

Abstract
The immediate-early regulatory protein ICP22 is required for efficient replication of herpes simplex virus type 1 in some cell types (permissive) but not in others (restrictive). In mice infected via the ocular route, the pathogenesis of an ICP22− virus, 22/n199, was altered relative to that of wild-type virus. Specifically, tear film titers of 22/n199-infected mice were significantly reduced at 3 h postinfection relative to those of mice infected with wild-type virus. Further, 22/n199 virus titers were below the level of detection in trigeminal ganglia (TG) during the first 9 days postinfection. On day 30 postinfection, TG from 22/n199-infected mice contained reduced viral genome loads and exhibited reduced expression of latency-associated transcripts and reduced reactivation efficiency relative to TG from wild-type virus-infected mice. Notably, the first detectable alteration in the pathogenesis of 22/n199 in these tests occurred in the eye prior to the onset of nascent virus production. Thus, ICP22− virions appeared to be degraded, cleared, or adsorbed more rapidly than wild-type virions, implying potential differences in the composition of the two virion types. Analysis of the protein composition of purified extracellular virions indicated that ICP22 is not a virion component and that 22/n199 virions sediment at a reduced density relative to wild-type virions. Although similar to wild-type virions morphologically, 22/n199 virions contain reduced amounts of two γ2 late proteins, US11 and gC, and increased amounts of two immediate-early proteins, ICP0 and ICP4, as well as protein species not detected in wild-type virions. Although ICP22− viruses replicate to near-wild-type levels in permissive cells, the virions produced in these cells are biochemically and physically different from wild-type virions. These virion-specific differences in ICP22− viruses add a new level of complexity to the functional analysis of this immediate-early viral regulatory protein.
Infected cell protein 22 (ICP22), a 420-amino-acid protein encoded by the US1 gene, is one of five herpes simplex virus type 1 (HSV-1) proteins expressed immediately following entry of viral genomes into the nuclei of susceptible cells. The US1 open reading frame contains an internal promoter that drives expression of the 249-amino-acid US1.5 protein, a largely uncharacterized protein that is in frame with the carboxyl terminus of ICP22 (5, 27). To date, the function(s) of the US1.5 protein has not been distinguished from that of ICP22 such that the reported functions of ICP22 may actually reflect the combined functions of the two proteins.
Although the role of ICP22 in HSV-1 replication has not been elucidated, evidence suggests that ICP22 alters the activities of cellular proteins required for efficient virus replication (1, 3, 4, 37). ICP22− viruses replicate to near-wild-type levels in Vero, HeLa, and HEp-2 cells (permissive cells), whereas in all other cell types tested, ICP22− viruses produce 2 to 3 logs less virus than wild-type virus (restrictive cells) (28, 31, 34, 37). In both permissive and restrictive cells, ICP22− viruses induce the synthesis of near-wild-type levels of immediate-early (IE), early (E), and γ1 late (L) proteins as well as viral DNA with kinetics similar to those of wild-type virus (31, 37). Notably, ICP22− viruses fail to induce efficient expression of a subset of γ2 L proteins upon infection (28, 31, 34, 37), a defect most evident in restrictive cells. Because of the cell-type-dependent growth properties of ICP22− viruses, it has been proposed (1, 28, 31, 34, 37) that the primary role of ICP22 during lytic replication is to alter the expression, activity, or posttranslational modification of cellular proteins to provide a suitable environment for expression of γ2 L genes whose expression is (i) dependent on newly synthesized viral DNA (19) and (ii) essential for virion assembly.
The role of ICP22 in vivo has not been well established because reduced quantities of ICP22− virus, its gene products, and viral genomes are detected during the first 30 days postinfection (dpi) relative to wild-type virus-infected animals. Acute replication of an ICP22− virus was examined following intravaginal inoculation of mice and determined to be reduced approximately 100-fold 1 dpi compared to wild-type virus-infected mice (30), and titers of the ICP22− virus in vaginal secretions were reduced relative to wild-type virus-infected animals during the first 9 days postinoculation (pi). Additionally, viral genome loads were reduced in the dorsal root ganglia of ICP22− virus-infected animals compared to wild-type virus-infected animals on day 30 pi and no reactivation was observed when ganglia from ICP22− virus-infected mice were explanted. In the mouse ocular model, two groups reported that more than half the total number of trigeminal ganglia (TG) explanted from mice infected with ICP22− viruses contained reactivatable virus using a similar explant model (30, 37). Although the aforementioned studies do not define the role of ICP22 in the establishment and reactivation of latency, they demonstrate that ICP22− viruses are attenuated in vivo.
To date, the functional properties of ICP22 have been elucidated largely through studies of two mutants of HSV-1 strain F, R325-βtk+ and del22Z (31, 32). In addition to mutations in the US1 gene, both of these viruses contain significant alterations in sequences surrounding the US1 gene. We therefore isolated an ICP22− virus, 22/n199, that is minimally altered in the US1 gene and contains no alterations in adjacent genes or cis-acting elements (28). In both permissive and restrictive cells, 22/n199 replicates in a manner similar to other ICP22− viruses (28, 31, 37). The primary purpose of the present study was to characterize the properties of 22/n199 in vivo. We show that the initial deficiency of 22/n199 in vivo occurs within the first 3 hours pi (hpi). Because this defect is evident prior to the onset of nascent virus production, we hypothesized that the physical properties of 22/n199 virions differ from those of wild-type virions. Although ICP22 was not a detectable component of wild-type virions, we show that it is required to produce virions of wild-type composition. Collectively, these findings demonstrate that ICP22 affects the composition of virions either directly or indirectly and that the abnormal composition of mutant virions, even when produced in permissive cells, affects the pathogenesis of ICP22− viruses in mice.
MATERIALS AND METHODS
Cells and viruses. (i) Cells.
The immortalized African green monkey kidney cell line Vero (ATCC CCL-81) and the human lung carcinoma cell line A549 (ATCC CCL-185) were obtained from the American Type Culture Collection (Manassas, VA).
To engineer ICP22-expressing Vero cells (V22 cells), a plasmid was first constructed in which the US1 gene was placed under the control of the human cytomegalovirus IE promoter. This plasmid, pCMV22, is a derivative of pRL-CMV (Promega, Madison, WI) and was constructed by replacing the luciferase gene in pRL-CMV with a PCR-generated, amino-terminally FLAG epitope-tagged ICP22 gene. To generate V22 cells, pCMV22 was cotransfected into Vero cells with a G418 resistance marker, and G418-resistant clones were isolated and tested for expression of FLAG-tagged ICP22 following HSV-1 infection (data not shown). One positive clone was designated V22. Immunoblot analysis indicated that V22 cells do not constitutively express detectable ICP22 but express it at high levels during HSV-1 infection (data not shown). Subsequent sequencing of pCMV22 revealed two coding mutations in the US1 gene relative to the parental KOS sequence. These changes are valine 10 to alanine and alanine 208 to threonine. Although the biological effect(s) of these alterations is unknown, ICP22 expressed in V22 cells is fully functional in its ability to complement the RNA polymerase II modification defect (34) of 22/n199 (K. Fraser and S. Rice, unpublished observation).
All cells were grown in Dulbecco's modified Eagle's medium (DMEM), supplemented with 10% fetal bovine serum, 100 units/ml penicillin, 100 μg/ml streptomycin, and 6 mM glutamine (complete DMEM) at 37°C in 5% CO2. V22 cells were passaged in the presence of 300 μg/ml G418. For virion preparation in V22 cells, cells were plated in the absence of G418.
(ii) Viruses.
The wild-type virus used in these studies was HSV-1 strain KOS passage 12 (38). The KOS-derived ICP22− mutant virus, 22/n199, and its rescuant, 22/n199R, have been described previously (28). Viral stocks were prepared in Vero or V22 cells and assayed on Vero cell monolayers (9).
Inoculation of mice.
Seven-week-old outbred male CD-1 mice (Charles River Laboratory Inc., Kingston, N.Y.) were infected with wild-type, 22/n199, or 22/n199R viruses by the ocular route as previously described (2, 16). Briefly, mice were anesthetized by intraperitoneal administration of ketamine (100 mg/kg of body weight) and xylazine (9 mg/kg). Corneas were scarified with a 26-gauge needle 10 times vertically and 10 times horizontally, and 3 μl of complete DMEM containing 5 × 106 PFU of virus was placed on each eye. Following inoculation, viral inoculum titers were confirmed by standard plaque assay and did not vary more than twofold from the calculated dose.
Viral replication in acutely infected mice.
To determine viral titers in the tear film of infected animals, tear film samples were collected each day for 9 days or every 30 min during the first 3 h following inoculation (2). Briefly, both eyes were swabbed with a single cotton-tipped applicator and placed in 0.5 ml of complete DMEM. To determine viral titers in TG of infected animals, mice were euthanized daily, and TG were removed and homogenized as previously described (2, 16). Viral titers in tear film and TG homogenates were determined by plaque assays on Vero cell monolayers (9).
Analysis of the efficiency of establishment and reactivation of latency in infected mice.
Infected mice were euthanized and TG were removed on day 30 postinoculation. The excised TG were used to examine viral genome loads, expression of latency-associated transcripts (LATs), and reactivation efficiency. The relative level of viral DNA in 100 ng of the ∼2.5 mg of total DNA in individual TG was determined by PCR analysis as previously described (20). In situ hybridization of ganglionic sections to assess the extent of LAT expression was performed as previously described (23, 40, 41). In vitro reactivation studies were carried out with TG explants by cocultivating explanted TG on Vero cell monolayers in growth medium and sampling the medium daily for reactivated virus (2, 15, 23). The presence of infectious virus was scored by inoculating 1-day-old Vero cell monolayers with TG cell culture medium and monitoring cells for the appearance of cytopathic effects by light microscopy.
Cellular lysates and extracellular and purified virion preparations.
Cellular lysates and virion preparations were obtained from the same infected cell monolayers. Briefly, 1.5 × 106 Vero or V22 cells were seeded in 150-mm dishes. After incubation for 24 h at 37°C, cells were infected at a multiplicity of infection of 10 PFU/cell. After 1 h of incubation at 37°C, inoculum was removed, monolayers were washed once with phosphate-buffered saline (PBS), and 15 ml of complete DMEM was added to each dish. At 24 hpi, medium containing extracellular virus was pooled and infected cells were collected. With the exception of 22/n199 virions produced in Vero cells, eight 150-mm dishes were used for each replicate virion preparation. Because 22/n199 produces considerably lower levels of extracellular virus per infected Vero cell than wild-type or 22/n199R viruses (data not shown), 24 150-mm dishes were used for 22/n199 virion preparations.
(i) Cellular lysates.
At 24 hpi, the virion-containing culture medium was pooled for extracellular virion isolation and infected cells from a single 150-mm dish were scraped into cold PBS containing 10 μg/ml leupeptin, 10 μg/ml aprotinin, 10 μg/ml pepstatin, and 1 mM benzamidine (protease inhibitor cocktail). Cells were collected by low-speed centrifugation at 1,600 × g for 5 min at 4°C and lysed in 500 μl of RIPA buffer (150 mM NaCl, 1% NP-40, 0.5% deoxycholate, 0.1% sodium dodecyl sulfate [SDS], 50 mM Tris [pH 8.0], and the protease inhibitor cocktail) for 30 min at 4°C. Lysates were clarified by low-speed centrifugation.
(ii) Extracellular virion preparation.
At 24 hpi, growth medium containing extracellular virus was collected from all dishes in each group and pooled. Cellular debris was removed by low-speed centrifugation for 5 min at 4°C. Extracellular virion preparations were obtained by subjecting clarified medium to high-speed centrifugation at 85,000 × g for 1 h at 4°C and resuspending the viral pellet in 100 μl TNE buffer (10 mM Tris [pH 7.5], 100 mM NaCl, 1 mM EDTA, and the protease inhibitor cocktail) for 24 h with gentle agitation at 4°C.
(iii) Purified virion preparations.
The protocols of Compton and Courtney (6), and Yao and Courtney (44) were modified to obtain purified virions. Extracellular virions were layered on sucrose-TNE step gradients (20%, 30%, 40%, 50%, and 60%) containing the protease inhibitor cocktail and centrifuged at 50,000 × g for 20 h at 4°C. Gradients were photographed with a Kodak DC500 digital camera and fractionated in 20 0.5-ml fractions, or the virion-containing bands were removed by side puncture. The density (percent sucrose) of each fraction or of each sample was determined using a refractometer (Bausch & Lomb, Rochester, NY). Virion preparations were diluted in PBS containing the protease inhibitor cocktail and centrifuged at 85,000 × g for 1 h at 4°C to remove sucrose. Virion pellets were resuspended in PBS containing the protease inhibitor cocktail by gentle agitation for 24 h at 4°C. Scanning electron microscopy of purified virion preparations was performed at the Harvard Conventional Electron Microscopy Facility.
Protein analysis.
Extracellular and purified virion preparations were denatured in equal volumes of RIPA buffer for 30 min at 4°C. Virion lysates were clarified by centrifugation at 1,600 × g for 5 min. The protein content of these samples and of cellular lysates was determined using the bicinchoninic acid protein assay kit (Pierce, Rockford, Ill.) per the manufacturer's instructions. Samples containing 5 μg of total protein were separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) using 10% polyacrylamide minigels (Bio-Rad MiniProtean 3; Hercules, CA) and either transferred to nitrocellulose membranes (Osmonics, Minnetonka, MN) for Western blot analysis or silver stained using the SilverQuest silver staining kit (Invitrogen, Carlsbad, CA) per the manufacturer's instructions. Western blot analysis was performed as previously described (28). Primary antibodies and the dilutions utilized were anti-VP5 (Abcam, Cambridge, MA), 1:1,000; anti-ICP27 (H1113; Goodwin Institute for Cancer Research, Plantation City, FL), 1:1,000; anti-UL42 (13, 14), 1:1,000; anti-gE (Abcam), 1:1,000; anti-US11 (10), 1:1,000; anti-ICP22 (28), 1:200; anti-ICP0 (US Biological, Swampscott, MA), 1:500; anti-ICP4 (Goodwin Institute for Cancer Research), 1:1,000; and anti-gC (Abcam), 1:500.
RESULTS
ICP22 is required for efficient infection in mice.
Two groups have examined the role of ICP22 during HSV-1 infection in vivo (30, 37). They utilized two different US1 mutants, both of which contain significant alterations in adjacent genes or cis-acting elements. Recently we reported the isolation and characterization of an ICP22− virus, 22/n199, containing a mutation in ICP22 similar to that of the best-characterized ICP22− virus, R325βtk+, but without partial deletions in US2 and sequences encoding the LS/T transcripts (28, 37, 45). Although 22/n199 replicates in a manner similar to that of the other ICP22− viruses in permissive and restrictive cells in vitro (28, 30, 37), it is unknown whether the phenotypes of the other ICP22− viruses in vivo can be attributed solely to the absence of ICP22. This work was initiated in an effort to assess the role of ICP22 in the establishment and reactivation of HSV-1 latency using the mouse ocular model. For this purpose, mice were infected following corneal scarification with 5 × 106 PFU/eye of wild-type, 22/n199, or 22/n199R (rescuant) viruses (28).
(i) Viral titers in tear film.
To determine the efficiency of viral replication and rate of viral clearance at the site of inoculation, we collected tear film at 3 hpi and on days 1 to 9 pi and determined viral titers by plaque assay on permissive Vero cell monolayers (9). All three viruses recovered from eclipse phase and replicated in a biphasic manner over the 9-day test period (Fig. 1A). Notably, on days 1 to 6 pi, tear film titers of 22/n199 were markedly lower than titers of the wild-type virus. Specifically, on days 1 to 3 pi, the tear film titers of 22/n199-infected mice were more than 100-fold lower than those of wild-type virus-infected mice (P = 0.003, two-way analysis of variance [ANOVA]). From days 4 through 6 pi, tear film titers of 22/n199-infected mice were reduced 10-fold compared to wild-type virus-infected mice (P = 0.02, two-way ANOVA). 22/n199 was cleared more rapidly than wild-type virus because tear film titers were below the level of detection 1 day earlier than in wild-type virus-infected mice in all six mice tested. 22/n199R replicated with wild-type efficiency and kinetics in these tests (P = 0.86, two-way ANOVA), demonstrating that the mutation in the US1 gene in 22/n199 is responsible for this phenotype.
FIG. 1.

Acute-phase replication in eyes and TG of wild-type, 22/n199, and 22/n199R viruses. (A) Tear film titers. Six mice per group were infected in both eyes with 5 × 106 PFU/eye following corneal scarification. At 3 hpi and on days 1 to 9 pi, both eyes were swabbed with a single cotton-tipped applicator and placed in complete DMEM, and titers were determined by plaque assay on Vero cell monolayers. The averages of six mice per time point are shown, and the error bars represent the standard deviations. (B) Viral titers in TG. On days 1 to 9 pi both TG were removed and homogenized and the titer of infectious virus was determined by plaque assay on Vero cell monolayers. The averages of 12 TG are shown; error bars represent the standard deviations.
(ii) Viral titers in TG.
To determine the efficiency of viral spread and replication in ganglionic cells, TG from wild-type-, 22/n199-, and 22/n199R-infected mice were excised on days 1 through 9 pi and homogenized. The amount of infectious virus was determined by plaque assay on Vero cell monolayers (Fig. 1B) (9). Levels of 22/n199R were similar to levels of wild-type virus (P = 0.50, two-way ANOVA) in TG in that infectious virus was first detected at 2 dpi, peaked with similar titers on day 3 pi, and declined progressively through day 6 pi. By contrast, no infectious virus was detected in TG of 22/n199-infected mice throughout the 9-day test period (P = 1.5 × 10−12, two-way ANOVA).
(iii) Gross pathology.
Physical examination of mice for 30 days pi revealed that those infected with the wild-type or 22/n199R viruses showed similar symptoms of disease. Specifically, on day 5 pi and thereafter, both wild-type- and 22/n199R-infected mice exhibited moderate to severe symptoms of blepharoconjunctivitis, ulcerative lesions, and periocular hair loss. Additionally, approximately 10% of mice infected with wild-type or 22/n199R viruses died by day 15 pi. By contrast, all mice infected with 22/n199 were disease free and survived the 30-day experiment.
22/n199 establishes and reactivates from latency inefficiently.
To determine whether ICP22 plays a role in the establishment and reactivation of latency, mice infected with wild-type, 22/n199, or 22/n199R viruses were euthanized on day 30 pi and TG were excised to determine (i) viral genome loads, (ii) the number of neurons expressing LATs, and (iii) the kinetics and efficiency of reactivation.
(i) Viral genome loads.
Because the TG of mice infected with 22/n199 contained no detectable infectious virus during the first 9 days pi, it seemed likely that TG from mice latently infected with 22/n199 would harbor reduced levels of viral DNA compared to the TG of wild-type virus-infected mice. To test this hypothesis, TG from mock- and latently infected mice were excised, total DNA was isolated, and a viral thymidine kinase (tk) gene-specific fragment was amplified by PCR as previously described (20). As expected, the tk-specific fragment was not amplified from TG DNA of four mock-infected mice whereas DNA from the TG of five wild-type virus-infected mice produced a 60-bp tk-specific fragment (Fig. 2A). Although DNA from all four 22/n199-infected mouse TG yielded a similar tk-specific fragment in the PCR, the quantity amplified was markedly reduced compared to the levels amplified from wild-type virus-infected TG (Fig. 2A).
FIG. 2.

Efficiency of establishment and reactivation of latency. Mice were infected as described in the legend to Fig. 1. On day 30 pi, mice were euthanized and both TG were removed. (A) Total DNA was isolated from individual TG from mock-, wild-type-, and 22/n199-infected mice, and a 60-bp HSV-1 tk gene-specific fragment was amplified from 100 ng of DNA by PCR. PCR products were separated on 10% polyacrylamide gels and visualized following ethidium bromide staining. The numbered lanes contain PCR products from individual ganglia. (B) Reactivation efficiencies of the wild-type, 22/n199, and 22/n199R viruses in TG explant cultures. Sixteen individual TG per group were cocultured on Vero cell monolayers. Culture medium was assayed daily for the presence of infectious virus as indicated by cytopathic effects on Vero cell monolayers. On day 10 postexplant, TG explant cultures were heat stressed by incubation at 43°C for 3 h as indicated by the arrow. The cumulative percentage of cultures containing reactivated virus is shown.
(ii) LAT expression.
Expression of the LATs is a hallmark of HSV-1 latent infection (8, 39, 41). As an alternative measure of the efficiency of the establishment of latency, TG latently infected with the wild-type, 22/n199, and 22/n199R viruses were examined for LAT expression by in situ hybridization as previously described (23, 40, 41). On day 30 pi, TG were excised, sectioned, probed with radiolabeled viral DNA fragments complementary to LAT or ICP0 (negative control) mRNA, and stained with photographic emulsion. Using a LAT-specific probe, dense grains were detected in all wild-type- and 22/n199R-infected TG by light microscopy; however, less than half of 22/n199 latently infected TG contained dense grains in neuronal nuclei (Table 1). The average number of neuronal nuclei that exhibited LAT-specific staining was approximately eightfold lower in TG from 22/n199-infected mice than in TG from wild-type- and 22/n199R-infected mice (Table 1). No signal was observed in latently infected TG from all mice when a probe specific for ICP0 mRNA was used (data not shown). Taken together, these results suggest that 22/n199 establishes latency less efficiently than wild type virus.
TABLE 1.
LAT expression in ganglia latently infected with wild-type, 22/n199, or 22/n199R viruses
Number of positive ganglia/number of ganglia examined.
Average number of LAT+ neurons/ganglion section.
Twenty-five sections were prepared for each TG.
(iii) Kinetics and efficiency of reactivation.
TG explants from wild-type-, 22/n199-, and 22/n199R-infected mice were cocultivated on Vero cell monolayers to determine the efficiency of ex vivo reactivation. Every day postexplant (dpe), an aliquot of tissue culture supernatant from each TG explant was removed and placed on fresh Vero cell monolayers and monolayers were examined daily for cytopathic effects by light microscopy (2, 16, 23). The mean times to detection of reactivated virus were similar for the wild-type (3.9 ± 0.2 dpe) and 22/n199R (3.4 ± 0.1 dpe) viruses (P = 0.28, two-sided t test) (Fig. 2B). By contrast, only 5% of TG explant cultures from 22/n199-infected mice contained infectious virus on day 4 postexplant and 12.5% by day 10. The remaining 22/n199 TG explants were heat stressed for 3 h at 43°C and tested daily for the presence of infectious virus through 16 dpe. Although heat stress is an effective means of inducing reactivation (17), no new reactivated virus was detected in these cultures. Thus, the kinetics and efficiency of reactivation of 22/n199 differed significantly from those of the wild-type virus (P < 1 × 10−6; Fisher's exact test).
22/n199 virions lose infectivity more rapidly than wild-type virions following ocular inoculation.
In vivo, ICP22− viruses are defective for all properties measured to date. At the site of inoculation, the infectivity of 22/n199 in tear film was reduced 500-fold by 3 hpi compared to wild-type virus-infected mice (Fig. 1A). To determine the kinetics of this reduction in viral infectivity, we determined viral titers in tear film every 30 min for the first 3 hours pi (Fig. 3). At 30 and 60 min pi, tear film titers of all three viruses decreased in a similar manner relative to inocula. By contrast, at 90 min pi, tear film titers of wild-type- and 22/n199R-infected mice declined less than 10-fold relative to input virus, whereas tear film titers from 22/n199-infected mice had dropped more than 200-fold. At 3 hpi, tear film titers were reduced 230- and 270-fold compared to input inoculum for wild-type and rescuant viruses. By contrast, at 3 hpi, the titer of 22/n199 in tear film was reduced 1.3 × 105-fold relative to inoculum (i.e., 560-fold lower than wild-type virus at 3 hpi). Because nascent virus production is not complete by 3 hpi, the reduction in 22/n199 titers in tear film cannot be a consequence of inefficient replication but, rather, likely results from differences in the rates of adsorption, clearance, or inactivation of mutant and wild-type virions.
FIG. 3.

Titers of wild-type, 22/n199, and 22/n199R viruses in tear film during the first 3 h postinoculation. Eight mice per group were infected; both eyes were swabbed at 30, 60, 90, 120, and 180 min pi with a single cotton-tipped applicator; and the titer of infectious virus was determined as described in the legend to Fig. 1. The average tear film titers from eight mice are shown, and error bars represent the standard deviations.
ICP22 is not detectable in purified wild-type virions.
A potential explanation for the biological differences between 22/n199 and wild-type viruses is that ICP22 is a virion component and 22/n199 virions lack this protein, rendering the virion unstable or a target for rapid clearance. This hypothesis is supported by the observations that two other HSV-1 immediate-early proteins, ICP0 and ICP4 (43, 44), and homologues of ICP22 from other α-neurotropic herpesviruses, orf63 of varicella-zoster virus and IR4 of equine herpesvirus (21), have been reported to be virion components (18, 21). To determine whether ICP22 is a component of the HSV-1 virion, we prepared partially purified extracellular and purified extracellular virions from two different cell types, Vero and A549 cells, infected with the wild-type virus. Virions were purified by equilibrium density sucrose gradient centrifugation. Cellular lysates and extracellular and purified extracellular virions were analyzed by SDS-PAGE followed by silver staining (Fig. 4A) and Western blot analysis (Fig. 4B). Extracellular and purified extracellular virion preparations derived from infected Vero cells exhibited similar, but not identical, protein profiles by silver staining (Fig. 4A). Notably, these profiles are similar to those previously reported for HSV-1 virions (43, 44). Purified HSV-1 virion preparations contained primarily the major recognized virion proteins (VPs, Fig. 4A).
FIG. 4.

Analysis of virion proteins. (A) Protein profiles of the lysates of wild-type-infected cells, extracellular virions, and purified extracellular virions. Vero cells were infected with the wild-type virus, and cellular lysates and extracellular and purified extracellular virions were isolated as described in Materials and Methods. These preparations were solubilized in RIPA buffer, and 5 μg of total protein was resolved using 10% SDS-polyacrylamide gels. Proteins were visualized by silver staining, and major virion proteins were labeled as VPs 1 to 23. (B) Western blot analysis of cellular lysates and virion preparations from A549 (A) and Vero (V) cells. Both mock- and wild-type virus-infected cell lysates as well as extracellular and purified virions were prepared and separated by SDS-PAGE. Proteins were transferred to nitrocellulose membranes and probed using antibodies specific for the proteins listed. MW, molecular weights in thousands.
Western blot analysis was performed to detect five recognized virion components (VP5, ICP0, ICP4, gE, and US11) (11, 29, 42-44), two viral proteins known to be excluded from virions (ICP27 and UL42) (43, 44), and ICP22 in mock-infected cell lysates, wild-type-infected cell lysates, and extracellular and purified virion preparations derived from both A549 and Vero cells (Fig. 4B). As expected, all viral proteins tested were expressed in infected cell lysates but not in lysates of mock-infected cells. The positive virion protein controls, VP5, ICP0, ICP4, gE, and US11, were detected in both extracellular and purified virion preparations whereas the negative controls, ICP27 and UL42, were absent from all virion preparations. Notably, ICP22, while present in infected cell lysates, was absent from all virion preparations shown in Fig. 4 or in virion preparations from five other independent experiments. These results support the notion that ICP22 is not a significant component of the HSV-1 virion. It remains possible, however, that ICP22 levels in virions are below the level of detection.
ICP22 influences the composition and physical properties of HSV-1 virions.
The rapid loss of infectivity of 22/n199 relative to the wild-type virus at the site of inoculation suggests that 22/n199 virions are functionally different from wild-type virions. To determine the nature of these differences, extracellular and purified virions were isolated from Vero cells infected with the wild-type, 22/n199, or 22/n199R viruses. As a control for ICP22 expression in trans, extracellular and purified 22/n199 virions were also prepared in ICP22-expressing V22 cells. Virions were not prepared from A549 cells because these cells are restrictive for ICP22− virus replication (data not shown).
Following centrifugation, sucrose gradients containing wild-type or 22/n199 virions prepared in Vero cells, as well as 22/n199 virions prepared in V22 cells, were photographed (Fig. 5). Three light-scattering bands, labeled light (l), medium (m), and heavy (h), were evident in each preparation. The intensity of each band differed among the virus types tested. In the gradients containing wild-type virions produced in Vero cells or 22/n199 virions produced in V22 cells, bands m and h were prominent; however, only band m was prominent in the gradient containing 22/n199 virions produced in Vero cells. Gradients containing purified 22/n199R virions produced in Vero cells resembled gradients containing wild-type virions (data not shown).
FIG. 5.
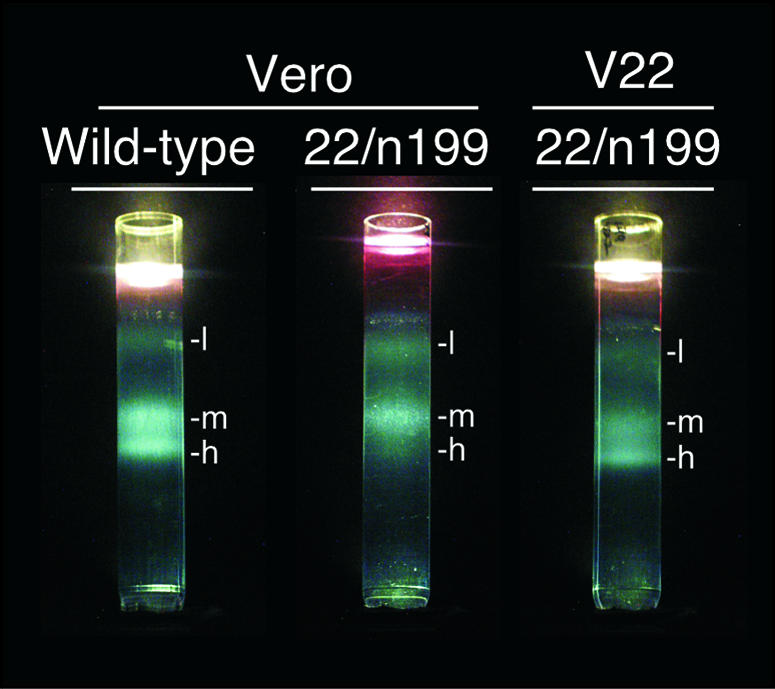
Photographs of sucrose gradients containing virion preparations. Extracellular virions from Vero cells infected with the wild-type or 22/n199 virus or V22 cells infected with 22/n199 were separated on sucrose step gradients (20 to 60%). Three light-scattering bands are labeled light (l), medium (m), and heavy (h).
To determine which of the three bands contained infectious virus, we prepared similar gradients containing wild-type, 22/n199, and 22/n199R virions produced in Vero cells and fractionated them. For each fraction we determined the sucrose gradient density by refractometry and viral infectivity by plaque assay. As shown in Fig. 6, sucrose gradient densities decreased linearly between fractions 5 and 13 in all three gradients. Viral infectivity in gradients containing wild-type or 22/n199R virions was concentrated in fraction 8, which corresponded to a sucrose content of 44% (Fig. 6A and C). By contrast, viral infectivity in gradients containing 22/n199 virions was concentrated in fraction 12, which corresponded to a sucrose content of 37% sucrose (Fig. 6B). Each fraction was then diluted in PBS, and virions were pelleted. Pellets were resuspended in RIPA buffer and the contents analyzed for VP5, the major capsid protein, by Western blotting. Consistently, VP5 was present in fraction 8 for wild-type and 22/n199R virions and in fraction 12 for 22/n199 virions, i.e., the same fractions in which viral infectivity was concentrated.
FIG. 6.

Determination of the percentage of sucrose in which wild-type (A), 22/n199 (B), and 22/n199R (C) virions sediment. Sucrose gradients similar to those shown in Fig. 5 were fractionated, and the infectivity of each fraction was determined by standard plaque assays on Vero cell monolayers. Closed circles indicate titers of each fraction, and the scale of PFU/ml is on the left of each plot. Open circles indicate the percent sucrose of each fraction, and the scale is on the right of each plot. Western blotting against VP5 was performed on each fraction and are shown beneath each graph.
All visible bands in additional gradients of wild-type, 22/n199, and 22/n199R virions produced in Vero cells and 22/n199 virions produced in V22 cells were harvested by side puncture and analyzed for infectivity and VP5 content. The infectivity and VP5 content in gradients containing 22/n199 virions produced in Vero cells were concentrated in the m band, which sedimented at an average sucrose content of 36.7% in five repeat experiments (Table 2). In these gradients the sedimentation of 22/n199 virions varied greatly from 33.8 to 39.7%. By contrast, the infectivity and VP5 content of all other virion preparations were concentrated in the h band, were less variable, and sedimented in approximately 44% sucrose (Table 2).
TABLE 2.
Percent sucrose in which virions sediment
| Virus | % Sucrose | na | SD |
|---|---|---|---|
| Wild type | 44.2 | 5 | 0.91 |
| 22/n199 | 36.7 | 5 | 2.51 |
| 22/n199R | 43.7 | 3 | 0.78 |
| 22/n199-V22 | 44.3 | 3 | 0.82 |
Number of preparations.
Because 22/n199 virions isolated from infected Vero cells were less dense than wild-type virions, the morphology of purified virion preparations was examined by scanning electron microscopy. Although two preparations of both types of virions contained large numbers of virions, no obvious differences in the size or morphology of virions were observed (data not shown). Additionally, these preparations were free of extraneous membrane-containing material, suggesting that purified extracellular virion preparations did not contain a large amount of contamination.
ICP22 affects the composition of HSV-1 virions.
22/n199 virions prepared in Vero cells were less dense than but morphologically similar to wild-type virions. This suggested that the composition of 22/n199 virions differs from that of wild-type virions. To test this hypothesis, extracellular and purified wild-type, 22/n199, and 22/n199R virions were solubilized in RIPA buffer and the protein content analyzed by silver staining (Fig. 7A) and Western blot analysis (Fig. 7B and C).
FIG. 7.

Comparison of the composition of wild-type 22/n199 and 22/n199R virions. Vero cell monolayers were infected with wild-type (wt), 22/n199 (9), and 22/n199R (R) viruses, or V22 cells were infected with 22/n199 (V). Cellular lysates, extracellular virions, and purified extracellular virions were prepared, solubilized in RIPA buffer, and resolved on 10% SDS-polyacrylamide gels. (A) Protein profile visualized by silver staining. Proteins unique to purified 22/n199 preparations are marked by asterisks. (B and C) Western blot analysis against the proteins indicated on the left. In the upper ICP0 Western blot a quantity of ICP0 was present that was sufficient to quench the signal in 22/n199 virion preparations. The bottommost ICP0 blot contains 1/10 the amount of protein loaded in each lane. MW, molecular weights in thousands.
Although the silver-stained protein profile of extracellular and purified 22/n199 virions resembled the profile of extracellular and purified wild-type or 22/n199R virions, several differences were evident. Specifically, the purified 22/n199 virion preparation contained reduced amounts of VPs 1 to 3 and 7 and increased amounts of VPs 15 and 22 compared to purified wild-type or 22/n199R virions. Moreover, preparations of 22/n199 virions contained proteins either not present or differentially modified compared to wild-type or 22/n199R virions. For example, 22/n199 virions contained additional proteins that migrate between VPs 9 and 10, VPs 20 and 21, and VPs 22 and 23 (Fig. 7A, asterisks).
To more specifically characterize differences in virion composition, we performed Western blot analysis on infected cell lysates, extracellular virions, and purified extracellular virions (Fig. 7B and C). Although no marked differences were observed in the amounts of VP5, ICP27, UL42, gE, or ICP22 in 22/n199 virions compared to wild-type or 22/n199R virions (Fig. 7B), the level of a γ2 protein, US11, was reduced in mutant virions. The amount of a second γ2 L protein, gC, was reduced below the level of detection in 22/n199 virions produced in Vero cells (Fig. 7C) but was present in wild-type amounts when 22/n199 virions were produced in ICP22-expressing V22 cells. Additionally, the amounts of ICP0 and ICP4 were enhanced in 22/n199 virions produced in Vero cells (Fig. 7C). (Two Western blots of ICP0 are shown in Fig. 7C because the predominant band in the upper blot was so intense that the signal was quenched. The lower blot contains 1/10 the amount of total protein.) By contrast, wild-type-like amounts of ICP0 and ICP4 were present in 22/n199 virions produced in V22 cells (Fig. 7C). Taken together, these findings indicate that ICP22 is required to incorporate wild-type amounts of at least two IE and two γ2 L proteins in HSV-1 virions either directly or indirectly and demonstrate that ICP22 is a major determinant of virion composition.
DISCUSSION
ICP22− virions lose infectivity rapidly following inoculation of mice via the ocular route. ICP22 was not detected in HSV-1 virions; however, it is a major determinant of virion composition. We postulate that this loss of infectivity is due to the aberrant composition of 22/n199 virions. Although ICP22 may affect virion composition directly, it is also possible that the ability of ICP22 to regulate gene expression is the basis for altered virion composition.
The behavior of ICP22− viruses in vivo.
Previous studies have shown that ICP22− viruses are highly attenuated in vivo (30, 37). We show that the initial defect in 22/n199 replication in mice infected via the ocular route occurs prior to the onset of nascent virus production (Fig. 3). Specifically, at 3 hpi, the titer of infectious virus in tear film decreased 230-fold relative to input in wild-type virus-infected animals, whereas tear film titers plummeted 1.3 × 105-fold in 22/n199-infected animals (Fig. 3). Although this rapid decline in viral infectivity limits what can be learned about downstream properties of 22/n199 in vivo, our studies suggest that ICP22 is not absolutely essential for the establishment or reactivation of latency. Thus, at 1 dpi, wild-type and ICP22− viruses recovered from eclipse phase as seen by an approximate 10-fold increase in tear film titers for both viruses compared to tear film titers at 3 hpi (Fig. 1A). Although the level of infectious virus at the site of inoculation was substantially reduced in 22/n199-infected mice compared to wild-type virus-infected mice on days 1 to 9 pi, the biphasic kinetics of virus replication for the two viruses were similar (Fig. 1A). These observations support the notion that 22/n199 is able to replicate in vivo, albeit at a reduced level compared to wild-type virus. By extension, it is likely that the reduced levels of infectious virus at the site of inoculation during the first 9 days postinoculation are largely responsible for the reduced establishment of latency as measured by reduced genome loads and reduced LAT expression (Fig. 2A; Table 1). Previous studies have shown that virus replication in vivo is a requirement for the establishment of latency (24), further suggesting that 22/n199 replicates to some degree in mice. Because genome loads are a determinant of reactivation efficiency (35), the reduced reactivation efficiency of 22/n199 is also likely due to early events in viral replication in mice. Collectively, these results support the notion that ICP22 is not absolutely essential for the establishment or reactivation of latency. Rather, deficiencies that occur prior to the onset of nascent virus production underlie the reduced pathogenicity of 22/n199 in the mouse ocular model.
22/n199 virions lose infectivity rapidly at the site of inoculation.
22/n199 virions rapidly become noninfectious during the first 3 h following infection in vivo. Two potential mechanisms for the decrease in infectivity of 22/n199 virions are (i) 22/n199 virions adsorb to cells of the eye and enter eclipse more rapidly than wild-type virions and (ii) 22/n199 virions are inactivated or cleared more rapidly than wild-type virions in tear film. In vitro, the length of eclipse phase of 22/n199 mirrors that of the wild-type virus in all cell types tested (28), suggesting that 22/n199 does not enter cells at a higher rate than wild-type virus. It seems more likely that 22/n199 virions are specifically inactivated or cleared in the eye, potentially because of their aberrant composition. One possible scenario underlying enhanced clearance involves gC. Glycoprotein C is an important virulence factor of HSV-1 because it is required to inactivate complement proteins (25, 26). Because gC levels are reduced in 22/n199 virions, we asked whether gC may be a factor responsible for the reduction in infectivity of these virions. Although there is a striking difference in gC levels of 22/n199 virions produced in ICP22-expressing V22 cells compared to Vero cells (Fig. 7), the tear film titers of both virus preparations decreased within the first 2 hpi in mouse eyes. In a preliminary experiment, the tear film titer of 22/n199 produced in Vero cells was reduced 50-fold compared to wild-type virus at 2 hpi of mice (Fig. 3), whereas the tear film titer of 22/n199 produced in V22 cells was reduced 10-fold relative to wild-type virus (data not shown). Since 22/n199 virions produced in V22 cells lost 80% of their infectivity at 2 hpi but contained normal gC levels compared to wild type, it is possible that gC levels are not the major factor in determining the infectivity of 22/n199 virions. Alternative possibilities include the following: (i) 22/n199 virions produced in V22 cells may lose infectivity in mouse eyes because the ICP22 expressed in V22 cells is not regulated in the same manner as during HSV-1 infection; (ii) the ICP22 gene in V22 cells contains two uncharacterized mutations as well as an amino-terminal tag, each of which may alter the function of ICP22; (iii) it is not known whether V22 cells express the US1.5 protein and whether the absence of the US1.5 protein affects virion stability; (iv) since ICP22 mRNA is present in wild-type virions (36) and ICP22 mRNA produced in V22 cells is expressed from the cytomegalovirus IE promoter, it is not known whether ICP22 mRNA produced in V22 cells is actually incorporated into virions and whether its absence affects virion stability; and (v) it is also a possibility that ICP22 expression in mouse eyes is required for cells to secrete a protein that stabilizes virions in tear film. We are currently examining these possibilities and the possibility that proteases or components of the innate immune response lead to the inactivation or clearance of 22/n199 virions.
ICP22 is undetectable in wild-type virions.
In cell-free preparations of wild-type virions, the level of ICP22 was below the limit of detection (Fig. 4), suggesting that ICP22 is not a component of the HSV-1 virion. This result was unexpected, since ICP22 homologues of other α-neurotropic herpesviruses (orf63 of varicella-zoster virus and IR4 of equine herpesvirus) have been reported to be associated with the virion tegument (18, 21). In each of the aforementioned studies, however, the starting material for virion preparation was lysates of whole infected cells. In preliminary studies, we too examined virion preparations obtained from lysed infected cells, and ICP22 was readily detected (data not shown). As shown in Fig. 4B and 7B, however, ICP22 was not detected in extracellular virion preparations derived from cell-free culture medium. Thus, we reasoned that the ICP22 initially detected in virion preparations from infected cell lysates was a contaminant of cell-associated virus. Thus, it remains a possibility that both orf63 and IR4 were contaminants of the purification procedure. It would therefore be of interest to determine whether orf63 and IR4 are present in extracellular virion preparations derived from cell-free culture medium. Alternatively, the homology between ICP22 and orf63 or IR4 is focused at the carboxy terminus of the protein, and it has been suggested that the US1.5 protein is the HSV-1 homologue of orf63 and IR4. Because our ICP22 antibody binds only to the amino terminus of the protein, we have not been able to determine whether the US1.5 protein is a virion component. We are currently attempting to address this question using an antibody directed against the carboxyl terminus of ICP22.
What is the molecular basis for the physical and biological differences in virions synthesized in the presence and absence of ICP22?
Purified 22/n199 virions synthesized in Vero cells sediment at a reduced density compared to wild-type virions produced in Vero cells or 22/n199 virions synthesized in ICP22-expressing V22 cells (Fig. 5 and 6; Table 2). Although 22/n199 virions resemble wild-type virions morphologically when examined by electron microscopy, the viral proteins in virions produced in the absence of ICP22 differ from those produced in the presence of ICP22 (Fig. 7). Of the proteins measured, the amounts of two IE proteins, ICP0 and ICP4, were enhanced and the amounts of two γ2 L proteins, US11 and gC, were reduced in 22/n199 virions synthesized in Vero cells. The pattern of incorporation of γ2 L proteins reflects the pattern of γ2 L gene expression in 22/n199-infected cells (28, 30, 37). Thus, ICP22− viruses induce the synthesis of reduced levels of γ2 L proteins in both permissive and restrictive cells compared to wild-type virus-infected cells (28, 31, 34, 37). Consequently, the availability of these γ2 L proteins for incorporation into virions is limited. By contrast, the observation that levels of ICP0 and ICP4 were increased in 22/n199 virions synthesized in Vero cells was unexpected, as reduced amounts of both proteins are evident in 22/n199-infected cell lysates compared to lysates of wild-type virus-infected cells at late times postinfection (Fig. 7). Because reduced levels of ICP0 and ICP4 are apparent at late but not early times in ICP22− mutant-infected cells relative to wild-type-infected cells (37), it is possible that the reduced levels seen at late times are due to enhanced incorporation of ICP0 and ICP4 into 22/n199 virions and exit of these virions from infected cells.
Properties of 22/n199 virions.
The composition of 22/n199 virions is altered relative to wild-type virions, yet these virions are infectious both in vitro and in vivo. Other HSV-1 mutants, e.g., ICP0− and UL13− viruses, produce aberrant virions but retain a degree of infectivity (7, 44). Not surprisingly, the virions produced by these mutant viruses possess altered biological properties relative to the wild-type virus. For example, the particle-to-PFU ratio of an ICP0− virus on Vero cell monolayers is 15 times greater than that of wild-type virus (12). Because equal amounts of wild-type and 22/n199 virion protein preparations contain similar amounts of VP5 (Fig. 7), we calculated PFU-to-VP5 ratios as a rough measure of particle-to-PFU ratio (26). We found that the VP5-to-PFU ratio was 24 times greater for 22/n199 than for wild-type virus. Thus, like ICP0− viruses, more virions produced in 22/n199-infected cells are defective.
The sedimentation of 22/n199 virions was concentrated in a single band in each preparation; however, 22/n199 virions sedimented variably in different preparations. By contrast, in the same experiments wild-type and 22/n199R virions sedimented reproducibly in 44% sucrose. Because gene regulation is altered in 22/n199-infected Vero cells relative to wild-type virus (30, 37), the availability of viral proteins for virion formation is also altered. To a degree, all kinetic classes of viral genes are affected during infection with ICP22− viruses. Although ICP22 is required to achieve wild-type levels of viral proteins in infected cells, it has been reported that ICP22 negatively regulates expression from viral promoters in transient-transfection assays (22, 33). Because ICP22 is able to both up- and down-regulate gene expression under different conditions, it is possible that its role may be to control the regulation of viral gene expression. Thus, the stochastic sedimentation of 22/n199 virions may be due to global alterations in viral gene expression. Taken together by whatever mechanism, ICP22 plays a significant role in controlling the composition of infectious HSV-1 virions.
Previous studies of ICP22− mutants in vivo have suggested that these viruses exhibit deficiencies in the establishment and reactivation of latency. Our studies suggest that ICP22 is not absolutely required for latency or reactivation in the mouse ocular model. Rather, ICP22 is required to retain infectivity in mouse eyes during the first 3 hours of infection, and the reduction in virus titers at the site of infection with ICP22− viruses reduces the efficiency of establishment of and, later, reactivation from latency. Because it is possible that the innate immune response is involved in the clearance of 22/n199 virions, we are currently asking whether ICP22 is required for reactivation when equal genome loads are present in TG. Because 22/n199 virions differ biochemically and physically from wild-type virions and because the loss in infectivity occurs prior to the onset of new virus production, virion-specific differences must underlie the loss of 22/n199 infectivity. Clearly, these differences in virion composition will add an unexpected level of complexity in the functional comparison of ICP22− mutant viruses with the wild-type virus.
Acknowledgments
We thank Jean-Jacques Diaz for the US11 antibody, Maria Ericsson at the Harvard Medical School Electron Microscopy Facility for technical service in both preparation and visualization of samples, Feng Yao for technical assistance with virion preparations, and other past and present members of the Schaffer lab for helpful comments and suggestions.
This work was supported by Public Health Service grant RO1 CA20260 from the National Cancer Institute to P.A.S. J.S.O. is the recipient of the National Research Service Award F32 CA99887 from the National Cancer Institute. S.A.R. was supported by Public Health Service grant RO1 AI50127 from the National Institute of Allergy and Infectious Diseases.
REFERENCES
- 1.Advani, S. J., R. Brandimarti, R. R. Weichselbaum, and B. Roizman. 2000. The disappearance of cyclins A and B and the increase in activity of the G2/M-phase cellular kinase cdc2 in herpes simplex virus 1-infected cells require expression of the α22/US1.5 and UL13 viral genes. J. Virol. 74:8-15. [PMC free article] [PubMed] [Google Scholar]
- 2.Balliet, J. W., and P. A. Schaffer. 2006. Point mutations in herpes simplex virus type 1 oriL, but not in oriS, reduce pathogenesis during acute infection of mice and impair reactivation from latency. J. Virol. 80:440-450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bruni, R., B. Fineschi, W. O. Ogle, and B. Roizman. 1999. A novel cellular protein, p60, interacting with both herpes simplex virus 1 regulatory proteins ICP22 and ICP0 is modified in a cell-type-specific manner and is recruited to the nucleus after infection. J. Virol. 73:3810-3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bruni, R., and B. Roizman. 1998. Herpes simplex virus 1 regulatory protein ICP22 interacts with a new cell cycle-regulated factor and accumulates in a cell cycle-dependent fashion in infected cells. J. Virol. 72:8525-8531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carter, K. L., and B. Roizman. 1996. The promoter and transcriptional unit of a novel herpes simplex virus 1 α gene are contained in, and encode a protein in frame with, the open reading frame of the α22 gene. J. Virol. 70:172-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Compton, T., and R. J. Courtney. 1984. Virus-specific glycoproteins associated with the nuclear fraction of herpes simplex virus type 1-infected cells. J. Virol. 49:594-597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coulter, L. J., H. W. Moss, J. Lang, and D. J. McGeoch. 1993. A mutant of herpes simplex virus type 1 in which the UL13 protein kinase gene is disrupted. J. Gen. Virol. 74:387-395. [DOI] [PubMed] [Google Scholar]
- 8.Deatly, A. M., J. G. Spivack, E. Lavi, and N. W. Fraser. 1987. RNA from an immediate early region of the type 1 herpes simplex virus genome is present in the trigeminal ganglia of latently infected mice. Proc. Natl. Acad. Sci. USA 84:3204-3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeLuca, N. A., A. M. McCarthy, and P. A. Schaffer. 1985. Isolation and characterization of deletion mutants of herpes simplex virus type 1 in the gene encoding immediate-early regulatory protein ICP4. J. Virol. 56:558-570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Diaz, J. J., D. Simonin, T. Masse, P. Deviller, K. Kindbeiter, L. Denoroy, and J. J. Madjar. 1993. The herpes simplex virus type 1 US11 gene product is a phosphorylated protein found to be non-specifically associated with both ribosomal subunits. J. Gen. Virol. 74:397-406. [DOI] [PubMed] [Google Scholar]
- 11.Diefenbach, R. J., M. Miranda-Saksena, E. Diefenbach, D. J. Holland, R. A. Boadle, P. J. Armati, and A. L. Cunningham. 2002. Herpes simplex virus tegument protein US11 interacts with conventional kinesin heavy chain. J. Virol. 76:3282-3291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Everett, R. D., C. Boutell, and A. Orr. 2004. Phenotype of a herpes simplex virus type 1 mutant that fails to express immediate-early regulatory protein ICP0. J. Virol. 78:1763-1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gallo, M. L., D. I. Dorsky, C. S. Crumpacker, and D. S. Parris. 1989. The essential 65-kilodalton DNA-binding protein of herpes simplex virus stimulates the virus-encoded DNA polymerase. J. Virol. 63:5023-5029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goodrich, L. D., F. J. Rixon, and D. S. Parris. 1989. Kinetics of expression of the gene encoding the 65-kilodalton DNA-binding protein of herpes simplex virus type 1. J. Virol. 63:137-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Halford, W. P., B. M. Gebhardt, and D. J. Carr. 1996. Mechanisms of herpes simplex virus type 1 reactivation. J. Virol. 70:5051-5060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Halford, W. P., and P. A. Schaffer. 2000. Optimized viral dose and transient immunosuppression enable herpes simplex virus ICP0-null mutants to establish wild-type levels of latency in vivo. J. Virol. 74:5957-5967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Halford, W. P., and P. A. Schaffer. 2001. ICP0 is required for efficient reactivation of herpes simplex virus type 1 from neuronal latency. J. Virol. 75:3240-3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holden, V. R., G. B. Caughman, Y. Zhao, R. N. Harty, and D. J. O'Callaghan. 1994. Identification and characterization of the ICP22 protein of equine herpesvirus 1. J. Virol. 68:4329-4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holland, L. E., K. P. Anderson, C. Shipman, Jr., and E. K. Wagner. 1980. Viral DNA synthesis is required for the efficient expression of specific herpes simplex virus type 1 mRNA species. Virology 101:10-24. [DOI] [PubMed] [Google Scholar]
- 20.Katz, J. P., E. T. Bodin, and D. M. Coen. 1990. Quantitative polymerase chain reaction analysis of herpes simplex virus DNA in ganglia of mice infected with replication-incompetent mutants. J. Virol. 64:4288-4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kinchington, P. R., D. Bookey, and S. E. Turse. 1995. The transcriptional regulatory proteins encoded by varicella-zoster virus open reading frames (ORFs) 4 and 63, but not ORF 61, are associated with purified virus particles. J. Virol. 69:4274-4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kwun, H. J., S. W. Yim, D. H. Lee, and K. L. Jang. 1999. Activation of the thymidine kinase promoter by herpes simplex virus type 1 immediate early proteins. Mol. Cells 9:277-280. [PubMed] [Google Scholar]
- 23.Leib, D. A., C. L. Bogard, M. Kosz-Vnenchak, K. A. Hicks, D. M. Coen, D. M. Knipe, and P. A. Schaffer. 1989. A deletion mutant of the latency-associated transcript of herpes simplex virus type 1 reactivates from the latent state with reduced frequency. J. Virol. 63:2893-2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leib, D. A., D. M. Coen, C. L. Bogard, K. A. Hicks, D. R. Yager, D. M. Knipe, K. L. Tyler, and P. A. Schaffer. 1989. Immediate-early regulatory gene mutants define different stages in the establishment and reactivation of herpes simplex virus latency. J. Virol. 63:759-768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lubinski, J., L. Wang, D. Mastellos, A. Sahu, J. D. Lambris, and H. M. Friedman. 1999. In vivo role of complement-interacting domains of herpes simplex virus type 1 glycoprotein gC. J. Exp. Med. 190:1637-1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lubinski, J. M., L. Wang, A. M. Soulika, R. Burger, R. A. Wetsel, H. Colten, G. H. Cohen, R. J. Eisenberg, J. D. Lambris, and H. M. Friedman. 1998. Herpes simplex virus type 1 glycoprotein gC mediates immune evasion in vivo. J. Virol. 72:8257-8263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ogle, W. O., and B. Roizman. 1999. Functional anatomy of herpes simplex virus 1 overlapping genes encoding infected-cell protein 22 and US1.5 protein. J. Virol. 73:4305-4315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Orlando, J. S., T. L. Astor, S. A. Rundle, and P. A. Schaffer. 2006. The products of the herpes simplex virus type 1 immediate-early US1/US1.5 genes downregulate levels of S-phase-specific cyclins and facilitate virus replication in S-phase Vero cells. J. Virol. 80:4005-4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Para, M. F., R. B. Baucke, and P. G. Spear. 1982. Glycoprotein gE of herpes simplex virus type 1: effects of anti-gE on virion infectivity and on virus-induced Fc-binding receptors. J. Virol. 41:129-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Poffenberger, K. L., A. D. Idowu, E. B. Fraser-Smith, P. E. Raichlen, and R. C. Herman. 1994. A herpes simplex virus type 1 ICP22 deletion mutant is altered for virulence and latency in vivo. Arch. Virol. 139:111-119. [DOI] [PubMed] [Google Scholar]
- 31.Poffenberger, K. L., P. E. Raichlen, and R. C. Herman. 1993. In vitro characterization of a herpes simplex virus type 1 ICP22 deletion mutant. Virus Genes 7:171-186. [DOI] [PubMed] [Google Scholar]
- 32.Post, L. E., and B. Roizman. 1981. A generalized technique for deletion of specific genes in large genomes: alpha gene 22 of herpes simplex virus 1 is not essential for growth. Cell 25:227-232. [DOI] [PubMed] [Google Scholar]
- 33.Prod'hon, C., I. Machuca, H. Berthomme, A. Epstein, and B. Jacquemont. 1996. Characterization of regulatory functions of the HSV-1 immediate-early protein ICP22. Virology 226:393-402. [DOI] [PubMed] [Google Scholar]
- 34.Rice, S. A., M. C. Long, V. Lam, P. A. Schaffer, and C. A. Spencer. 1995. Herpes simplex virus immediate-early protein ICP22 is required for viral modification of host RNA polymerase II and establishment of the normal viral transcription program. J. Virol. 69:5550-5559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sawtell, N. M., D. K. Poon, C. S. Tansky, and R. L. Thompson. 1998. The latent herpes simplex virus type 1 genome copy number in individual neurons is virus strain specific and correlates with reactivation. J. Virol. 72:5343-5350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sciortino, M. T., M. Suzuki, B. Taddeo, and B. Roizman. 2001. RNAs extracted from herpes simplex virus 1 virions: apparent selectivity of viral but not cellular RNAs packaged in virions. J. Virol. 75:8105-8116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sears, A. E., I. W. Halliburton, B. Meignier, S. Silver, and B. Roizman. 1985. Herpes simplex virus 1 mutant deleted in the α22 gene: growth and gene expression in permissive and restrictive cells and establishment of latency in mice. J. Virol. 55:338-346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith, K. O. 1964. Relationship between the envelope and the infectivity of herpes simplex virus. Proc. Soc. Exp. Biol. Med. 115:814-816. [DOI] [PubMed] [Google Scholar]
- 39.Spivack, J. G., and N. W. Fraser. 1987. Detection of herpes simplex virus type 1 transcripts during latent infection in mice. J. Virol. 61:3841-3847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spivack, J. G., and N. W. Fraser. 1988. Expression of herpes simplex virus type 1 latency-associated transcripts in the trigeminal ganglia of mice during acute infection and reactivation of latent infection. J. Virol. 62:1479-1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stroop, W. G., D. L. Rock, and N. W. Fraser. 1984. Localization of herpes simplex virus in the trigeminal and olfactory systems of the mouse central nervous system during acute and latent infections by in situ hybridization. Lab. Investig. 51:27-38. [PubMed] [Google Scholar]
- 42.Trus, B. L., W. W. Newcomb, F. P. Booy, J. C. Brown, and A. C. Steven. 1992. Distinct monoclonal antibodies separately label the hexons or the pentons of herpes simplex virus capsid. Proc. Natl. Acad. Sci. USA 89:11508-11512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yao, F., and R. J. Courtney. 1989. A major transcriptional regulatory protein (ICP4) of herpes simplex virus type 1 is associated with purified virions. J. Virol. 63:3338-3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yao, F., and R. J. Courtney. 1992. Association of ICP0 but not ICP27 with purified virions of herpes simplex virus type 1. J. Virol. 66:2709-2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yeh, L., and P. A. Schaffer. 1993. A novel class of transcripts expressed with late kinetics in the absence of ICP4 spans the junction between the long and short segments of the herpes simplex virus type 1 genome. J. Virol. 67:7373-7382. [DOI] [PMC free article] [PubMed] [Google Scholar]