labeled DNA probe prepared using the DECA Prime kit (Ambion). Bands were detected by autoradiography.
Cell-cell fusion assays.
For “fusion from within,” BS-C-1 cells were infected with 5 PFU/cell of vF9Li in the presence or absence of IPTG. After 18 h, cells were exposed to pH 5.3 or pH 7.5 buffer for 3 min at 37°C and then incubated in Eagle's minimal essential medium. After 3 h, cells were fixed with 4% paraformaldehyde, made permeable with 0.05% Triton X-100, and stained with DAPI. For “fusion from without,” BS-C-1 cells were incubated with 300 PFU per cell of purified F9+ virions or the same number of F9− virions for 1 h at 4°C. The cells were incubated with a pH 5.3 or pH 7.4 buffer for 5 min at 37°C and then in medium containing 300 μg of cycloheximide per ml. After 3 h, cells were fixed, made permeable, and stained with Alexa 568-phalloidin (Molecular Probes) and DAPI.
Immunoaffinity purification.
Infected cells were Dounce homogenized, and the postnuclear supernatant was isolated by low-speed centrifugation. Proteins were extracted with NP-40 detergent from a membrane-enriched fraction isolated by flotation over a sucrose cushion or a 100,000 × g pellet. The extracted proteins were bound to agarose beads coupled to anti-V5 antibody (Sigma) and analyzed by Western blotting as described previously (31).
RESULTS
Conservation of the VACV F9 protein.
The VACV F9L gene (VACVWR048) is predicted to encode a 23.8-kDa protein with a transmembrane domain separating a long N-terminal segment from a 16-amino-acid carboxy-terminal tail. The six cysteines were previously shown to form three intramolecular disulfide bonds, a process dependent on the VACV-encoded redox system (34). An F9 ortholog is present in each sequenced poxvirus, and a multiple alignment reveals conservation of the cysteines and the putative carboxy-terminal transmembrane domain (Fig. 1A).
FIG. 1.

F9 is conserved throughout the Poxviridae. (A) Multiple amino acid sequence alignment of VACV F9 and orthologs from representatives of each poxvirus genus. The bar denotes the predicted transmembrane (TM) domain. Identical amino acids are shaded gray; invariant cysteines are white on a black background. Abbreviations: VAC, vaccinia virus (Orthopoxvirus); YAB, Yaba monkey tumor virus (Yatapoxvirus); MYX, myxoma virus (Leporipoxvirus); LSD, lumpy skin disease virus (Capripoxvirus); SPV, swinepox virus (Suipoxvirus); MCV, molluscum contagiosum virus (Molluscipoxvirus); FPV, fowlpox virus (Avipoxvirus); ORF, ORF virus (Parapoxvirus); MSV, Melanoplus sanguinipes entomopoxvirus (Entomopoxvirus B); AMS, Amsacta moorei entomopoxvirus (Entomopoxvirus B). (B) Sequence alignment of VACV WR F9 and L1 proteins. Identical amino acids are denoted as in panel A.
F9 and L1 share the same domain organization and are homologous (29); the only notable difference in their organization is the presence of a myristoylation signal in L1 not found in F9. The myristoylation of L1 and its importance for protein function in virus infection were demonstrated previously (24). Figure 1B shows the alignment of F9 and L1 from the WR strain of vaccinia virus with 20% identical amino acids including the six invariant cysteines known to be organized in three disulfide bonds. Based on the high level of similarity, F9 can be modeled using the atomic structure determined for L1 (35).
The F9L gene encodes a late protein that is incorporated into MV membranes.
A predicted viral late promoter (9) upstream of the F9L open reading frame (ORF) suggested that the encoded protein would be expressed following viral DNA replication. To analyze the kinetics of F9 expression, VACV-infected cells were harvested at various times after infection and analyzed by Western blotting with an anti-F9 polyclonal antibody. Both the late synthesis of the protein and inhibition by the DNA replication inhibitor AraC (Fig. 2) confirmed that a late class promoter regulates F9. The migration of the unreduced F9 is more rapid than estimated from its mass due to intramolecular disulfide bonds (34).
FIG. 2.
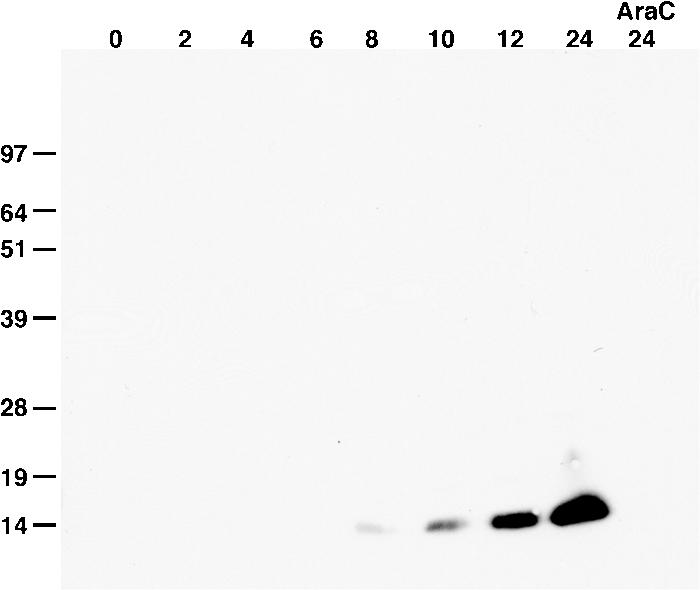
Kinetics of F9 expression. BS-C-1 cells were infected with VACV for the hours indicated at the top of the figure, harvested in the presence of N-ethylmaleimide, and analyzed by nonreducing SDS-PAGE and Western blotting with a rabbit polyclonal antibody specific for F9 followed by horseradish peroxidase-conjugated anti-rabbit antibody. One infection was performed in the presence of AraC, and cells were harvested at 24 h. Bands were detected by chemiluminescence. Numbers at left show molecular masses in kilodaltons.
The predicted transmembrane domain suggested that F9 would be incorporated into viral or cellular membranes. Initial experiments indicated that the protein was associated with MVs purified by sucrose density gradient centrifugation. Compared to L1, F9 was poorly soluble upon extraction with NP-40 or NP-40 plus DTT (data not shown) as found for some components of the entry/fusion complex (22, 23). Evidence for the location of F9 in the MV membrane was obtained by surface biotinylation of purified MVs. Virions were incubated with sulfo-NHS-SS-biotin, a membrane-nonpermeating reagent that selectively labels exposed primary amines of lysine residues. Excess biotin was removed, and the virions were solubilized with SDS in the absence of reducing agent. The biotinylated proteins were bound to immobilized NutrAvidin and eluted by incubation with detergent containing DTT, which cleaves the SS bond of the sulfo-NHS-SS-biotin. Bound and unbound proteins were analyzed by Western blotting with antibodies to F9 and other viral proteins. Biotinylated F9 and L1 bound the NeutrAvidin beads to similar extents (Fig. 3A). The absence of significant binding of proteins from mock-treated unbiotinylated virions confirmed the specificity of the affinity purification (Fig. 3A). In addition, only a small amount of the core protein A10 was biotinylated and bound NeutrAvidin, indicating that the virions were mostly intact. These data indicated that F9 is exposed on the surface of the MV.
FIG. 3.
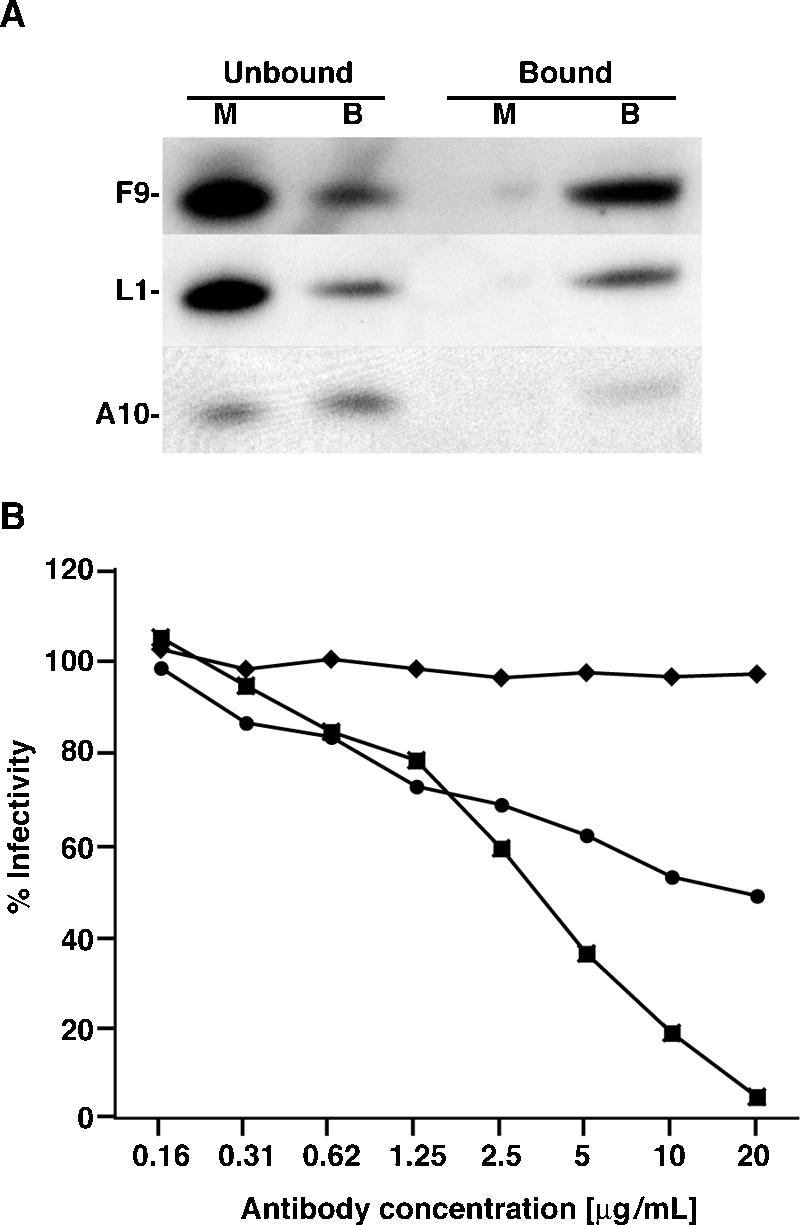
F9 is an MV membrane protein. (A) Biotinylation of MV membrane proteins. Purified VACV virions were mock treated (lanes M) or treated with 1 mg/ml of sulfo-NHS-SS-biotin (lanes B) for 30 min at 4°C. Virions were lysed with 0.5% SDS, and biotinylated proteins were isolated by incubation with NutrAvidin resin. Bound proteins were eluted with 0.5% SDS containing 50 mM DTT. Western blot analysis was performed using antibodies specific for F9, L1, and A10. (B) MV neutralization (11). Purified WR-NPsiinfeklEGFP MVs were mixed with various concentrations of polyclonal antibody to F9 (circles), L1 (squares), or A33 (diamonds) for 1 h at 37°C. The mixture was then added to HeLa cells in the presence of AraC and incubated for 16 h. EGFP-expressing cells were quantified by flow cytometry. Data are presented as the percentage of EGFP-expressing cells normalized to cells incubated with virus that was mock treated with antibody.
Rabbit antibody prepared against a soluble protein lacking the transmembrane and short C-terminal segment of F9 was tested for its ability to neutralize MVs using a sensitive and quantitative flow cytometry assay (11). As positive and negative controls, we analyzed similarly prepared rabbit polyclonal antibodies to soluble forms of the MV membrane protein L1 and the EV membrane protein A33, respectively. MV neutralization by anti-F9 antibody was observed, though it was less complete than that with anti-L1 antibody at the same IgG concentrations (Fig. 3B). As expected, antibody to the EV membrane protein A33 did not neutralize MV infection (Fig. 3B). These data confirmed the MV biotinylation data and showed that the long N-terminal domain of F9 is exposed on the surface of the MV.
F9 is required for VACV infectivity.
Conservation of the F9L gene throughout the Poxviridae suggested that the encoded protein has an essential role. To test this hypothesis, a recombinant virus, vF9Li, was constructed in which expression of the F9L gene was inducibly controlled by IPTG (Fig. 4A). An early-late VACV promoter ensured continuous synthesis of the E. coli lac repressor, and a VACV late promoter and lac operator regulated expression of the bacteriophage T7 RNA polymerase. The F9L gene was reversed in orientation to prevent read-through transcription from neighboring genes and placed under the control of the T7 promoter and lac operator. In addition an EGFP reporter was introduced into the genome of vF9Li to distinguish plaques containing the recombinant virus from its parent. In the absence of IPTG, the lac repressor binds to the lac operator upstream of both the T7 RNA polymerase gene and the F9L gene, resulting in transcriptional repression. In the presence of IPTG, the lac repressor is inactivated, allowing expression of the T7 RNA polymerase and transcription of the F9L gene.
FIG. 4.

F9L is essential for replication of infectious virus. (A) Diagram of vF9Li. A segment of the viral genome shows the open reading frames for the bacteriophage T7 RNA polymerase (T7 Pol), E. coli lac repressor (LacI), EGFP, VACV F9L, F8L, and F10L. Also indicated are the E. coli Lac operator (LacO) and the VACV promoters P11 and P7.5 and T7 promoter PT7. (B) Effect of IPTG concentration on virus yield and F9 expression. BS-C-1 cells were infected with 10 PFU per cell of vT7LacOI (squares) or vF9Li (circles) in the presence of indicated concentrations of IPTG. At 24 h postinfection, cells were harvested, subjected to three cycles of freezing-thawing, and sonicated. Virus yields were determined by plaque assay on BS-C-1 cells in the presence of 100 μM IPTG. The inset shows F9 protein at 24 h as a function of IPTG concentration assayed by Western blotting with anti-F9 antibody. (C) One-step virus growth. BS-C-1 cells were infected with 10 PFU per cell of vT7LacOI (filled squares) or vF9Li in the presence (filled circles) or absence (open circles) of 100 μM IPTG. At the indicated times, virus was harvested from infected cells and plaque assayed as described above.
The dependence of vF9Li replication on IPTG concentration was determined: cells were infected with vF9Li in the presence of various concentrations of IPTG and 24-h virus yields were determined by plaque assay in the presence of IPTG. The yields of vF9Li increased with IPTG concentrations from 10 to 50 μM and plateaued at 100 μM, whereas that of the parental virus was unaffected (Fig. 4B). F9 synthesis, as determined by Western blotting, was barely detected at 25 μM IPTG and increased up to 200 μM IPTG (Fig. 4B). The greater synthesis of F9 at 200 μM IPTG did not increase virus replication over that of lower amounts, and therefore, 100 μM IPTG was used for subsequent experiments.
A single-step growth experiment was performed to determine the kinetics of IPTG-induced virus replication. Cells were infected either with vF9Li in the presence or absence of IPTG or with the parental virus vT7LacOI. At successive intervals, virus titers were measured by plaque assay in the presence of IPTG. The growth kinetics of vF9Li in the presence of IPTG and vT7LacOI were similar, whereas there was only a slight rise in vF9Li titer in the absence of IPTG (Fig. 4C). These data demonstrated that F9 is needed for production of infectious virus.
Virion morphogenesis occurs in the absence of F9.
Based on the role of L1 and its sequence similarity to F9, we considered that F9 might also be necessary for virion morphogenesis. Cleavage of certain core proteins is dependent on changes that occur during the conversion of the IV to the MV. Consequently inhibition of proteolysis is an indicator of a developmental block. To assay for core protein processing, cells were infected with vF9Li or vT7LacOI in the presence or absence of IPTG or rifampin, respectively. Rifampin is a specific inhibitor of morphogenesis and serves to demonstrate a block in processing (16). At 12 h after infection, a time at which host protein synthesis has been shut down, cells were pulsed with [35S]methionine and cysteine for 30 min and lysed either immediately or after an additional 12-h chase in unlabeled medium. The lysates were analyzed by SDS-PAGE and autoradiography. In each case, virus infection resulted in inhibition of host protein synthesis at 12 h (Fig. 5). The positive and negative controls of untreated and rifampin-treated vT7LacOI-infected cells confirmed the dependence of processing of the precursor proteins p4a and p4b to 4a and 4b on morphogensis (Fig. 5). Importantly, processing of p4a and p4b occurred to similar extents in cells infected with vF9Li in the presence or absence of IPTG (Fig. 5), indicating that F9 expression was not required for this step. These results contrasted with the finding of a block in core protein processing when expression of L1 was repressed (24), providing the first indication that F9 is not required for virion development.
FIG. 5.

Processing of viral core proteins. BS-C-1 cells were infected with 5 PFU per cell of vF9Li in the presence or absence of IPTG or vT7LacOI in the presence or absence of 100 μg/ml of rifampin. After 11.5 h, cells were incubated in methionine- and cysteine-deficient medium for 30 min and then with 100 μCi of a [35S]methionine and [35S]cysteine mixture for 30 min. The cells were washed and harvested either immediately after the pulse (P) or after incubation in unlabeled medium for an additional 12-h chase (C). Cell lysates were analyzed by SDS-PAGE and autoradiography. Core precursor proteins p4a and p4b as well as processed proteins 4a and 4b are indicated. Positions of marker proteins are shown at the left with masses in kilodaltons.
Transmission electron microscopy was used to directly examine the effect of F9 repression on virion morphogenesis. Images of cells infected with vF9Li in the absence of IPTG showed the entire array of immature (Fig. 6A) and mature forms including MVs (Fig. 6B) and EVs (Fig. 6C), which were indistinguishable from those formed in the presence of IPTG (not shown). Thus, F9, unlike L1, is not needed for morphogenesis.
FIG. 6.
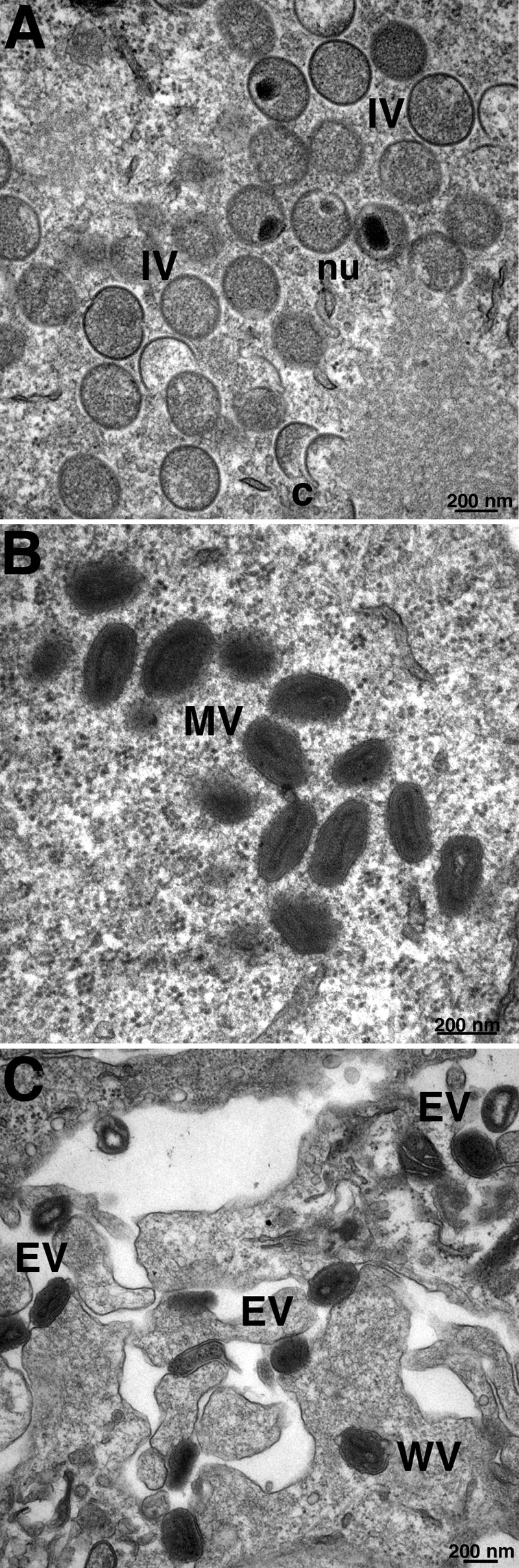
Electron microscopy of cells infected with vF9Li in the absence of IPTG. BS-C-1 cells were infected with vF9Li at a multiplicity of 4 PFU per cell in the absence of IPTG. After 20 h, the cells were processed for transmission electron microscopy as previously described (41). (A) Crescents (c) and IVs with visible nucleoids (nu). (B) MVs. (C) Wrapped virions (WV) and EVs. Bars, 200 nm.
Analysis of the infectivity and polypeptide composition of purified virions.
MVs were purified from cells infected with vF9Li in the absence or presence of IPTG to produce virions deficient in F9 (F9−) and containing F9 (F9+), respectively. The specific infectivity of F9+ virions was approximately 150-fold greater than that of F9− virions. Nevertheless, negatively stained F9+ and F9− virions were indistinguishable by electron microscopy (not shown). Furthermore, there was no apparent difference in the polypeptide compositions of wild-type, F9+, and F9− virions as determined by SDS-PAGE and silver staining (Fig. 7A). Western blotting confirmed the absence of F9 and the presence of other representative membrane proteins including L1 and two representative components of the putative entry-fusion complex, A21 and A28, in MVs made without IPTG (Fig. 7B).
FIG. 7.
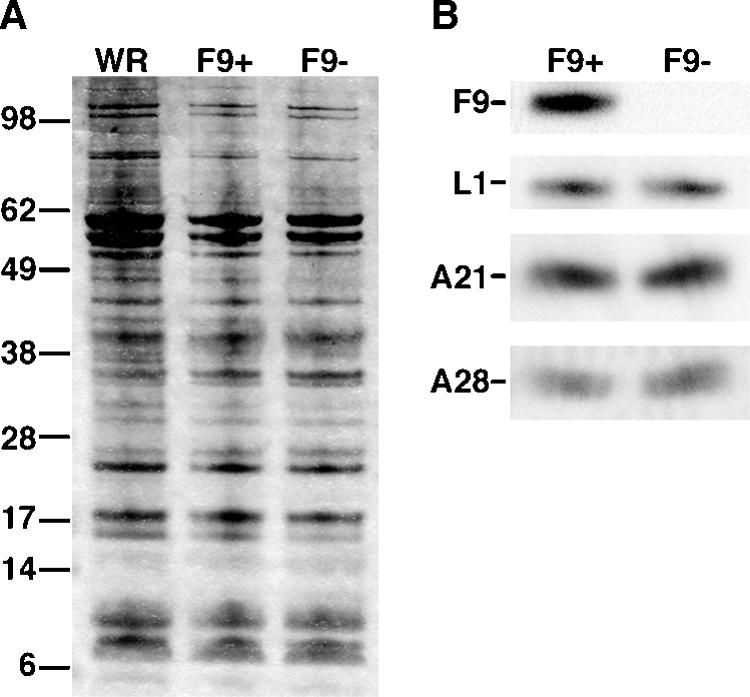
Polypeptide composition of purified F9+ and F9− virions. Purified MVs were obtained by sedimentation through two successive 36% sucrose cushions and a 25 to 40% sucrose gradient. Equal numbers of VACV WR, F9+, and F9− virions were solubilized with SDS and separated by SDS-PAGE in 4 to 12% polyacrylamide Bis-Tris gels. (A) Silver stain. The masses in kilodaltons are indicated on the left. (B) Western blot probed with anti-F9, anti-L1, anti-A21, and anti-A28 antibodies as indicated.
Virus binding and penetration.
Considering that F9 is an MV membrane protein that is essential for formation of infectious virus but is not required for virion morphogenesis, we considered that F9 might have a role in virus attachment or entry. The presence of a complete transcription system within VACV cores that is activated upon entry into the cytoplasm provides a highly sensitive assay for an early postentry event. Cells were infected with equal numbers of F9+ or F9− virions in the presence of AraC, which prevents viral DNA replication and late gene transcription. At 3 h after infection, total RNA was extracted and a Northern blot assay was performed with a radiolabeled probe complementary to the early transcript of the C11R gene. The autoradiograph in Fig. 8 shows an intense C11R band from the F9+-infected cells but none from the F9−- or mock-infected cells. The glyceraldehyde-3-phosphate dehydrogenase RNA loading control indicated that similar amounts of RNA were analyzed in all samples. Thus, F9− virions are defective in entry or an early postentry step.
FIG. 8.
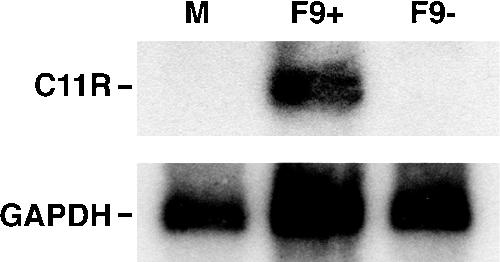
Early transcription in vF9Li-infected cells. BS-C-1 cells were either mock infected (M) or infected with an equal number of purified F9+ or F9− MV particles in the presence of AraC. At 3 h after infection, total RNA was harvested and a Northern blot assay was performed using a [32P]dCMP-labeled DNA probe for VACV early C11R and cellular glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA. An autoradiogram is shown.
A cell binding and penetration assay (39) was performed to further evaluate the defect of MVs lacking F9. This assay is dependent on the inability of an antibody to a core protein to bind prior to removal of the viral membrane, even when the particles are fixed and permeabilized. Cells were incubated with an equal number of F9+ or F9− virions at 4°C for 1 h, washed extensively to remove unbound or loosely bound virus, and shifted to 37°C for 2 h to allow penetration and release of cores into the cytoplasm. Following fixation and permeabilization, cells were incubated with antibodies specific for the L1 MV membrane protein and for the A4 core protein. Both F9+ and F9− virions bound to cells, as indicated by staining with the anti-L1 antibody after adsorption of virus at 4°C (not shown). After incubation at 37°C for 2 h, staining of virus cores with the anti-A4 antibody was detected only in the cytoplasm of cells infected with F9+ virions, whereas numerous anti-L1 staining particles remained on the outside of the cells infected with F9− virions (Fig. 9). Thus, F9 is needed for penetration but not cell binding.
FIG. 9.

Cell binding and penetration assay. HeLa cells were incubated with 10 PFU of purified F9+ virions or the same number of F9− virions at 4°C for 1 h and then shifted to 37°C for 2 h. Subsequently, cells were fixed, permeabilized, and stained for indirect immunofluorescence microscopy. The L1 MV membrane protein was stained with a mouse monoclonal anti-L1 antibody and a goat anti-mouse secondary antibody conjugated to Alexa Fluor 488 (green). The A4 core protein was stained with a rabbit primary antibody and a goat anti-rabbit IgG secondary antibody conjugated to Alexa Fluor 568 (red). Cellular DNA was stained with DAPI (blue).
F9 is required for low-pH-triggered cell-cell fusion.
Previous experiments showed that proteins required for virus entry are also needed to mediate cell-cell fusion (23, 30, 32, 36, 37). One type of fusion, called “fusion from within,” is induced by briefly incubating infected cells with progeny EVs on the surface in medium at a pH below 6; the second type, “fusion from without,” is induced by adsorbing MVs to the surface of uninfected cells and then lowering the pH. In a fusion-from-within assay, syncytia were observed when cells infected with vF9Li in the presence of IPTG were pulsed with low pH at 18 h after infection but not when IPTG was omitted (Fig. 10). Syncytia did not form in either case under neutral-pH conditions (Fig. 10). Similarly, in a fusion-from-without assay, large syncytia formed when cells inoculated with F9+ virions were shocked with low pH but not when cells inoculated with F9− virions were treated in the same way (not shown). Thus, F9− virions are unable to mediate low-pH-triggered cell-cell fusion.
FIG. 10.

Low-pH-triggered cell-cell fusion assays. BS-C-1 cells were infected with vF9Li in the presence or absence of IPTG. At 18 h after infection, the cells were treated for 5 min with pH 7.4 or pH 5.3 buffer at 37°C and then returned to normal medium. After 18 h, the cells were fixed, permeabilized, and stained with DAPI. Cells were visualized by phase-contrast microscopy, while DNA (blue) and GFP (green) staining were visualized by fluorescence microscopy.
Interaction of F9 with the entry-fusion complex.
Since F9 is required for entry and fusion, we examined whether it associates with other known entry-fusion proteins. BS-C-1 cells were infected with the recombinant vF9-V5, which has the V5 epitope tag at its C terminus, or with wild-type VACV WR. After 24 h, the membrane pellet fraction was solubilized with NP-40 detergent and F9-V5 and any associated proteins were captured on beads coupled to anti-V5 monoclonal antibody. The bound proteins were analyzed by SDS-PAGE and Western blotting with antibodies to four representative components of the entry/fusion complex: A16, A21, A28, and L5. Each of those proteins from the lysates of cells infected with vF9-V5 but not of cells infected with the control virus lacking the epitope tag was captured by antibody to V5 (Fig. 11A). The association of F9 with all four proteins suggests that it interacts with the multiprotein complex.
FIG. 11.

Interaction of F9 with the entry/fusion complex. (A) BS-C-1 cells were infected with vF9-V5 or VACV WR. After 24 h, the cells were disrupted with a Dounce homogenizer and postnuclear supernatants were centrifuged at 100,000 × g for 1 h. The pellet was extracted with PBS-NP-40, and the soluble fraction was immunopurified on agarose beads conjugated to V5 antibody. Proteins were resolved by SDS-PAGE, transferred to a nitrocellulose membrane, and incubated with anti-V5 antibody conjugated to peroxidase and primary rabbit antibodies to the A16, A21, A28, and L5 proteins followed by peroxidase-conjugated secondary antibody to rabbit IgG. (B) BS-C-1 cells were infected with vA28-HAi/H2-V5 in the presence or absence of IPTG. The NP-40-PBS-soluble extract of the membrane-enriched fraction was immunopurified on agarose beads conjugated to V5 antibody and resolved by SDS-PAGE. Proteins were transferred to a nitrocellulose membrane and detected by Western blotting with peroxidase-conjugated anti-V5 or anti-HA antibody or with anti-F9 rabbit antibody followed by peroxidase-conjugated anti-rabbit IgG.
To confirm the association of F9 with the entry/fusion complex, we infected cells with a recombinant VACV (vA28-HAi/H2-V5) that expresses an inducible HA-tagged A28 protein and a constitutively expressed V5-tagged H2 protein. H2 and A28 are components of the entry-fusion complex, formation of which is dependent on each of these proteins (30). The proteins in a membrane-enriched fraction from the infected cells were extracted and allowed to bind agarose beads conjugated to V5 antibody. The bound proteins were analyzed by SDS-PAGE and Western blotting. A28-HA and F9 were associated with H2-V5 in the presence of inducer, whereas the anti-V5 beads captured only H2-V5 in the absence of inducer (Fig. 11B). Taken together, these results indicated that F9 binds to proteins of the entry/fusion complex but not directly to H2. F9 could bind to A28 or to other components of the complex.
DISCUSSION
The product of the VACV F9L gene is expressed late in infection and incorporated into the MV membrane with the long N-terminal domain on the exterior, as shown by biotinylation and antibody neutralization of infectivity. The latter topology implies that the N-terminal domain is cytoplasmic during morphogenesis, which is consistent with a previous demonstration that the poxvirus-encoded redox proteins form the three intramolecular disulfide bonds of F9 (34) and with the known topology of the related L1 protein (2, 35), which has 20% sequence similarity with F9. Given the conservation of F9 in all sequenced poxviruses, even those infecting insects, we were not surprised to find that repression of F9 expression interfered with formation of infectious virus. In view of the relationship between L1 and F9, however, we had presumed that the phenotypes would be similar. Although repression of L1 results in a block at a late stage of morphogenesis, repression of F9 did not. Core proteins were processed normally, and virions lacking F9 were indistinguishable from wild-type virions by electron microscopy, SDS-PAGE analysis, and Western blotting of representative membrane proteins. Nevertheless, the infectivity of the F9-deficient virions made in the absence of inducer was 150-fold less than that of virions made in the presence of inducer. This degree of reduction is similar to that found for inducible mutants of other entry/fusion proteins. The low residual infectivity may be due to a combination of factors including incomplete removal of the inoculum virus, which was made in the presence of IPTG and therefore contains F9, and slight expression of F9 in the absence of inducer.
Synthesis of viral early RNAs was not detected in cells inoculated with F9-deficient MVs, suggesting an entry or early postentry block. The defect was more clearly defined by confocal microscopy. We found that F9-deficient MVs could bind to cells but that cytoplasmic cores were not detected by antibody labeling. A similar phenotype was found for other protein components of the putative entry/fusion complex (22, 23, 30, 32, 36, 37). F9 shares many characteristics with components of the entry/fusion complex including (i) conservation among all sequenced poxviruses, (ii) postreplicative expression, (iii) association with the MV membrane, (iv) formation of intramolecular disulfide bonds by the poxvirus-encoded redox pathway, and (v) absence of an essential role in morphogenesis. Furthermore, immunoaffinity purification with an epitope-tagged F9 indicated that the latter protein associates with the entry/fusion complex. The reverse experiment was also positive: antibody to an epitope-tagged component of the entry/fusion complex also captured F9. Although F9 associates with the complex, it may not be an integral component for the following reasons. First, the amount of F9 associated with the complex is low, as it was not detected in Coomassie blue-stained gels in which the other components appeared to be present in approximately stoichiometric quantities, accounting for the failure to detect it by mass spectroscopy (31). Second, the complex does not form or is unstable when expression of any one of the previously characterized components of the complex is repressed, but the complex still formed when F9 was repressed (T. Senkevich, unpublished). If F9 is not directly involved in fusion, it might have another role in entry that would require interaction with the entry/fusion complex. One intriguing possibility is that F9 is involved in receptor binding. Moreover, because of its structural relationship to F9, L1 may also have such a role in addition to its participation in morphogenesis. Experiments to test this hypothesis are in progress.
Acknowledgments
We thank Andrea Weisberg for electron microscopy and assistance with the illustrations, Norman Cooper for tissue culture cells, and Wolfgang Resch and Alan Townsley for helpful discussions.
The work was supported by the intramural program of the NIAID, NIH. The F9 antibody kindly provided by Gary Cohen and Roselyn Eisenberg was prepared with support to them from USPHS grant NIH RCE-U54-AI57168 from the NIAID, NIH.
REFERENCES
- 1.Alcami, A. 2003. Viral mimicry of cytokines, chemokines and their receptors. Nat. Rev. Immunol. 3:36-50. [DOI] [PubMed] [Google Scholar]
- 2.Aldaz-Carroll, L., J. C. Whitbeck, M. Ponce de Leon, H. Lou, L. K. Pannell, J. Lebowitz, C. Fogg, C. White, B. Moss, G. H. Cohen, and R. J. Eisenberg. 2005. Physical and immunological characterization of a recombinant secreted form of the membrane protein encoded by the vaccinia virus L1R gene. Virology 341:59-71. [DOI] [PubMed] [Google Scholar]
- 3.Alexander, W. A., B. Moss, and T. R. Fuerst. 1992. Regulated expression of foreign genes in vaccinia virus under the control of bacteriophage T7 RNA polymerase and the Escherichia coli lac repressor. J. Virol. 66:2934-2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Armstrong, J. A., D. H. Metz, and M. R. Young. 1973. The mode of entry of vaccinia virus into L cells. J. Gen. Virol. 21:533-537. [DOI] [PubMed] [Google Scholar]
- 5.Broyles, S. S. 2003. Vaccinia virus transcription. J. Gen. Virol. 84:2293-2303. [DOI] [PubMed] [Google Scholar]
- 6.Carter, G. C., M. Law, M. Hollinshead, and G. L. Smith. 2005. Entry of the vaccinia virus intracellular mature virion and its interactions with glycosaminoglycans. J. Gen. Virol. 86:1279-1290. [DOI] [PubMed] [Google Scholar]
- 7.Chang, A., and D. H. Metz. 1976. Further investigations on the mode of entry of vaccinia virus into cells. J. Gen. Virol. 32:275-282. [DOI] [PubMed] [Google Scholar]
- 8.Condit, R. C., N. Moussatche, and P. Traktman. 2006. In a nutshell: structure and assembly of the vaccinia virion. Adv. Virus Res., 66:31-124. [DOI] [PubMed]
- 9.Davison, A. J., and B. Moss. 1989. The structure of vaccinia virus late promoters. J. Mol. Biol. 210:771-784. [DOI] [PubMed] [Google Scholar]
- 10.Demkowicz, W. E., J. S. Maa, and M. Esteban. 1992. Identification and characterization of vaccinia virus genes encoding proteins that are highly antigenic in animals and are immunodominant in vaccinated humans. J. Virol. 66:386-398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Earl, P. L., J. L. Americo, and B. Moss. 2003. Development and use of a vaccinia virus neutralization assay based on flow cytometric detection of green fluorescent protein. J. Virol. 77:10684-10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Earl, P. L., N. Cooper, S. Wyatt, B. Moss, and M. W. Carroll. 1998. Preparation of cell cultures and vaccinia virus stocks, p. 16.16.1-16.16.3. In F. M. Ausubel, R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl (ed.), Current protocols in molecular biology, vol. 2. John Wiley and Sons, New York, N.Y. [Google Scholar]
- 13.Earl, P. L., B. Moss, L. S. Wyatt, and M. W. Carroll. 1998. Generation of recombinant vaccinia viruses, p. 16.17.1-16.17.19. In F. M. Ausubel, R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl (ed.), Current protocols in molecular biology, vol. 2. Greene Publishing Associates & Wiley Interscience, New York, N.Y. [Google Scholar]
- 14.Hsiao, J. C., C. S. Chung, and W. Chang. 1999. Vaccinia virus envelope D8L protein binds to cell surface chondroitin sulfate and mediates the adsorption of intracellular mature virions to cells. J. Virol. 73:8750-8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Janeczko, R. A., J. F. Rodriguez, and M. Esteban. 1987. Studies on the mechanism of entry of vaccinia virus into animal cells. Arch. Virol. 92:135-150. [DOI] [PubMed] [Google Scholar]
- 16.Katz, E., and B. Moss. 1970. Formation of a vaccinia virus structural polypeptide from a higher molecular weight precursor: inhibition by rifampicin. Proc. Natl. Acad. Sci. USA 6:677-684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Law, M., G. C. Carter, K. L. Roberts, M. Hollinshead, and G. L. Smith. 2006. Ligand-induced and non-fusogenic dissolution of a viral membrane. Proc. Natl. Acad. Sci. USA 103:5989-5994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin, C. L., C. S. Chung, H. G. Heine, and W. Chang. 2000. Vaccinia virus envelope H3L protein binds to cell surface heparan sulfate and is important for intracellular mature virion morphogenesis and virus infection in vitro and in vivo. J. Virol. 74:3353-3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moss, B. 2001. Poxviridae: the viruses and their replication, p. 2849-2883. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 4th ed., vol. 2. Lippincott Williams & Wilkins, Philadelphia, Pa. [Google Scholar]
- 20.Moss, B. 2006. Poxvirus entry and membrane fusion. Virology 344:48-54. [DOI] [PubMed] [Google Scholar]
- 21.Moss, B., and F. De Silva. 2006. Poxvirus, p. 707-728. In M. L. DePamphilis (ed.), DNA replication and human disease. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 22.Ojeda, S., A. Domi, and B. Moss. 2006. Vaccinia virus G9 protein is an essential component of the poxvirus entry-fusion complex. J. Virol. 80:9822-9830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ojeda, S., T. G. Senkevich, and B. Moss. 2006. Entry of vaccinia virus and cell-cell fusion require a highly conserved cysteine-rich membrane protein encoded by the A16L gene. J. Virol. 80:51-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ravanello, M. P., and D. E. Hruby. 1994. Conditional lethal expression of the vaccinia virus L1R myristylated protein reveals a role in virus assembly. J. Virol. 68:6401-6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rodríguez, D., M. Esteban, and J. R. Rodríguez. 1995. Vaccinia virus A17L gene product is essential for an early step in virion morphogenesis. J. Virol. 69:4640-4648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rodriguez, J. F., and G. L. Smith. 1990. Inducible gene expression from vaccinia virus. Virology 177:239-250. [DOI] [PubMed] [Google Scholar]
- 27.Rodriguez, J. R., C. Risco, J. L. Carrascosa, M. Esteban, and D. Rodriguez. 1998. Vaccinia virus 15-kilodalton (A14L) protein is essential for assembly and attachment of viral crescents to virosomes. J. Virol. 72:1287-1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sanderson, C. M., M. Hollinshead, and G. L. Smith. 2000. The vaccinia virus A27L protein is needed for the microtubule-dependent transport of intracellular mature virus particles. J. Gen. Virol. 81:47-58. [DOI] [PubMed] [Google Scholar]
- 29.Senkevich, T. G., E. V. Koonin, J. J. Bugert, G. Darai, and B. Moss. 1997. The genome of molluscum contagiosum virus: analysis and comparison with other poxviruses. Virology 233:19-42. [DOI] [PubMed] [Google Scholar]
- 30.Senkevich, T. G., and B. Moss. 2005. Vaccinia virus H2 protein is an essential component of a complex involved in virus entry and cell-cell fusion. J. Virol. 79:4744-4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Senkevich, T. G., S. Ojeda, A. Townsley, G. E. Nelson, and B. Moss. 2005. Poxvirus multiprotein entry-fusion complex. Proc. Natl. Acad. Sci. USA 102:18572-18577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Senkevich, T. G., B. M. Ward, and B. Moss. 2004. Vaccinia virus entry into cells is dependent on a virion surface protein encoded by the A28L gene. J. Virol. 78:2357-2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Senkevich, T. G., A. Weisberg, and B. Moss. 2000. Vaccinia virus E10R protein is associated with the membranes of intracellular mature virions and has a role in morphogenesis. Virology 278:244-252. [DOI] [PubMed] [Google Scholar]
- 34.Senkevich, T. G., C. L. White, E. V. Koonin, and B. Moss. 2000. A viral member of the ERV1/ALR protein family participates in a cytoplasmic pathway of disulfide bond formation. Proc. Natl. Acad. Sci. USA 97:12068-12073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Su, H. P., S. C. Garman, T. J. Allison, C. Fogg, B. Moss, and D. N. Garboczi. 2005. The 1.51-A structure of the poxvirus L1 protein, a target of potent neutralizing antibodies. Proc. Natl. Acad. Sci. USA 102:4240-4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Townsley, A., T. G. Senkevich, and B. Moss. 2005. The product of the vaccinia virus L5R gene is a fourth membrane protein encoded by all poxviruses that is required for cell entry and cell-cell fusion. J. Virol. 79:10988-10998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Townsley, A., T. G. Senkevich, and B. Moss. 2005. Vaccinia virus A21 virion membrane protein is required for cell entry and fusion. J. Virol. 79:9458-9469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Traktman, P., K. Liu, J. DeMasi, R. Rollins, S. Jesty, and B. Unger. 2000. Elucidating the essential role of the A14 phosphoprotein in vaccinia virus morphogenesis: construction and characterization of a tetracycline-inducible recombinant. J. Virol. 74:3682-3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vanderplasschen, A., M. Hollinshead, and G. L. Smith. 1998. Intracellular and extracellular vaccinia virions enter cells by different mechanisms. J. Gen. Virol. 79:877-887. [DOI] [PubMed] [Google Scholar]
- 40.Ward, B. M. 2005. Visualization and characterization of the intracellular movement of vaccinia virus intracellular mature virions. J. Virol. 79:4755-4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wolffe, E. J., D. M. Moore, P. J. Peters, and B. Moss. 1996. Vaccinia virus A17L open reading frame encodes an essential component of nascent viral membranes that is required to initiate morphogenesis. J. Virol. 70:2797-2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wolffe, E. J., S. Vijaya, and B. Moss. 1995. A myristylated membrane protein encoded by the vaccinia virus L1R open reading frame is the target of potent neutralizing monoclonal antibodies. Virology 211:53-63. [DOI] [PubMed] [Google Scholar]
- 43.Yeh, W. W., B. Moss, and E. J. Wolffe. 2000. The vaccinia virus A9L gene encodes a membrane protein required for an early step in virion morphogenesis. J. Virol. 74:9701-9711. [DOI] [PMC free article] [PubMed] [Google Scholar]