

Abstract
Awareness of the metabolism of second-generation antipsychotics by the cytochrome P450 (CYP) system can inform the clinician about how to avoid and manage drug–drug interactions involving these enzymes. Clozapine is metabolized primarily by CYP1A2, with additional contributions by CYP2C19, CYP2D6 and CYP3A4. Risperidone is metabolized primarily by CYP2D6 and to a lesser extent by CYP3A4. Olanzapine is metabolized primarily by CYP1A2 and to a lesser extent by CYP2D6. Quetiapine and ziprasidone are metabolized by CYP3A4. At the usual clinical doses, these drugs appear not to significantly affect the metabolism of other medications. There is, however, a lack of in vivo metabolic data, especially for the 3 newest second-generation antipsychotics: olanzapine, quetiapine and ziprasidone.
Medical subject headings: antipsychotic agents, biotransformation, cytochrome P-450 enzyme system, drug interactions, metabolism, schizophrenia
Abstract
La connaissance du métabolisme des neuroleptiques de deuxième génération par le système cytochrome P450 (CYP) peut informer le clinicien sur la façon d'éviter et de prendre en charge les interactions entre médicaments qui mettent en cause ces enzymes. La clozapine est métabolisée principalement par l'enzyme CYP1A2 et les enzymes CYP2C19, CYP2D6 et CYP3A4 contribuent aussi au métabolisme. La rispéridone est métabolisée principalement par la CYP2D6 et, à un degré moindre, par la CYP3A4. L'olanzapine est métabolisée principalement par la CYP1A2 et, à un degré moindre, par la CYP2D6. La quétiapine et la ziprasidone sont métabolisées par la CYP3A4. Aux doses cliniques habituelles, ces médicaments ne semblent pas avoir d'effet significatif sur le métabolisme d'autres médicaments. On manque toutefois de données métaboliques in vivo, particulièrement dans le cas des trois neuroleptiques de deuxième génération les plus récents, soit l'olanzapine, la quétiapine et la ziprasidone.
Introduction
Schizophrenia is a chronic illness affecting approximately 1% of the population.1 Its pharmacologic management has changed over the past decade, in part owing to the acceptance of a new class of medications, generally termed atypical, novel or second-generation antipsychotics. These medications are reported to have a lower incidence of side effects than the older antipsychotics, and their use has arguably become first-line.2,3 In this review, we will use the term second-generation antipsychotics to describe these medications.
Coincidentally, there has been increased awareness among clinicians of the potential for pharmacokinetic drug–drug interactions and a greater understanding of the role that cytochrome P450 (CYP) enzymes may play in these interactions. Most individuals with schizophrenia take medications indefinitely and at some point take additional medications. Therefore, the risk of polypharmacy-related complications is very real in this population.
This article reviews the available data on the CYP-mediated metabolism of the commercially released second-generation antipsychotics clozapine, risperidone, olanzapine, quetiapine and ziprasidone. Non-CYP-mediated pathways, such as the phase II reaction glucuronidation, are involved in the metabolism of some second-generation antipsychotics4 but are not a focus of this review. We emphasize the potential for metabolic drug–drug interactions and, as much as possible, rely on data from in vivo studies of humans. Drug metabolism is summarized; comprehensive reviews are available elsewhere.5,6,7,8,9 We discuss the difficulties of interpreting the in vivo data, especially resolving conflicting data and generalizing the findings from controlled in vivo studies and case reports to clinical practice.
Drug metabolism
Pharmacologically active agents can interact at several levels, including drug metabolism. Drug metabolism can be defined as the chemical modification of the drug that occurs in a biologic environment. The primary role of drug metabolism is to facilitate the deactivation and excretion of the xenobiotic from the body. In addition, metabolism may result in the formation of active metabolites that have pharmacologic activities similar to those of the parent compound or altogether different biologic actions, including the ability to alter the metabolism of other chemical compounds.
The liver is the primary site of drug metabolism; other tissues, such as kidney, brain, skin, blood, lung and gastrointestinal mucosa, also contribute.10 Most medications are transformed in phase I or II reactions. Phase I reactions result in conversion of the parent drug, usually by oxidation, reduction or hydrolysis, to a more hydrophilic metabolite. Phase II reactions result in inactivation of the medication through chemically coupling to an endogenous substance, such as glucuronic acid, glycine, glutathione or glutamine, or conjugation with acetate, sulfate or methyl groups, thus facilitating renal excretion.
Oxidative reactions are among the most important of the phase I reactions. They are mediated by the family of enzymes that constitute the CYP system. At least 17 mammalian CYP gene families have been defined, and more than 30 human gene products have been identified.11,12 CYPs with 40% or greater DNA sequence homology are classified in the same family, and those with 55% or greater sequence homology are classified in the same subfamily. Thus, CYP3A4 is the designation of the CYP enzyme in family 3, subfamily A, gene product 4. Any drug may be a substrate for one or more CYPs.
Individual CYPs may be present in different forms or amounts in different people.13,14,15,16,17 Thus, people may be described as poor metabolizers (PMs) or extensive metabolizers (EMs) of certain drugs. It is generally well accepted that CYP2D6 and CYP2C19 exist as genetic polymorphisms.13,14,15,16,17 Ultrarapid metabolizers, with multiple copies of the CYP2D6 gene, have been identified.15,16 There is less support for polymorphisms for CYP1A2,14,15,16,17 CYP2A616,17 and CYP3A4.14,17
Inhibition and induction of CYPs are among the most common causes of drug interactions.18 The role of polypharmacy in the development of medication-related side effects is not limited to simple competition for a specific CYP. A drug (or its metabolites) may be a substrate for, and an inhibitor or inducer of, the same CYP. Further, a drug (or its metabolites) may alter the activity of a specific CYP without being a substrate. Thus, concomitantly prescribed medications may affect the serum levels of substrates of those CYPs.
Drug interaction at the CYP level has been reviewed recently.19 Direct drug (or metabolite) interaction with CYPs may cause inhibition by 1 of 3 broad mechanisms: reversible inhibition, quasi-irreversible inhibition and irreversible inhibition. In vivo, the last 2 inhibition subtypes are, for practical purposes, equivalent, causing a loss of a CYP's activity through formation of a covalently (irreversibly) modified enzyme–drug complex or a drug–enzyme complex that is bound so tightly that dissociation is unlikely (quasi-irreversible); in both cases, there is lowered Vmax and capacity. In either case, restoration of normal CYP activity requires synthesis of new enzyme. With reversible inhibition, normal function can be restored rapidly after the inhibiting agent is removed from the body; there is competition between the inhibitor and the second-generation antipsychotic for the CYP, with an increase in Km but an unaltered Vmax.
CYP activity also can be increased by medications, usually through increased production of the enzyme. Known to be inducible are CYP1A1, CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2E1 and CYP3A4.20 In contrast to inhibition, induction via increased CYP levels is slow. Allosteric interaction, binding of a drug (or metabolite) to a site on the enzyme removed from the active site, may result in a more active CYP and would occur more quickly.
It is not always possible to definitively identify whether a specific CYP is involved in the metabolism of a particular drug. A number of in vitro approaches have been used to investigate and predict drug–CYP interactions.13,21,22,23 Generally, the drug is incubated in the presence of human liver tissue (liver slices, microsomes, subcellular fractions of hepatic tissues or cultured hepatocytes) or microsomes prepared from various cell systems in which specific cDNAs for human CYPs are expressed,13,21,24 and the production of specific metabolites or a decrease in the amount of parent drug is measured. Refinements include the use of selective CYP inhibitors, probe substrates (drugs known to be metabolized by a specific CYP) or monoclonal antibodies to a particular CYP. Interpretation of the results, particularly as to their predictive clinical value, has been reviewed.13,25,26,27 Similarly, a number of approaches have been used in vitro to assess the ability of drugs to induce CYPs.28 However, although in vitro investigations can provide information on whether a drug (or metabolite) is a substrate, inhibitor or inducer of a particular CYP, the contribution of each CYP to the drug's metabolism in vivo is speculative.
There have been few controlled in vivo studies of drug metabolism. In some, human volunteers have received a known inhibitor or inducer of a specific CYP along with the drug under investigation. The rates of clearance of the drug in the presence and absence of the inhibitor or inducer are compared to determine whether the CYP is involved in the drug's metabolism.24
Another in vivo approach has been to exploit known genetic polymorphisms and their effects on drug metabolism, clearance, inhibition and induction. The subject is identified as being a PM or an EM, and the rate of metabolism of the specific drug is measured.24 This type of study is limited to the CYPs for which there are known polymorphisms that affect activity.
For the most part, data on the in vivo metabolism of a drug by a specific CYP have been derived from case reports on individuals who have received medications concurrently. The role of a particular CYP can only be inferred from these studies. It is possible, for example, that the observed clinical effects resulted from interactions other than CYP-mediated metabolism in the liver. Results in case reports should be taken as suggestive, not conclusive. Case reports are by definition noncontrolled. Factors such as diet, other illnesses and polymorphic status can profoundly affect a drug's metabolism.19,29 Furthermore, results are often from a single or very few individuals, and blood samples may have been drawn without due consideration of the time since the last dose.
Recently, a number of case series have been published in which individuals receiving a second-generation antipsychotic were treated with a second medication known to be metabolized by, or to inhibit or induce, a particular CYP. The effect on the serum level of the antipsychotic was measured. As discussed by Lin and Lu,19 results from these studies can be difficult to interpret and generalize from, given the interindividual variability of CYP activity in humans. This variability stems from a number of factors, including the distribution of CYPs in other tissues, the polymorphic status of the individual (with respect to absolute CYP amounts, CYP activity and sensitivity to inhibitors and inducers), age, health status and environmental factors such as diet and cigarette smoking. For example, EMs are more susceptible to enzyme inhibition than are PMs.19 Furthermore, a particular drug may be metabolized by more than one CYP; thus, inhibiting one CYP may not result in a significant change in serum levels.
Clozapine (Table 1)
Table 1

Clozapine was the first second-generation antipsychotic released for clinical use. Consequently, there are more data available on its metabolism than on other medications in this class. Byerly and DeVane,30 as well as Taylor,31 have reviewed the pharmacokinetics of clozapine.
CYP1A1
We did not identify any in vivo studies that implicated CYP1A1 in the metabolism of clozapine. However, Fang and colleagues,32 in an in vitro study, failed to find evidence supporting a role for this enzyme in the metabolism of clozapine.
CYP1A2
It seems well established that CYP1A2 is involved in the metabolism of clozapine. Pirmohamed and associates,33 using enzyme-selective inhibitors in vitro, showed that CYP1A2 catalyzes the demethylation of clozapine. Using a variety of in vitro techniques, including human liver microsomes, chemical inhibitors, CYP-specific antibodies and a yeast expression system to express CYP1A2, Eiermann and coworkers34 also were able to demonstrate involvement of this enzyme. Linnet and Olesen35 obtained similar results using commercial microsomes prepared by means of cDNA expression, although they suggested that CYP1A2 was likely to play only a minor role in the in vivo metabolism of clozapine at therapeutic concentrations. More recently, the same authors,36 using a lower, more therapeutic concentration of clozapine, found that CYP1A2 and CYP2C19 were most responsible for clozapine demethylation in vitro, CYP2C9 and CYP2D6 playing a more modest role. At higher concentrations of clozapine, CYP3A4 becomes more relevant. Fang and colleagues32 demonstrated that CYP1A2 could catalyze both demethylation of clozapine and the formation of clozapine-N-oxide, although its role in the formation of the latter is likely minor. Similarly, Tugnait et al37 reported that inhibition of CYP1A2 in vitro by antibodies or chemical inhibitors affected both demethylation and oxidation of clozapine significantly but that the effect on the former was greater in magnitude.
Fluvoxamine, a selective serotonin reuptake inhibitor (SSRI), inhibits both CYP1A238,39 and CYP2C19.39,40 Using human hepatocyte microsystem preparations, Chang and colleagues41 noted that fluvoxamine caused 48.5% inhibition of clozapine metabolism; 42.0% inhibition was observed with the chemical CYP1A2 furafyline. Similarly, Olesen and Linnet42 noted significant inhibition of clozapine demethylation in vitro after incubation with fluvoxamine, a reaction catalyzed in vitro by CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3A4.35,36 However, since fluvoxamine has a lower Ki for CYP1A2 and CYP2C19, the result is probably due primarily to inhibition of 1 of these 2 enzymes.
One method of determining CYP1A2 polymorphism is to measure the clearance rate of caffeine, a substrate of this enzyme. Bertilsson and associates43 observed that individuals who rapidly cleared caffeine had lower serum levels of clozapine after receiving a single dose of this medication. Bender and Eap44 observed that in 2 individuals who were not responding clinically to clozapine, there was a similar correlation between rapid clearance of caffeine and low serum levels of clozapine; a third individual who was not responding to clozapine did not have rapid caffeine clearance. After reviewing drug-monitoring data (dose and serum levels), Jerling and coworkers45 demonstrated, using nonparametric statistical methods, a similarity in the distribution of clozapine clearance rates and CYP1A2 phenotype in the population.
There is growing evidence that interactions between clozapine and caffeine may be clinically relevant. Vanier and Chouinard46 described an individual being treated with clozapine who showed signs of increased arousal and extrapyramidal symptoms after consuming caffeinated beverages; the signs and symptoms resolved when noncaffeinated drinks were substituted. Clozapine levels were not measured, but, because the reaction was acute, the authors concluded that it was due to potentiation of dopamine D2-receptor stimulation secondary to the dopamine D1-agonist and adenosine A2-antagonist activity of caffeine. Odom-White and de Leon47 observed an increase in serum levels of both clozapine and norclozapine in a patient who consumed an excessive daily amount of caffeine (more than 1200 mg). Carrillo et al48 reported a case series in which serum levels of clozapine, norclozapine and clozapine-N-oxide were measured in 7 patients initially, after 5 days without caffeine and 2 weeks after reintroduction of caffeine. They noted a statistically significant reduction in levels of clozapine (47%) and clozapine-N-oxide (31%) and a significant increase (185%) in the ratio of norclozapine to clozapine levels after caffeine was removed from the diet; the ratio correlated with CYP1A2 activity, as measured by the caffeine clearance rate. All serum levels returned to the original values after caffeine was reintroduced.
Cigarette smoking induces CYP1A2.49 Smokers have been observed to have lower serum levels of clozapine than nonsmokers, sometimes significantly lower50,51,52 and sometimes not.53 This interaction can have clinical significance. Two patients receiving clozapine had seizures after they stopped smoking.54,55 The seizures were attributed to elevated serum levels of clozapine, although the levels were not measured. After recovery, 1 of the patients required a 40% reduction in the dose of clozapine.55
Increased serum levels of clozapine (and norclozapine) after the start of fluvoxamine treatment have been reported from several cases,56,57,58,59,60,61,62,63,64 and some of the patients had side effects indicative of elevated clozapine levels.60,62,63,64 The clozapine levels returned to normal when fluvoxamine was discontinued.58,61,62,63,64 The effects of fluvoxamine on clozapine serum levels have also been studied prospectively. Wetzel and associates51 treated individuals who had stable serum clozapine levels with fluvoxamine and found significant increases in the serum levels of clozapine, N-desmethylclozapine and clozapine-N-oxide; they estimated that the elimination half-life of clozapine tripled. Chang and colleagues41 treated 9 men with schizophrenia with fluvoxamine for 2 weeks. Immediately before and on the last day of fluvoxamine treatment, they administered a single dose of clozapine and measured the serum levels of the antipsychotic and its metabolites over a 48-hour period. They observed a significant increase in clozapine level and patient sedation with the second dose. The serum levels of the metabolites decreased transiently after fluvoxamine treatment.
Although the interaction between clozapine and fluvoxamine has been viewed primarily in terms of negative outcomes resulting from excessive serum levels of clozapine, it could have a benefit. Lammers and coworkers65 treated patients concomitantly with fluvoxamine and a titrating dose of clozapine, obtaining therapeutic serum levels of clozapine with relatively low daily doses (i.e., 96.9 ± 37.2 mg); the patients had clinically significant reductions of symptoms while avoiding the sedation associated with the usual initial doses of clozapine. In a similar study, Lu et al66 treated patients with clozapine at a daily dose of 100 mg; after adding fluvoxamine to the regimen, they observed a significant increase in clozapine serum levels that coincided with clinical relief in two-thirds of the patients, without significant side effects.
Further data supporting a role for CYP1A2 in the metabolism of clozapine comes from publications on the effects of treatment with antibiotics. Joos, Frank and Kaschka67 described a patient in whom the serum clozapine level decreased and symptoms re-emerged upon treatment with rifampin (an inducer of both CYP1A2 and CYP3A68), isoniazid (a general inducer of CYPs69) and pyrazinamide. The symptoms resolved when the clozapine dose was increased. When the rifampin was replaced with ciprofloxacin (an inhibitor of CYP1A270), the serum clozapine level increased 60% despite lowering of the dose of clozapine. Similarly, Fuhr and colleagues71 noted an 80% increase in serum clozapine levels resulting from treatment with the CYP1A2 inhibitor fluoroquinolone.72 In a randomized double-blind crossover study, Raaska and Neuvonen73 found that individuals with schizophrenia who were clinically stable with clozapine treatment had a significant increase in plasma clozapine and N-desmethylclozapine levels when treated additionally with ciprofloxacin as compared with placebo; the increases correlated with the concentration of ciprofloxacin in the blood.
CYP2C subfamily
Compared with CYP1A2, relatively few data are available on the role of the CYP2C subfamily in the metabolism of clozapine. Eiermann and coworkers34 reported that CYP2C8 is not involved in the metabolism of clozapine in vitro. In contrast, the study of Fang and colleagues32 indicated that this enzyme could catalyze the N-demethylation of clozapine and the formation of clozapine-N-oxide in vitro, although to a lesser extent than CYP1A2 and CYP3A4. In vitro studies have shown that CYP2C9 can demethylate clozapine,32,35,36 although clinically, its role is likely minor. The same authors independently reported that CYP2C19 could metabolize clozapine in vitro, although they disagreed about this enzyme's in vivo relevance; Linnet and Olesen35,36 concluded that it might account for up to 35% of in vivo metabolism. As discussed earlier, fluvoxamine can cause clinically relevant increases in serum clozapine levels, although it is a more potent inhibitor of CYP1A238,39 than of CYP2C19.39,40 The in vivo role of CYP1A2 is supported by reports not involving fluvoxamine; confirmation of an in vivo role for CYP2C19 in the metabolism of clozapine requires additional study.
CYP2D6
The evidence concerning the role of CYP2D6 in the metabolism of clozapine is contradictory. In vitro studies using microsomal preparations of cDNA-expressed CYP2D632,35,36,37 have indicated that this enzyme can catalyze the metabolism of clozapine. However, there is some question about whether the degree of metabolism seen is clinically relevant.32,35,36,37 Eiermann and coworkers34 reported that this enzyme was not involved in the in vitro metabolism of clozapine. Using liver preparations from PMs and EMs, Pirmohamed and associates33 found no correlation between the CYP2D6 phenotype and the in vitro degradation of clozapine.
A number of SSRIs inhibit CYP2D6. Eggert, Crismon and Dovjon74 reported a case in which there was no change in serum levels of clozapine after the individual taking this medication was started on fluoxetine therapy. In contrast, Joos and associates75 observed a clinically significant increase in clozapine levels 19 days after the start of paroxetine treatment in a patient who was an EM. As Lin and Lu19 noted, such individuals are more susceptible to the effects of concomitantly prescribed inhibitors. In a larger, naturalistic study, Centorrino et al76 reported that individuals treated with clozapine alone had significantly lower serum levels than patients who were concomitantly treated with paroxetine, fluoxetine or sertraline. Previously, they had reported that only 4 of 8 patients had elevated levels of clozapine after being treated with fluoxetine.77 In a prospective study, Spina and coworkers78 observed significant increases in the serum levels of clozapine and its metabolites in 9 of 10 patients treated with fluoxetine for 8 weeks. By contrast, Wetzel and associates51 found no such increase in 13 of 14 patients concomitantly treated with paroxetine in their prospectively study.
Two case reports have been published79,80 describing patients who experienced a 2-fold increase in serum clozapine level after taking the CYP2D6 substrate risperidone.
In all but 1 of the studies concerning CYP2D6, the patients were not characterized by polymorphic status, and it is possible that some of the discrepancies reflect this fact. Two studies attempted to correlate polymorphic status of CYP2D6 with clozapine levels;81,82 neither was able to do so. The in vivo metabolism of clozapine by CYP2D6 may not generally be clinically significant. However, individuals who are CYP2D6 EMs, and therefore more susceptible to inhibition of this enzyme,19 and have a PM phenotype for another CYP involved in clozapine metabolism (such as CYP1A2) may be more sensitive to drug–drug interactions related to CYP2D6.
CYP2E1
From in vitro studies, CYP2E1 does not appear to be involved in the metabolism of clozapine.32,35,36,37
CYP3A4
Several in vitro studies have indicated that clozapine metabolism can be catalyzed by CYP3A4.32,35,36,37 From in vitro stimulation studies, Linnet and Olesen35 concluded that this enzyme may be responsible for up to 35% of clozapine metabolism in vivo. However, more recently, they reported that at therapeutic concentrations of clozapine the role of CYP3A4 may be minor.36 Tugnait et al37 reported that, whereas CYP1A2 was more important in N-demethylation of clozapine, CYP3A4 was more important in N-oxidation.
Few in vivo studies have investigated the role of CYP3A4 in the metabolism of clozapine. After taking erythromycin, an inhibitor of CYP3A4,83 2 patients had side effects consistent with clozapine toxicity and elevated serum levels;84,85 after the antibiotic was discontinued, the side effects resolved and the serum levels of clozapine fell. However, human volunteers showed no significant changes in the serum concentration or rate of renal clearance of clozapine or its metabolites after a single dose when also treated with erythromycin.86
Concomitant administration of nefazodone, an inhibitor of CYP3A4,87 has been shown to result in a dose-dependent increase in the serum clozapine level.88 However, in 6 patients with schizophrenia, Taylor et al89 observed only minor increases in the serum levels of clozapine (4%) or norclozapine (16%) after nefazodone was introduced. They did not report the serum levels of clozapine-N-oxide but did note that some subjects had more significant increases in the levels of clozapine and norclozapine; perhaps these subjects were CYP1A2 PMs and therefore more dependent on alternative pathways of clozapine metabolism. Raaska and Neuvonen90 had similar negative results using the CYP3A4 inhibitor itraconazole. They too did not report clozapine-N-oxide serum levels but noted that a patient who was also receiving fluoxetine experienced a moderate elevation in levels of both clozapine and desmethyl-N-clozapine. Thus, it seems likely that CYP3A4 is involved in the metabolism of clozapine, although clinically significant interactions may occur only in individuals with impaired alternative pathways of metabolism, such as via CYP1A2.
Inhibition or induction of CYPs by clozapine
Clozapine may affect the CYP-mediated metabolism of other medications. However, in vivo data are lacking. Ring and colleagues91 reported from in vitro studies that clozapine can inhibit CYP2C9, CYP2C19, CYP2D6 and CYP3A, the Ki values being 31 μM, 69 μM, 19 μM and 99 μM, respectively. Shin, Soukhova and Flockhart92 reported Ki values from studies with different metabolic substrates of CYP1A2 (> 300 μM), CYP2C9 (31 μM), CYP2C19 (> 300 μM), CYP2D6 (39 μM) and CYP3A (> 300 μM) and predicted from these results that there would be no inhibition by clozapine of these CYPs in vivo. However, Smith and Risken93 reported a doubling of the serum levels of nortriptyline (a CYP2D6 substrate) after a patient was started on clozapine therapy. Clozapine may accumulate in the liver and thus inhibit CYPs. Alternatively, perhaps clozapine's metabolites are more potent inhibitors than the parent drug and are responsible for any observed clinical interactions; the in vitro studies were performed only with clozapine.
The only reported investigations into the potential of clozapine to induce CYPs have been in vitro studies using nonhuman tissues. Incubation of rat tissues with clozapine resulted in increased levels of CYP1A, CYP2B and CYP3A but not CYP2D4.94 The clinical relevance of these findings is unclear. Nonetheless, the lack of reports indicating an effect on the metabolism of other prescribed drugs since clozapine was introduced for clinical use suggests that any inhibition or induction is insignificant.
Risperidone (Table 2)
Table 2
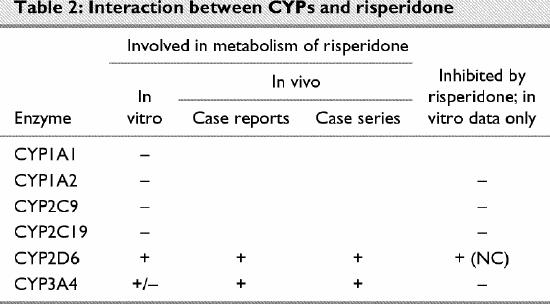
Compared with clozapine, few publications have reported potential drug–drug interactions involving risperidone. The paucity of data may reflect a relative lack of clinical experience with this medication, although given the time the drug has been available, this seems unlikely. Rather, the lack of publications on adverse events likely reflects either greater safety or a lack of recognition that symptoms are due to interactions with CYPs. Caccia95 recently reviewed the metabolism of risperidone.
CYP1A subfamily
In vitro studies using human liver microsomes96 and recombinant human97 CYP1A1 and CYP1A2 failed to find evidence of the involvement of either enzyme in the metabolism of risperidone.
CYP2C subfamily
Risperidone is not metabolized by CYP2C9 or CYP2C19 in vitro.96,97
CYP2D6
In vitro studies have shown that CYP2D6 catalyzes the metabolism of risperidone96,97 to its major metabolite, 9-hydroxyrisperidone.98
Several studies have attempted to correlate CYP2D6 polymorphic status with serum levels of risperidone and 9-hydroxyrisperidone. Using a single-dose model in human volunteers, Huang and associates99 observed a correlation between polymorphic phenotype and the formation of 9-hydroxyrisperidone. In a cross-sectional study, Olesen and coworkers100 noted that patients who were CYP2D6 PMs had an increased ratio of risperidone to 9-hydroxyrisperidone but no difference in total concentration of the active moiety (risperidone plus 9-hydroxyrisperidone).100 Further, they found no correlation between side effects and polymorphic status. Similarly, Scordo et al101 and Bork and colleagues102 reported a correlation between polymorphic status and serum risperidone but not 9-hydroxyrisperidone levels. Bork and colleagues observed that patients who were genotypically CYP2D6 PMs were more likely to stop treatment with risperidone, although they noted limitations to the study design. Spina and associates,103 in a prospective study of 10 patients stabilized on risperidone therapy who were concomitantly given the CYP2D6 inhibitor paroxetine, found a significant increase in the serum level of risperidone but not 9-hydroxyrisperidone; only 1 patient had side effects (parkinsonian symptoms) that might have been attributable to the increase.
Recently, there has been an increased interest and awareness of the potential differences in clinical efficacy of enantiomers (nonsuperimposable mirror images, akin to the right and left hand) of drugs.104,105 Risperidone does not have a chiral centre, but one is formed when it is metabolized to 9-hydroxyrisperidone, the result being stereoisomers. Yasui-Furukori and coworkers106 have demonstrated that CYP2D6 plays a predominant role in the formation of (+)-9-hydroxyrisperidone in vivo.
CYP3A4
Initial in vitro investigations did not indicate a role for CYP3A4 in the metabolism of risperidone.96 However, Fang, Bourin and Baker97 noted that ketoconazole (an inhibitor of CYP3A4) could inhibit the formation of 9-hydroxyrisperidone in vitro. Bork and colleagues102 noted in their study a number of cases in which inhibitors and inducers of CYP3A4 affected the serum levels of risperidone and had potential clinical significance. One patient receiving the CYP3A4 inducer carbamazepine had a doubling in the serum level of 9-hydroxyrisperidone and severe akathisia after the anticonvulsant was discontinued. Another patient had akathisia, parkinsonian symptoms and a significant elevation in the serum level of 9-hydroxyrisperidone after the CYP3A4 inducer mesotidazine was discontinued. A patient receiving the CYP3A4 inhibitor nefazodone experienced similar symptoms, although the serum levels of risperidone and 9-hydroxyrisperidone in the absence of nefazodone treatment were not reported. De Leon and Bork107 reported a case in which the 9-hydroxyrisperidone level doubled after carbamazepine was discontinued. Spina et al108 observed that individuals receiving carbamazepine but not sodium valproate had lower serum levels of risperidone and 9-hydroxyrisperidone than controls, although only the difference in the levels of the metabolite was statistically significant. In all the reports involving CYP3A4, only the level of 9-hydroxyrisperidone seemed to be affected by alterations in CYP3A4 activity; this may indicate that CYP3A4 is involved predominantly in the metabolism of 9-hydroxyrisperidone. Interestingly, Spina and colleagues109 noted a decrease in the plasma levels of both risperidone and 9-hydroxyrisperidone and an exacerbation in psychotic symptoms after carbamazepine was taken by a patient who was a CYP2D6 PM.
Formation of 9-hydroxyrisperidone has the potential to result in the production of enantiomeric isomers. Ketoconazole inhibits the formation of (-)-9-hydroxyrisperidone,106 suggesting that the metabolism of risperidone by CYP3A4 results in the synthesis of this specific enantiomer. The clinical significance of this finding remains to be determined.
Inhibition of CYPs by risperidone
Risperidone apparently does not inhibit CYP1A2, CYP2C9, CYP2C19 or CYP3A. Its minor inhibition of CYP2D6 is likely not clinically relevant at therapeutic doses of the antipsychotic.92,110
Olanzapine (Table 3)
Table 3

The role of CYPs in the metabolism of olanzapine has become clearer in recent years. Callaghan and associates111 reviewed the pharmacokinetics and pharmacodynamics of olanzapine. Markowitz and coworkers112 recently found that human volunteers treated with the glucuronidation inhibitor probenecid showed statistically significant changes in plasma pharmacokinetic parameters that were not reflected in the serum clearance of olanzapine.
CYP1A2
From in vitro studies, Ring et al113 suggested that CYP1A2 was responsible for the formation of N-desmethylolanzapine. Callaghan and associates111 noted that the formation of this metabolite correlated significantly with olanzapine clearance rates in vitro.
In a placebo-controlled study, Mäenpää and colleagues114 observed that concomitant treatment with the CYP1A2 inhibitor fluvoxamine resulted in increased serum levels and decreased rates of clearance of olanzapine, along with decreased serum levels of N-desmethylolanzapine.114 Similarly, in a single patient, Markowitz and DeVane115 noted that concomitant treatment with the CYP1A2 inhibitor ciprofloxacin resulted in a doubling of the serum olanzapine levels that reversed when the antibiotic was discontinued;115 the patient did not have side effects attributable to the increased levels of the antipsychotic. Further support for an in vivo role for CYP1A2 in the metabolism of olanzapine comes from the observation by Callaghan and associates111 that smokers had lower serum levels and higher clearance rates than nonsmokers.
Carbamazepine induces both CYP1A2 and CYP3A4.116 Two groups of investigators117,118 reported a decrease in serum olanzapine levels when patients were treated concomitantly with this anticonvulsant. Owing to a lack of evidence supporting a role for CYP3A4 in the metabolism of olanzapine, they concluded that the effects of carbamazepine on olanzapine metabolism were via induction of CYP1A2.
CYP2D6
CYP2D6 appears to be responsible for the formation of the metabolite 2-hydroxyolanzapine in vitro.113 Callaghan and associates111 found that concomitant treatment with the CYP2D6 inhibitor fluoxetine resulted in statistically significant but “clinically insignificant” changes in the serum levels and rates of clearance of olanzapine.
CYP3A4
There is no evidence, including in vitro data,113 supporting a role for CYP3A4 in the metabolism of olanzapine.
Inhibition of CYPs by olanzapine
Olanzapine does not inhibit CYP1A2, CYP2C9, CYP2D6 or CYP3A111,113 in vitro at Ki values that appear to be clinically relevant. Macias and associates119 reported that olanzapine did not affect the metabolism of the CYP1A2 substrate theophylline in vitro.
Quetiapine (Table 4)
Table 4
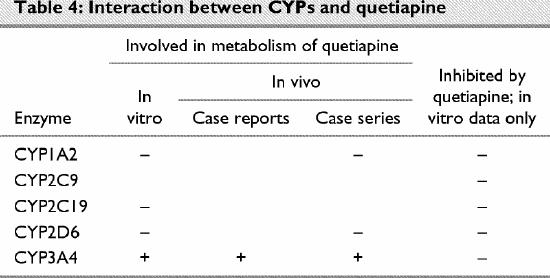
Quetiapine has been available commercially for a relatively short time. Consequently, clinical data on potential drug–drug interactions are lacking. Dev and Raniwalla120 have reviewed quetiapine's clinical safety.
CYP1A2
According to the manufacturer, quetiapine is not metabolized by CYP1A2.120 Goldstein121 reported that cigarette smoking does not affect the pharmacokinetics of this drug.
CYP2C19
According to the manufacturer, quetiapine is not metabolized by CYP2C19.122
CYP2D6
According to the manufacturer, the metabolism of quetiapine is altered insignificantly by concomitant administration of imipramine or fluoxetine.120 Thus, it is unlikely that CYP2D6 is involved in the in vivo metabolism of quetiapine.
CYP3A4
According to the manufacturer, quetiapine is primarily metabolized by CYP3A4.120 In a multiple-dose study, concomitant administration of the CYP3A4 inhibitor ketoconazole resulted in a significant increase in serum quetiapine levels and half-life.120 Concomitant administration of the CYP inducer phenytoin resulted in a significant decrease in serum quetiapine levels that corresponded with an increase in clearance rates;122 the investigators concluded that since quetiapine is primarily metabolized by CYP3A4, the observed pharmacokinetic changes were due to induction of CYP3A4. Savasi, Millson and Owen123 reported a case in which the serum levels of quetiapine increased 24-fold after concomitant phenytoin administration was stopped and another case in which the serum levels of quetiapine decreased with concomitant administration of the CYP3A4 inducer carbamazepine.
Inhibition or induction of CYPs by quetiapine
According to the manufacturer, quetiapine does not affect the metabolism of compounds known to be metabolized by CYP1A2, CYP2C9, CYP2D6 or CYP3A4.120
Ziprasidone (Table 5)
Table 5

Ziprasidone has only recently been released to the market. Using human liver microsomes, CYP-specific inhibitors and recombinant enzymes, Prakash and coworkers110 found no evidence supporting a role for CYP1A2, CYP2C9, CYP2C19 or CYP2D6 in the metabolism of this drug in vitro.
CYP3A4
In vitro data indicate that CYP3A4 is the primary CYP involved in the metabolism of ziprasidone.110 As expected, Prakash and coworkers110 found that concomitant administration of CYP3A4 inhibitors altered the drug's pharmacokinetics. In an open-label, randomized, crossover study, Micelli et al124 observed a statistically significant increase in serum levels of this antipsychotic when administered along with ketoconazole.
Inhibition of CYPs by ziprasidone
In vitro data indicate that, although ziprasidone can inhibit CYP2D6 and CYP3A4, the Ki values are such that in vivo inhibition is unlikely at clinical doses of the drug.110 This finding is supported by the observation that ziprasidone did not alter the metabolism of the CYP2D6 substrate dextromethorphan in vivo.125
Conclusions
Clozapine is metabolized by both CYP1A2 and CYP3A4. Concomitant use of this medication and inhibitors of these enzymes should be carried out with caution and adequate supervision. In addition, individuals who are EMs because of multiple copies of the CYP2D6 gene may be susceptible to side effects when treated concomitantly with clozapine and CYP2D6 inhibitors.
Risperidone is metabolized primarily by CYP2D6 and to a lesser degree by CYP3A4. Although polypharmacy can result in elevated serum levels of the antipsychotic, the risks of inhibiting its metabolism appear to be minimal. Among the second-generation antipsychotics, risperidone seems to have the greatest ability to inhibit the metabolism of other medications, but the risk is low at the usual clinical doses.
The 3 newest antipsychotics were approved for commercial release in a regulatory environment in which data about the drug's metabolism and the potential risks of polypharmacy, particularly in relation to the CYP enzymes, are required and reviewed before approval. This fact, combined with the relatively short period of availability, may account for the lack of data in the scientific literature on the metabolism of these medications. Olanzapine is metabolized primarily by CYP1A2, with lesser contributions from CYP2D6. Quetiapine and ziprasidone are metabolized by CYP3A4. In contrast to the 2 previously released second-generation antipsychotics, insufficient data are available to permit comment on the clinical safety of the 3 newest drugs in terms of metabolic drug–drug interactions.
Laboratory and in vivo metabolic studies of second-generation antipsychotics need to be expanded. In particular, both controlled studies and case reports are required to confirm the in vitro data and provide clinicians with indications and contraindications for concomitant therapy with these medications.
Acknowledgments
We are grateful to the Bebensee Foundation and the Canadian Institutes of Health Research (CIHR) for funding support. Dr. Prior is a CIHR Centennial Fellow.
Footnotes
Competing interests: None declared.
Correspondence to: Dr. Trevor Prior, Alberta Hospital Edmonton, Box 307, Edmonton AB T5J 2J7; fax 780 472-5411; trevor.prior@amhb.ab.ca
Submitted Apr. 17, 2002 Revised Sept. 18, 2002 Accepted Sept. 24, 2002
References
- 1.Kessler RC, McGonagle KA, Zhao S. Lifetime 12-month prevalence of DSM-III-R psychiatric disorders in the United States: results from the National Comorbidity Survey. Arch Gen Psychiatry 1994;51:8-19. [DOI] [PubMed]
- 2.American Psychiatric Association. Practice guidelines for the treatment of patients with schizophrenia. Am J Psychiatry 1997; 154 (Suppl):1-63. [DOI] [PubMed]
- 3.Canadian Psychiatric Association. Canadian clinical practice guidelines for the treatment of schizophrenia. Can J Psychiatry 1998; 43 (Suppl 2):25S-40S. [PubMed]
- 4.Shen WW. The metabolism of atypical antipsychotic drugs: an update. Ann Clin Psychiatry 1999;11:145-58. [DOI] [PubMed]
- 5.Parkinson A. Biotransformation of xenobiotics. In: Klaassen CD, editor. Casarett and Doull's toxicology: the basic science of poisons. 5th ed. New York: McGraw-Hill; 1996. p. 113-86.
- 6.Coutts RT, Fang J, Bourin M, Baker GB. Principles of drug metabolism, with an emphasis on psychiatric drugs. In: Boulton AA, Baker GB, Bateson AN, editors. Neuromethods. Vol. 33. Totowa (NJ): Humana Press; 1999. p. 255-86.
- 7.Ring BJ, Wrighton SA. Industrial viewpoint: application of in vitro drug metabolism in various phases of drug development. In: Levy RH, Thummel KE, Trager WF, Hansten PD, Eichelbaum M, editors. Metabolic drug interactions. Baltimore (MD): Lippincott, Williams and Wilkins; 2000. p. 29-39.
- 8.Janicak PG, Davis JM, Preskorn SH, Ayd FJ Jr. Pharmacokinetics. In: Principles and practice of psychopharmacotherapy. 3rd ed. Baltimore (MD): Lippincott, Williams & Wilkins; 2001. p. 51-72.
- 9.Greenblatt DJ, von Moltke LL, Harmatz JS, Shader RI. Pharmacokinetics, pharmacodynamics and drug disposition. In: Davis KL, Charney D, Coyle JL, Nemeroff C, editors. Neuropsychopharmacology: the fifth generation of progress. Baltimore (MD): Lippincott, Williams and Wilkins; 2002. p. 507-24.
- 10.Krishna DR, Klotz U. Extrahepatic metabolism of drugs in humans. Clin Pharmacokinet 1994;26:144-60. [DOI] [PubMed]
- 11.Nelson DR, Kaymans L, Kamataki T, Stegeman JJ, Feyereison R, Waxman DJ, et al. P450 superfamily: update on new sequences, gene mapping, accession numbers and nomenclature. Pharmacogenetics 1996;6:1-42. [DOI] [PubMed]
- 12.Nelson DR. Cytochrome P450 and individuality of species. Arch Biochem Biophys 1999;369:1-10. [DOI] [PubMed]
- 13.Glue P, Clement RP. Cytochrome P450 enzymes: in vitro assessment and clinical implications. In: Boulton AA, Baker GB, Bateson AN, editors. Neuromethods. Vol. 34. Totowa (NJ): Humana Press; 1998. p. 195-211.
- 14.Aitchison KJ, Jordan BD, Sharon T. The relevance of ethnic influences of pharmacogenetics to the treatment of psychosis. Drug Metabol Drug Interact 2000;16:15-38. [DOI] [PubMed]
- 15.Poolsup N, Li Wan Po A, Knight TL. Pharmacogenetics and psychopharmacotherapy. J Clin Pharm Ther 2000;25:197-220. [DOI] [PubMed]
- 16.Meisel C, Roots I, Cascorbi I, Brinkmann U, Brockmoller J. How to manage individualized drug therapy: application of pharmacogenetic knowledge of drug metabolism and transport. Clin Chem Lab Med 2000;38:869-76. [DOI] [PubMed]
- 17.Hiratsuka M, Mizugaki M. Genetic polymorphisms in drug-metabolising enzymes and drug targets. Mol Genet Metab 2001; 73: 298-305. [DOI] [PubMed]
- 18.Lin JH, Lu AYH. Inhibition and induction of cytochrome P450 and the clinical implications. Clin Pharmacokinet 1998;35:361-90. [DOI] [PubMed]
- 19.Lin JH, Lu AYH. Interindividual variability in inhibition and induction of cytochrome P450 enzymes. Annu Rev Pharmacol Toxicol 2001;41:535-67. [DOI] [PubMed]
- 20.Ronis MJJ, Ingelmann-Sundberg M. Induction of human drug-metabolizing enzymes: mechanisms and implications. In: Wolff TE, editor. Handbook of drug metabolism. New York: Marcel Dekker; 1999. p. 239-62.
- 21.Shen WW. The metabolism of psychotic drugs: a review of enzymatic biotransformation and inhibition. Biol Psychiatry 1997; 41 :814-26. [DOI] [PubMed]
- 22.Kern A, Bader A, Pichlmayr R, Sewing KF. Drug metabolism in hepatocyte sandwich cultures of rats and humans. Biochem Pharmacol 1997;54:761-72. [DOI] [PubMed]
- 23.Guillouzo A. Liver cell models in in vitro toxicology. Environ Health Perspect 1998;106:511-32. [DOI] [PMC free article] [PubMed]
- 24.Glue P, Banfield C. Psychiatry, psychopharmacology and P-450s. Hum Psychopharmacol 1996;11:97-114.
- 25.Iwatsubo T, Hirota N, Ooie T, Suzuki H, Shimada N, Ohiba K, et al. Predictions of in vivo drug metabolism in the human liver from in vitro metabolism data. Pharmacol Ther 1997;73:147-51. [DOI] [PubMed]
- 26.Bertz RJ, Grannerman GR. Use of in vitro and in vivo data to estimate the likelihood of metabolic pharmacokinetic interactions. Clin Pharmacokinet 1997;32:210-58. [DOI] [PubMed]
- 27.Wright SA, Ring BJ. Predicting drug interactions and pharmacokinetic variability with in vitro methods: the olanzapine experience. Drug Metab Rev 1999;31:15-28. [DOI] [PubMed]
- 28.LeCluyse EL. Human hepatocyte culture systems for the in vitro evaluation of cytochrome P450 expression and regulation. Eur J Pharm Sci 2001;13:343-68. [DOI] [PubMed]
- 29.Lane RM. Pharmacokinetic drug interaction potential of selective-serotonin reuptake inhibitors. Int Clin Psychopharmacol 1996; 11:31-61. [DOI] [PubMed]
- 30.Byerly MJ, DeVane CL. Pharmacokinetics of clozapine and risperidone: a review of the literature. J Clin Psychopharmacol 1996; 16:177-87. [DOI] [PubMed]
- 31.Taylor D. Pharmacokinetic interactions involving clozapine. Br J Psychiatry 1997;171:109-12. [DOI] [PubMed]
- 32.Fang J, Coutts RT, McKenna KF, Baker GB. Elucidation of individual cytochrome P450 enzymes involved in the metabolism of clozapine. Naunyn-Schmiedebergs Arch Pharmacol 1998; 358: 592-9. [DOI] [PubMed]
- 33.Pirmohamed M, Williams D, Madden S, Templeton E, Park BK. Metabolism and bioactivation of clozapine by human liver in vitro. J Pharmacol Exp Ther 1995;272:984-90. [PubMed]
- 34.Eiermann B, Engel G, Johansson I, Zanger UM, Bertilsson L. The involvement of CYP1A2 and CYP3A4 in the metabolism of clozapine. Br J Clin Pharmacol 1997;44:439-46. [DOI] [PMC free article] [PubMed]
- 35.Linnet K, Olesen OV. Metabolism of clozapine by cDNA-expressed human cytochrome P450 enzymes. Drug Metab Dispos 1997; 25:1379-82. [PubMed]
- 36.Olesen OV, Linnet K. Contributions of five human cytochrome P450 isoforms to the N-demethylation of clozapine in vitro at low and high concentrations. J Clin Pharmacol 2001;41:823-32. [DOI] [PubMed]
- 37.Tugnait M, Hawes EM, McKay G, Eichelbaum M, Midha KK. Characterization of the human hepatic cytochromes P450 involved in the in vitro oxidation of clozapine. Chem Biol Interact 1999; 118:171-89. [DOI] [PubMed]
- 38.Brøsen K. The pharmacogenetics of the selective serotonin reuptake inhibitors. Clin Investig 1993;71:1002-9. [DOI] [PubMed]
- 39.Jeppessen U, Gram LF, Vistisen K, Loft S, Poulsen HE, Brøsen K. Dose-dependent inhibition of CYP1A2, CYP2C19 and CYP2D6 by citalopram, fluoxetine, fluvoxamine and paroxetine. Eur J Clin Pharmacol 1996;51:73-8. [DOI] [PubMed]
- 40.Jeppesen U, Rasmussen BB, Brøsen K. Fluvoxamine inhibits the CYP2C19-catalyzed bioactivation of chloroguanide. Clin Pharmacol Ther 1997;62:279-86. [DOI] [PubMed]
- 41.Chang WH, Augustin B, Lane HY, ZumBrunnen T, Liu HC, Kazmi Y, et al. In-vitro and in-vivo evaluation of the drug–drug interaction between fluvoxamine and clozapine. Psychopharmacology 1999;145:91-8. [DOI] [PubMed]
- 42.Olesen OV, Linnet K. Fluvoxamine–clozapine drug interaction inhibition in vitro of five cytochrome P450 isoforms involved in clozapine metabolism. J Clin Psychopharmacol 2000;20:35-42. [DOI] [PubMed]
- 43.Bertilsson L, Carrillo JA, Dahl ML, Llerena A, Alm C, Bondesson U, et al. Clozapine disposition covaries with CYP1A2 activity determined by the caffeine test. Br J Clin Pharmacol 1994; 38: 471-3. [DOI] [PMC free article] [PubMed]
- 44.Bender S, Eap CB. Very high cytochrome P450 1A2 activity and non-response to clozapine. Arch Gen Psychiatry 1998;55: 1048-50. [DOI] [PubMed]
- 45.Jerling M, Merlè Y, Mentrè F, Mallet A. Population pharmacokinetics of clozapine evaluated with the nonparametric maximum likelihood method. Br J Clin Pharmacol 1997;44:447-53. [DOI] [PMC free article] [PubMed]
- 46.Vanier JL, Chouinard G. Interaction between caffeine and clozapine. J Clin Psychopharmacol 1994;14:284-5. [DOI] [PubMed]
- 47.Odom-White JA, de Leon J. Clozapine levels and coffee. J Clin Psychiatry 1996;57:175-6. [PubMed]
- 48.Carrillo JA, Herraiz AG, Ramos SI, BenÍtez MD. Effects of caffeine withdrawal from the diet on the metabolism of clozapine in schizophrenic patients. J Clin Psychopharmacol 1998;18:311-6. [DOI] [PubMed]
- 49.Desai HD, Seabolt J, Jann MW. Smoking in patients receiving psychotropic medications: a pharmacokinetic perspective. CNS Drugs 2000;15:469-94. [DOI] [PubMed]
- 50.Haring C, Meise U, Humpel C, Saria A, Fleischacker WW, Hinterhuber H. Dose-related plasma levels of clozapine: influence of smoking behaviour, sex and age. Psychopharmacology (Berl) 1989;99(Suppl):S38-40. [DOI] [PubMed]
- 51.Wetzel H, Anghelescu I, Szegedi A, Wiesner J, Weigmann H, Hartter S, et al. Pharmacokinetic interactions of clozapine with selective serotonin reuptake inhibitors: differential effects of fluvoxamine and paroxetine in a prospective study. J Clin Psychopharmacol 1998;18:2-9. [DOI] [PubMed]
- 52.Seppälä NH, Leinonen EVJ, Lehtonen ML, Kivistö KT. Clozapine serum concentrations are lower in smoking than in non-smoking schizophrenic patients. Pharmacol Toxicol 1999;85,244-6. [DOI] [PubMed]
- 53.Hasegawa M, Gutierrez-Esteinou R, Way L, Meltzer HY. Relationship between clinical efficacy and clozapine concentrations in plasma in schizophrenia: effect of smoking. J Clin Psychopharmacol 1993;13:383-90. [PubMed]
- 54.McCarthy R. Seizure following smoking cessation in a clozapine responder. Pharmacopsychiatry 1994;27:210-1. [DOI] [PubMed]
- 55.Skogh E, Bengtsson F, Nordin C. Could discontinuing smoking be hazardous for patients administered clozapine medication? A case report. Ther Drug Monit 1999;21:580-2. [DOI] [PubMed]
- 56.Weigmann H, Muller H, Dahmer W, Wetzel H, Hiemke C. Interaction of fluvoxamine with the metabolism of clozapine. Pharmacopsychiatry 1993;26:209.
- 57.Hiemke C, Weigmann H, Härtter S, Dahmen N, Wetzel H, Muller H. Elevated level of clozapine in serum after addition of fluvoxamine. J Clin Psychopharmacol 1994;14:279-81. [PubMed]
- 58.Jerling M, Lindström L, Bondesson U, Bertilsson L. Fluvoxamine inhibition and carbamazepine induction of the metabolism of clozapine: evidence from a therapeutic drug monitoring service. Ther Drug Monit 1994;16:368-74. [DOI] [PubMed]
- 59.Szegedi A, Weisner J, Hiemke C. Improved efficacy and fewer side effects under clozapine therapy after addition of fluvoxamine. J Clin Psychopharmacol 1995;15:141-3. [DOI] [PubMed]
- 60.Dequardo JR, Roberts M. Elevated clozapine levels after fluvoxamine initiation. Am J Psychiatry 1996;153:840-1. [DOI] [PubMed]
- 61.DuMartier G, Lochu A, Colon de Melo CP, Ghribi O, Roche-Rabreau D, DeGrassat K, et al. Elevated clozapine plasma concentrations after fluvoxamine initiation. Am J Psychiatry 1996; 153: 738-9. [DOI] [PubMed]
- 62.Koponen HJ, Leinonen E, Lepola U. Fluvoxamine increases the serum clozapine levels significantly. Eur Neuropsychopharmacol 1996;6:69-71. [DOI] [PubMed]
- 63.Armstrong SC, Stephans JR. Blood clozapine levels elevated by fluvoxamine: potential for side effects and lower clozapine dosage. J Clin Psychiatry 1997;58:499. [DOI] [PubMed]
- 64.Heeringa M, Beurskens R, Schouten W, Verduijn MM. Elevated plasma levels of clozapine after concomitant use of fluvoxamine. Pharm World Sci 1999;21:243-4. [DOI] [PubMed]
- 65.Lammers CH, Deuschle M, Weigmann H, Härtter S, Hiemke C, Heese C, et al. Coadministration of clozapine and fluvoxamine in psychotic patients — clinical experience. Pharmacopsychiatry 1999;32:76-7. [DOI] [PubMed]
- 66.Lu ML, Lane HY, Chen KP, Jann MW, Su MH, Chang WH. Fluvoxamine reduces clozapine dosage needed in refractory schizophrenic patients. J Clin Psychiatry 2000;61:594-9. [DOI] [PubMed]
- 67.Joos AAB, Frank UG, Kaschka WP. Pharmacokinetic interaction of clozapine and rifampicin in a forensic patient with an atypical mycobacterial infection. J Clin Psychopharmacol 1998;18:83-5. [DOI] [PubMed]
- 68.Wietholtz H, Zysset T, Marschall HU, Generet K, Matern S. The influence of rifampin treatment on caffeine clearance in healthy man. J Hepatol 1995;22:78-81. [DOI] [PubMed]
- 69.Baciewicz AM, Self TH. Isoniazid interactions. South Med J 1985; 78:714-8. [DOI] [PubMed]
- 70.Batty KY, Davis TM, Ilett KF, Dusci LJ, Langton SR. The effect of ciprofloxacin on theophylline pharmacokinetics in healthy subjects. Br J Clin Pharmacol 1995;39:305-11. [DOI] [PMC free article] [PubMed]
- 71.Fuhr U, Anders EM, Mahr G, Sorgel F, Staib AH. Inhibitory potency of quinolone antibacterial agents against cytochrome P450 1A2 activity in vivo and in vitro. Antimicrob Agents Chemother 1992;36:942-8. [DOI] [PMC free article] [PubMed]
- 72.Markowitz JS, Gill HS, Devane CL, Mintzer JE. Fluoroquinolone inhibition of clozapine metabolism. Am J Psychiatry 1997; 154:881. [DOI] [PubMed]
- 73.Raaska K, Neuvonen PJ. Ciprofloxacin increases serum clozapine and N-desmethylclozapine: a study in patients with schizophrenia. Eur J Clin Pharmacol 2000;56:585-9. [DOI] [PubMed]
- 74.Eggert AC, Crismon ML, Dovjon PG. Lack of effect of fluoxetine on plasma clozapine concentration. J Clin Psychiatry 1994; 55: 454-5. [PubMed]
- 75.Joos AAB, König F, Frank UG, Kaschka WP, Mörike KE, Ewald R. Dose-dependent pharmacokinetic interaction of clozapine and paroxetine in an extensive metabolizer. Pharmacopsychiatry 1997;30:266-70. [DOI] [PubMed]
- 76.Centorrino F, Baldessarini RJ, Frankenburg FR, Kando J, Volpicelli SA, Flood JG. Serum levels of clozapine and norclozapine in patients treated with selective serotonin reuptake inhibitors. Am J Psychiatry 1996;153:820-2. [DOI] [PubMed]
- 77.Centorrino F, Baldessarini RJ, Kando J, Frankenburg F, Volpicelli SA, Puopolo PR, et al. Serum concentration of clozapine and its major metabolite: effect with fluoxetine and valproate. Am J Psychiatry 1994;151:123-5. [DOI] [PubMed]
- 78.Spina E, Avenoso A, Facciolà G, Fabrazzo M, Mateleone P, Maj M, et al. Effect of fluoxetine on the plasma concentrations of clozapine and its major metabolites in patients with schizophrenia. Int Clin Psychopharmacol 1998;13:141-5. [DOI] [PubMed]
- 79.Tyson SC, Devane CL, Risch SC. Pharmacokinetic interaction between risperidone and clozapine. Am J Psychiatry 1995;152:1401-2. [DOI] [PubMed]
- 80.Koreen AR, Lieberman JA, Kronig M, Cooper TB. Cross-tapering clozapine and risperidone. Am J Psychiatry 1995;152:1690. [DOI] [PubMed]
- 81.Dahl ML, Llerena A, Bondesson U, Lindström L, Bertilsson L. Disposition of clozapine in man: lack of association with debrisoquine and S-mephenytoin hydroxylation polymorphisms. Br J Pharmacol 1994;37:71-4. [DOI] [PMC free article] [PubMed]
- 82.Arranz MJ, Dawson E, Shaikh S, Sham P, Sharma T, Aitchison K, et al. Cytochrome P450 2D6 genotype does not determine response to clozapine. Br J Clin Pharmacol 1995;39:417-20. [DOI] [PMC free article] [PubMed]
- 83.Murray M. P450 enzymes: inhibition mechanisms, genetic regulation and effects of liver disease. Clin Pharmacokinet 1992;23: 132-46. [DOI] [PubMed]
- 84.Funderburg L, Vertrees J, True J, Miller A. Seizure following addition of erythromycin to clozapine therapy. Am J Psychiatry 1994; 151:1840-1. [DOI] [PubMed]
- 85.Cohen LG, Chesley S, Eugenio L, Flood JG, Fisch J, Goff DC. Erythromycin-induced clozapine toxic reaction. Arch Intern Med 1996;156:675-7. [PubMed]
- 86.Hägg S, Spigset O, Mjörndal T, Granberg K, Persbo-Lundqvist G, Dahlqvist R. Absence of interaction between erythromycin and a single dose of clozapine. Eur J Clin Pharmacol 1999;55:221-6. [DOI] [PubMed]
- 87.von Moltke LL, Greenblatt DJ, Granda BW, Grassi JM, Schmider J, Harmatz JS, et al. Nefazodone, meta-chlorophenylpiperazine, and their metabolites in vitro: cytochromes mediating transformation, and P450-3A4 inhibitory actions. Psychopharmacologia 1999; 145:113-22. [DOI] [PubMed]
- 88.Khan AY, Preskorn SH. Increase in plasma levels of clozapine and norclozapine after administration of nefazodone. J Clin Psychiatry 2001;62:375-6. [DOI] [PubMed]
- 89.Taylor D, Bodani M, Hubbeling A, Murray R. The effect of nefazodone on clozapine plasma concentrations. Int Clin Psychopharmacol 1999;14:185-7. [PubMed]
- 90.Raaska K, Neuvonen PJ. Serum concentrations of clozapine and N-desmethylclozapine are unaffected by the potent CYP3A4 inhibitor itraconazole. Eur J Clin Pharmacol 1998;54:167-70. [DOI] [PubMed]
- 91.Ring BJ, Binkley SN, Vandenbranden M, Wrighton SA. In vitro interaction of the antipsychotic agent olanzapine with human cytochromes P450 CYP2C9, CYP2C19, CYP2D6 and CYP3A. Br J Clin Pharmacol 1996;41:181-6. [DOI] [PubMed]
- 92.Shin JG, Soukhova N, Flockhart DA. Effect of antipsychotic drugs on human liver cytochrome P-450 (CYP) isoforms in vitro: preferential inhibition of CYP2D6. Drug Metab Dispos 1999; 27:1078-84. [PubMed]
- 93.Smith T, Risken J. Effect of clozapine on plasma nortriptyline concentration. Pharmacopsychiatry 1994;27:41-2. [DOI] [PubMed]
- 94.Hedlund E, Wyas A, Kainu T, Backlund M, Köhler C, Pelto-Huikko M, et al. Cytochrome P4502D4 in the brain: specific neuronal regulation by clozapine and toluene. Mol Pharmacol 1996; 50:342-50. [PubMed]
- 95.Caccia S. Biotransformation of post-clozapine antipsychotics: pharmacological implications. Clin Pharmacokinet 2000;38:393-414. [DOI] [PubMed]
- 96.Lavrijsen K, Meuldermans W, Heykants J. In vitro inhibition study in human liver microsomes to investigate the interaction potential of various drugs with the metabolism of risperidone. Janssen Preclinical Research Report R64 776/FK1257. Bruges, Belgium: Janssen; 1993.
- 97.Fang J, Bourin M, Baker GB. Metabolism of risperidone to 9-hydroxyrisperidone by human cytochrome P450 2D6 and 3A4. Naunyn-Schmiedebergs Arch Pharmacol 1999;359:147-51. [DOI] [PubMed]
- 98.Mannens G, Huang ML, Meuldermanns W, Hendricks J, Woestenborghs R, Heykants J. Absorption, metabolism and excretion of risperidone in humans. Drug Metab Dispos 1993; 21: 1134-41. [PubMed]
- 99.Huang ML, van Peer A, Woestenborghs R, DeCoster R, Heykants J, Jansen AAJ, et al. Pharmacokinetics of the novel antipsychotic agent risperidone and the prolactin response in healthy subjects. Clin Pharmacol Ther 1993;54:251-68. [DOI] [PubMed]
- 100.Olesen OV, Licht RW, Thomsen E, Bruun T, Viftrup JE, Linnet K. Serum concentrations and side effects in psychiatric patients during risperidone therapy. Ther Drug Monit 1998;20:380-4. [DOI] [PubMed]
- 101.Scordo MG, Spina E, Facciolà G, Avenoso A, Johansson I, Dahl ML. Cytochrome P450 2D6 genotype and steady state plasma levels of risperidone and 9-hydroxyrisperidone. Psychopharmacology 1999;147:300-5. [DOI] [PubMed]
- 102.Bork JA, Rogers T, Wedlund PJ, de Leon J. A pilot study on risperidone metabolism: the role of cytochromes P450 2D6 and 3A. J Clin Psychiatry 1999;60:469-76. [PubMed]
- 103.Spina E, Avenoso A, Facciolà G, Scordo MG, Ancione M, Madia A. Plasma concentrations of risperidone and 9-hydroxyrisperidone during combined treatment with paroxetine. Ther Drug Monit 2001;23:223-7. [DOI] [PubMed]
- 104.Leonard BE. An introduction to enantiomers in psychopharmacology. Hum Psychopharmacol 2001;16:S79-S84. [DOI] [PubMed]
- 105.Baker GB, Prior TI. Stereochemistry and drug efficacy and development: relevance of chirality to antidepressant and antipsychotic drugs. Ann Med 2002;34:537-43. [DOI] [PubMed]
- 106.Yasui-Furukori N, Hidestrand M, Spina E, Facciolà G, Scordo MG, Tybring G. Different enantioselective 9-hydroxylation of risperidone by the two human CYP2D6 and CYP3A4 enzymes. Drug Metab Dispos 2001;29:1263-8. [PubMed]
- 107.de Leon J, Bork J. Risperidone and cytochrome P450 3A. J Clin Psychiatry 1997;58:10. [DOI] [PubMed]
- 108.Spina E, Avenoso A, Facciolà G, Salemi M, Scordo MG, Giacobello T, et al. Plasma concentrations of risperidone and 9-hydroxyrisperidone: effect of comedication with carbamazepine or valproate. Ther Drug Monit 2000;22:481-5. [DOI] [PubMed]
- 109.Spina E, Scordo MG, Avenoso A, Perucca E. Adverse drug interaction between risperidone and carbamazepine in a patient with chronic schizophrenia and deficient CYP2D6 activity. J Clin Psychopharmacol 2001;21:108-9. [DOI] [PubMed]
- 110.Prakash C, Kamel A, Cui D, Whalen RD, Miceli JJ, Tweedie D. Identification of the major human liver cytochrome P450 isoform(s) responsible for the formation of the primary metabolites of ziprasidone and prediction of possible drug interactions. Br J Clin Pharmacol 2000;49:35S-42S. [DOI] [PMC free article] [PubMed]
- 111.Callaghan JT, Bergstrom RF, Ptak LR, Beasley CM. Olanzapine: pharmacokinetic and pharmacodynamic profile. Clin Pharmacokinet 1999;37:177-93. [DOI] [PubMed]
- 112.Markowitz JS, Devane CL, Liston HL, Boulton DW, Risch SC. The effects of probenecid on the disposition of risperidone and olanzapine in healthy volunteers. Clin Pharmacol Ther 2002; 71: 30-8. [DOI] [PubMed]
- 113.Ring BJ, Catlow J, Lindsay TJ, Gillespie T, Roskos LK, Cerimele BJ, et al. Identification of the human cytochromes P450 responsible for the in vitro formation of the major oxidative metabolites of the antipsychotic agent olanzapine. J Pharmacol Exp Ther 1996;276:658-66. [PubMed]
- 114.Mäenpää J, Wrighton S, Bergstrom R, Cerimele B, Tatum D, Hatcher B, et al. Pharmacokinetic and pharmacodynamic interactions between fluvoxamine and olanzapine. Clin Pharmacol Ther 1997;61:225.
- 115.Markowitz JS, DeVane CL. Suspected ciprofloxacin inhibition of olanzapine resulting in increased plasma concentration. J Clin Psychopharmacol 1999;19:289-91. [DOI] [PubMed]
- 116.Parker AC, Pritchard P, Preston T, Choonara I. Induction of CYP1A2 activity by carbamazepine in children using the caffeine breath test. Br J Clin Pharmacol 1998;45:176-8. [DOI] [PMC free article] [PubMed]
- 117.Lucas RA, Gilfillan DJ, Bergstrom RF. A pharmacokinetic interaction between carbamazepine and olanzapine: observations on possible mechanism. Eur J Clin Pharmacol 1998;54:639-43. [DOI] [PubMed]
- 118.Olesen OV, Linnet K. Olanzapine serum concentrations in psychiatric patients given standard doses: the influence of comedication. Ther Drug Metab 1999;21:87-90. [DOI] [PubMed]
- 119.Macias WL, Bergstrom RF, Cerimele BJ, Kassahm K, Tatum DE, Callaghan JT. Lack of effect of olanzapine on the pharmacokinetics of a single aminophylline dose in healthy men. Pharmacotherapy 1998;18:1237-48. [PubMed]
- 120.Dev V, Raniwalla J. Quetiapine: a review of its safety in the management of schizophrenia. Drug Safety 2000;23:296-307. [DOI] [PubMed]
- 121.Goldstein JM. Quetiapine fumarate (Seroquel®): a new atypical antipsychotic. Drugs Today 1999;35:193-210. [DOI] [PubMed]
- 122.Wong YWJ, Yeh C, Thyrum PT. The effects of concomitant phenytoin administration on the steady-state pharmacokinetics of quetiapine. J Clin Psychopharmacol 2001;21:89-93. [DOI] [PubMed]
- 123.Savasi I, Millson RC, Owen JA. Quetiapine blood level variability. Can J Psychiatry 2002;47:94. [PubMed]
- 124.Miceli JJ, Smith M, Robarge L, Morse T, Laurent A. The effects of ketoconazole on ziprasidone pharmacokinetics — a placebo-controlled crossover study in healthy volunteers. Br J Clin Pharmacol 2000;49:71S-76S. [DOI] [PMC free article] [PubMed]
- 125.Wilner KD, Demattos SB, Anziano RJ, Apseloff G, Gerber N. Ziprasidone and the activity of cytochrome P450 2D6 in healthy extensive metabolizers. Br J Clin Pharmacol 2000; 49: 43S-47S. [DOI] [PMC free article] [PubMed]