

Abstract
Hepatitis C virus (HCV) nonstructural protein 5A (NS5A) is a component of viral replicase and is well known to modulate the functions of several host proteins. Here, we show that NS5A specifically interacts with FKBP8, a member of the FK506-binding protein family, but not with other homologous immunophilins. Three sets of tetratricopeptide repeats in FKBP8 are responsible for interactions with NS5A. The siRNA-mediated knockdown of FKBP8 in a human hepatoma cell line harboring an HCV RNA replicon suppressed HCV RNA replication, and this reduction was reversed by the expression of an siRNA-resistant FKBP8 mutant. Furthermore, immunoprecipitation analyses revealed that FKBP8 forms a complex with Hsp90 and NS5A. Treatment of HCV replicon cells with geldanamycin, an inhibitor of Hsp90, suppressed RNA replication in a dose-dependent manner. These results suggest that the complex consisting of NS5A, FKBP8, and Hsp90 plays an important role in HCV RNA replication.
Keywords: FK506-binding protein, geldanamycin, hepatitis C virus, Hsp90, RNA replication
Introduction
Hepatitis C virus (HCV) persistently infects approximately 170 million people worldwide, and it is responsible for most cases of severe chronic liver diseases, including cirrhosis and hepatocellular carcinoma (Wasley and Alter, 2000). Although treatment with interferon (IFN) alpha and ribavirin is available for about half of the population of HCV patients (Manns et al, 2001), therapeutic and preventative vaccines are still necessary for more effective treatment; however, such vaccines have not yet been developed. HCV belongs to the Flaviviridae family and possesses a positive-sense single-stranded RNA with a nucleotide length of 9.6 kb. The HCV genome encodes a single large precursor polyprotein composed of about 3000 amino acids, and the polyprotein is processed by cellular and viral proteases into at least 10 structural and nonstructural (NS) proteins (Moriishi and Matsuura, 2003).
The development of efficient therapies for hepatitis C has been hampered by the lack of a reliable cell-culture system, as well as by the absence of a non-primate animal model. The HCV replicon consists of an antibiotic selection marker and a genotype 1b HCV RNA, which replicates autonomously in the intracellular compartments in a human hepatoma cell line, Huh7 (Lohmann et al, 1999). This replicon system has functioned as an important tool in the investigation of HCV replication and it has served as a cell-based assay system for the evaluation of antiviral compounds. Recently, cell culture systems for in vitro replication and infectious viral production were established based on the full-length HCV genome of genotype 2a, which was isolated from an HCV-infected patient who developed fulminant hepatitis (Lindenbach et al, 2005; Wakita et al, 2005; Zhong et al, 2005). However, no robust in vitro culture systems for the 1a and 1b genotypes, which are the most prevalent HCV genotypes in the world, have been established to date.
Several viruses require viral and host molecular chaperones for entry, replication, and assembly, as well as for other steps in viral production (Maggioni and Braakman, 2005; Mayer, 2005). Cyclosporine A has been found to effectively inhibit viral replication in hepatitis C patients and in HCV replicon cells (Inoue et al, 2003; Watashi et al, 2003). Recently, it was shown that cyclophilin (Cyp) B specifically binds to NS5B and promotes association with the genomic RNA; furthermore, cyclosporine A was shown to disrupt interactions between NS5B and CypB (Watashi et al, 2005). CypB belongs to the immunophilin family, which shares peptidyl propyl cis/trans isomerase (PPIase) activity and an affinity for the immunosuppressive drug (Fischer and Aumuller, 2003). Furthermore, blockades of CypA, CypB, and CypC, as well as the induction of cellular stress responses, have been suggested to be involved in cyclosporine A-induced reduction of HCV RNA replication (Nakagawa et al, 2005). However, the involvement of other immunophilins in HCV RNA replication is not yet well understood.
HCV nonstructural protein 5A (NS5A) is a membrane-anchored phosphoprotein that possesses multiple functions in viral replication, IFN resistance, and pathogenesis (Macdonald and Harris, 2004). NS5A contains a zinc metal-binding motif within the N-terminal domain, and this zinc-binding ability is known to be essential for HCV replication (Tellinghuisen et al, 2004, 2005). Adaptive mutations frequently mapped in the coding region of NS5A have been shown to increase RNA replication (Yi and Lemon, 2004; Appel et al, 2005) and they are known to affect the hyperphosphorylation of NS5A by an unknown host kinase (Koch and Bartenschlager, 1999; Neddermann et al, 1999; Pietschmann et al, 2001). RNA replication in HCV replicon cells has been shown to be inhibited by treatment with lovastatin, a drug that decreases the production of mevalonate by inhibiting 3-hydroxy-3-methylglutaryl CoA reductase; this inhibition of RNA replication was reversed by the addition of geranylgeraniol, which suggests that HCV RNA replication requires geranylgeranylated proteins (Ye et al, 2003; Kapadia and Chisari, 2005). A NS5A-pull-down assay identified a geranylgeranylated protein, FBL2, as a NS5A-binding protein (Wang et al, 2005). Although several host proteins could potentially interact with NS5A, little is known about NS5A function.
To gain a better understanding of the functional role of NS5A in HCV replication, we screened human libraries by employing a yeast two-hybrid system and using NS5A as bait. We thereby successfully identified FKBP8 as an NS5A-binding protein. FKBP8 is classified as a member of the FK506-binding protein family, but it lacks several amino-acid residues thought to be important for PPIase activity and FK506 binding (Lam et al, 1995). We demonstrated here that FKBP8 forms a complex with Hsp90 and NS5A, and that this complex is critical for HCV replication, as based on the finding that treatment of the HCV replicon cells with geldanamycin, an inhibitor of Hsp90, suppressed RNA replication. These results therefore suggest that protein complex formation with NS5A, FKBP8, and Hsp90 plays a crucial role in HCV RNA replication.
Results
Identification of human FKBP8 as an HCV NS5A-binding partner
To identify host proteins that specifically interact with NS5A, we screened human brain and liver libraries using a yeast two-hybrid system that employs NS5A as bait. One positive clone was isolated from among 2 million colonies of the human fetal brain library, and the nucleotide sequence of this clone was determined. Several positive clones were isolated from the human liver library, but most of these clones included exon fragments of other than FKBP and/or noncoding regions. A BLAST search revealed that the positive clone encodes a full-length coding region of FKBP38, human FK506-binding protein 38 kDa. Although FKBP38 has been isolated from human and mouse mRNA (Lam et al, 1995), an additional sequence at the N-terminus of FKBP38 was revealed based on an analysis of the transcriptional start site in the genomic sequences of FKBP38 (Nielsen et al, 2004). The isoforms of FKBP38 were designated as FKBP8, which includes splicing variants of 44 and 46 kDa in mice, and 45 kDa in humans corresponds to the 44 kDa of the mouse FKBP8 (Nielsen et al, 2004). Human FKBP8 is identical to FKBP38 except for the extra 58 amino-acid residues at the N-terminus, and the FK506-binding domain in the N-terminal half, followed by three sets of tetratricopeptide repeats (TPRs), a calmodulin binding site, and a transmembrane domain (Figure 1A). Because the levels of expression of FKBP8 and FKBP38 have not been well characterized in human cell lines, we generated a mouse monoclonal antibody against human FKBP8, and we designated it as clone KDM19. This antibody recognizes a 50-kDa of endogenous FKBP8 in 293T cells, as well as exogenous HA-tagged FKBP8 (HA-FKBP8), which has slightly greater molecular weight (Figure 1B). Although the KDM19 antibody detected an exogenous HA-tagged FKBP38 (HA-FKBP38) in 293T cells, no protein band corresponding to endogenous FKBP38 was detected. Similar results were obtained in human liver tissue and in the hepatoma cell lines Huh7, HepG2, and FLC-4 (data not shown). These findings suggest that FKBP8, but not FKBP38, is a major product in human cells. In order to examine whether or not FKBP8 binds to NS5A protein in mammalian cells, Flag-tagged NS5A (Flag-NS5A) was expressed together with HA-FKBP8 in 293T cells. Cells transfected with the expression plasmids were harvested at 48-h post-transfection, lysed, and subjected to immunoprecipitation. Flag-NS5A was co-precipitated with HA-FKBP8 by anti-HA antibody (Figure 1C). Flag-NS5A was also immunoprecipitated together with HA-FKBP38, suggesting that the extra N-terminal sequence of FKBP8 is not critical for NS5A binding (data not shown). To further confirm the specific interaction of HCV NS5A with endogenous FKBP8, this interaction was examined in Huh7(9–13) cells harboring subgenomic HCV RNA replicon. Endogenous FKBP8 was co-precipitated with HCV NS5A by anti-FKBP8 antibody (Figure 1D). To determine the direct interaction between FKBP8 and NS5A, His6-tagged FKBP8 (His-FKBP8) and thioredoxin-fused domain 1 of NS5A (Trx-NS5A) prepared in Escherichia coli were examined by pull-down analysis. Trx-NS5A was co-precipitated with His-FKBP8 by anti-FKBP8 antibody (Supplementary Figure 1), suggesting that FKBP8 can directly bind to NS5A domain I.
Figure 1.
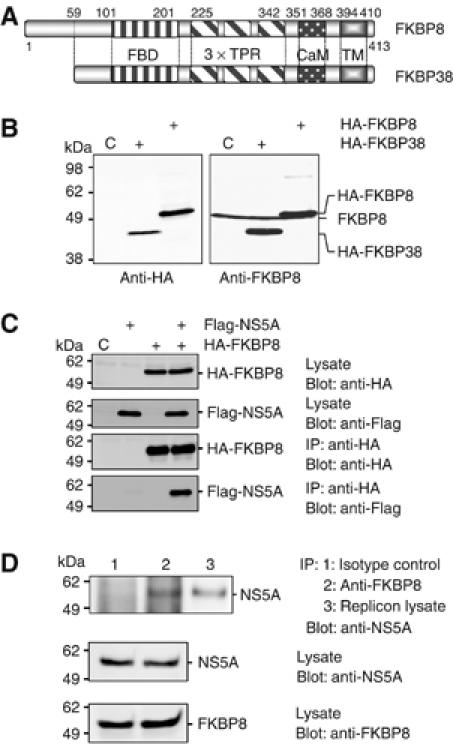
Expression of FKBP8 and FKBP38 in mammalian cells. (A) Schematic representation of FKBP8 and FKBP38. The FK506-binding domain (FBD), tetratricopeptide repeat (TPR), putative calmodulin binding motif (CaM), and transmembrane domain (TM) are shown. (B) N-terminally HA-tagged FKBP8 and FKBP38 were expressed in 293T cells and visualized by immunoblotting using mouse monoclonal antibody to FKBP8 or the HA tag. (C) HA-FKBP8 was expressed together with Flag-NS5A of genotype 1b (J1) in 293T cells and was immunoprecipitated with anti-HA antibody. Immunoprecipitated proteins were subjected to immunoblot with anti-Flag or HA antibody. (D) Endogenous FKBP8 in HCV replicon (9–13) cells was immunoprecipitated with isotype control (lane 1) or anti-FKBP8 antibody, KDM-11 (lane 2). Endogenous FKBP8 was co-immunoprecipitated with HCV NS5A. The data shown in each panel are representative of three independent experiments.
In order to investigate the interaction of FKBP8 with the NS5A of other HCV genotypes, HA-tagged NS5A (HA-NS5A) proteins of genotype 1a (H77C), 1b (Con1 and J1), or 2a (JFH1) were expressed together with Flag-tagged FKBP8 (Flag-FKBP8) in 293T cells (Figure 2A). Flag-FKBP8 was co-immunoprecipitated with the HA-NS5As of all of the genotypes examined here by anti-HA antibody, although it should be noted that the interaction between Flag-FKBP8 and the HA-NS5A of genotype 2a was weaker than that of the other genotypes tested. Furthermore, the HA-NS5As were co-precipitated with Flag-FKBP8 by anti-Flag antibody (Figure 2A, bottom panel). The TPR domain of FKBP8 is known to be responsible for protein–protein interactions. Among the immunophilins, FKBP8 shares high homology with CypD and FKBP52, both of which contain three tandem repeats of TPR, as does FKBP8 (Boguski et al, 1990; Hirano et al, 1990). However, co-immunoprecipitation of Flag-NS5A with HA-FKBP52 and HA-CypD by anti-Flag or anti-HA antibody was not successful (Figure 2B). These results indicate that FKBP8 specifically interacts with NS5A.
Figure 2.
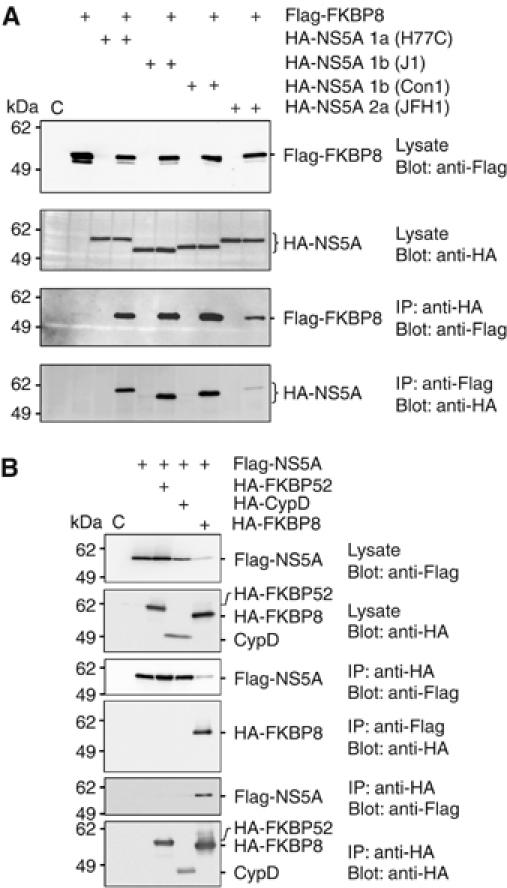
Specific interaction between FKBP8 and NS5A. (A) HA-NS5As were obtained from several genotypes of HCV and were expressed with Flag-FKBP8 in 293T cells. Proteins immunoprecipitated with anti-HA or Flag antibody were subjected to Western blotting. (B) Flag-NS5A was coexpressed with HA-FKBP8, -CypD, or -FKBP52 in 293T cells. Proteins immunoprecipitated with anti-HA or -Flag tag antibody were subjected to Western blotting. The data shown in each panel are representative of three independent experiments.
The TPR domain is required for the interaction between NS5A and FKBP8
FKBP8, CypD, and FKBP52 have high similarity and identity to each other within the TPR domain (Lam et al, 1995). Several FKBP8 mutants lacking the transmembrane region, the calmodulin-binding region, the TPR domains, and/or the FK506-binding domain were generated in order to identify the region responsible for the interaction with NS5A (Figure 3A). HA-tagged FKBP8 mutants were coexpressed with Flag-NS5A in 293T cells and were immunoprecipitated with anti-HA antibody. Flag-NS5A was co-immunoprecipitated with the FKBP8 mutants, except in the case of a dTPR mutant lacking the transmembrane, calmodulin binding, and TPR domains (Figure 3B). Although the level of expression of dFBD, an FKBP8 mutant with a deletion in the N-terminal region containing the FK506-binding domain, was lower than that of dTPR, co-immunoprecipitated NA5A was clearly detected. These findings suggested that the lack of an association of dTPR with NS5A was not due to the relatively low level of expression of dTPR, as compared to those of the other FKBP8 mutants. A specific interaction of NS5A with the TPR domain, but not with the transmembrane, calmodulin binding, or FK506-binding domains of FKBP8, was also observed using the yeast two-hybrid system (data not shown). These results indicated that FKBP8 interacts with HCV NS5A through the TPR domain.
Figure 3.
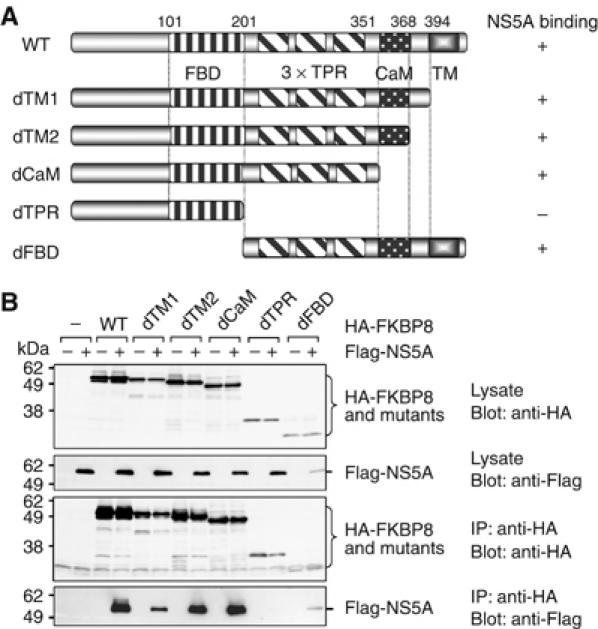
Determination of the NS5A-binding region in FKBP8. (A) Schematic representation of FKBP8 and deleted mutants. (B) Flag-NS5A was coexpressed with HA-FKBP8 and its mutants in 293T cells. Proteins immunoprecipitated with anti-HA antibody were subjected to Western blotting. The data shown in each panel are representative of three independent experiments.
FKBP8 forms a homomultimer and a heteromultimer with NS5A
FKBP8 is similar to FKBP52 and CypD with respect to their amino-acid sequences and functional domains. In order to examine the interactions among FKBP8, FKBP52, and CypD, Flag-FKBP8 was coexpressed with HA-FKBP52, HA-CypD, or HA-FKBP8 in 293T cells and it was immunoprecipitated with anti-Flag or anti-HA antibody. Flag-FKBP8 and HA-FKBP8 were co-immunoprecipitated with each antibody, but not with HA-FKBP52 or HA-CypD. It is known that Hsp90 forms a homodimer and also interacts with FKBP52 through TPR domain as FKBP8 (Chadli et al, 2000). If homodimer of FKBP8 is due to intermediating of Hsp90 as FKBP8-Hsp90-Hsp90-FKBP8 complex, FKBP52 would be co-precipitated with FKBP8 as FKBP8-Hsp90-Hsp90-FKBP52. However, we could not detect any association of FKBP8 and FKBP52 in the immunoprecipitation analysis (Figure 4A). These data suggest that FKBP8 can form a homomultimer without Hsp90 and associate with neither FKBP52 nor CypD through Hsp90. To examine the effects of the interaction with NS5A on the homomultimerization of FKBP8, HA-NS5A was coexpressed with Flag-FKBP8 and Glu-Glu-tagged FKBP8 (EE-FKBP8) in 293T cells, and was then immunoprecipitated with anti-Flag or anti-EE antibody. HA-NS5A was co-immunoprecipitated with Flag-FKBP8 and EE-FKBP8 by anti-Flag or anti-EE antibody (Figure 4B). Although multimerization of EE-FKBP8 and Flag-FKBP8 was increased about 2 times in the presence of HA-NS5A, but no further increase of the multimerization of FKBP8 was observed by the increase of HA-NS5A expression (Figure 4C). These results further support the notion that NS5A binds to FKBP8 via the TPR domain and slightly influence homomultimerization exerted by the FK506-binding domain.
Figure 4.
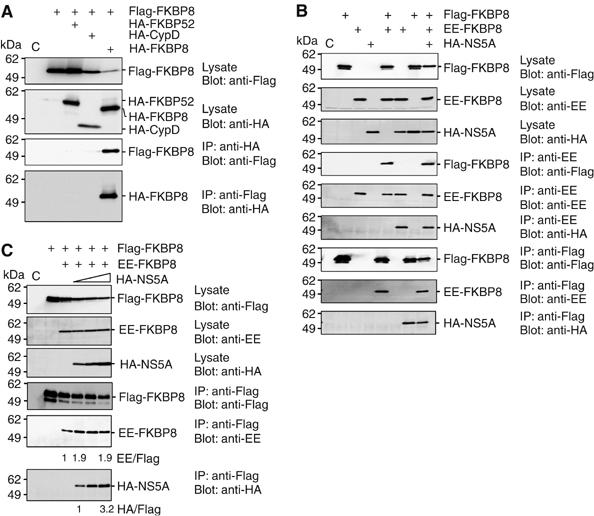
Homomultimerization of FKBP8. (A) Flag-FKBP8 was coexpressed with HA-FKBP52, -CypD, or -FKBP8 in 293T cells, and was immunoprecipitated with anti-HA or Flag antibody. Precipitates were analyzed by Western blotting. (B) Flag- or EE-tagged FKBP8 was coexpressed with HA-NS5A in 293T cells and was immunoprecipitated with anti-EE or Flag antibody. Precipitates were analyzed by Western blotting. (C) Flag- and EE-tagged FKBP8 were coexpressed with increasing amounts of HA-NS5A (0.1, 0.2, and 0.4 μg of expression plasmid/well) in 293T cells. Immunoprecipitates with anti-Flag antibody were analyzed by Western blotting. The data shown in each panel are representative of three independent experiments.
Knockdown of FKBP8 decreases RNA replication in HCV replicon cells
In order to determine the role of endogenous FKBP8 on HCV RNA replication, 80 nM of small interfering RNA (siRNA) targeted to FKBP8 or control siRNA was transfected into Huh7 (9–13) cells harboring subgenomic HCV replicon RNA. To verify the specificity of the knockdown of FKBP mRNA, we synthesized three siRNAs targeted to different regions of FKBP8 (Targets 1–3). The total RNA was extracted from the transfected cells, and HCV RNA and FKBP8 mRNA levels were determined by real-time polymerase chain reaction (PCR). HCV subgenomic RNA and FKBP8 mRNA levels in the cells transfected with each of the FKBP8 siRNAs were reduced by more than 60%, as compared to the levels in cells treated with the control siRNA at 72 h post-transfection (Figure 5A). The levels of expression of FKBP8 and the HCV proteins (i.e., NS4B, NS5A, and NS5B) decreased in HCV replicon cells transfected with 80 or 160 nM of the FKBP8 siRNA (Target 1), but this was not observed in the cells with the control siRNA (Figure 5B). To further confirm the specificity of the reduction in HCV RNA replication in the replicon cells putatively achieved by the knockdown of FKBP8, a plasmid encoding Flag-FKBP8 containing either a silent mutation within the siRNA target sequence (Flag-rFKBP8) or empty plasmid was transfected into the HCV replicon cells and then selection was carried out with the appropriate antibiotics. The remaining cells, that is, Huh7rFKBP8 and Huh7c cells, harboring the Flag-rFKBP8 and empty plasmid, respectively, were pooled and then transfected with the FKBP8 siRNA (Target 1) or control siRNA. Although transfection of the FKBP8 siRNA led to a 60% reduction of HCV RNA and FKBP8 mRNA in Huh7c cells, in comparison with levels in cells transfected with the control siRNA, no reduction in HCV RNA, and only a slight reduction in FKBP8 mRNA levels were observed in Huh7rFKBP8 cells (Figure 5C). Flag-rFKBP8 expression was clearly detected in Huh7rFKBP8 cells after transfection with the FKBP8 siRNA or control siRNA, whereas the endogenous FKBP8 decreased in both Huh7rFKBP8 and Huh7c cells with the FKBP8 siRNA (Figure 5D). These findings suggest that the slight reduction of FKBP8 mRNA in the Huh7rFKBP8 cells was due to a loss of endogenous FKBP8. Knockdown of FKBP8 by siRNA induce no apoptosis in a hepatoma cell line (Supplementary Figure 2). These results therefore confirmed that the inhibition of HCVRNA replication by FKBP8 siRNA was due to a specific reduction in the mRNA of FKBP8, but was not due to a nonspecific reduction of any other host mRNA.
Figure 5.

Decrease in HCV RNA by FKBP8-targeted siRNA. (A) HCV replicon cells (9–13 cells) were transfected with each of three kinds of siRNA targeted to FKBP8 or nontargeted siRNA at a final concentration of 80 nM. Transfected cells were collected at 72 h post-transfection, and FKBP8 mRNA and HCV RNA levels were determined by real-time PCR after being normalized with β-actin mRNA. (B) HCV replicon cells transfected with 80 and 160 nM of Target 1 or nontargeted siRNA were harvested at 72 h post-transfection, and the samples were analyzed by immunoblotting. (C) HCV replicon cells expressing Flag-rFKBP8 mutant (Huh7rFKBP8) or control cells (Huh7c) were transfected with Target 1 (gray bars) or nontargeted (white bars) siRNA at a concentration of 80 nM. Transfected cells were harvested at 72 h post-transfection, and HCV RNA (left) and FKBP8 mRNA (right) were measured by real-time PCR and expressed as % increase after being normalized with the expression of β-actin mRNA. (D) Levels of expression of endogenous FKBP8, exogenous Flag-rFKBP8, and β-actin in the replicon cells after transfection of the siRNAs were determined by immunoblotting using specific antibodies. The data shown in each panel are representative of three independent experiments.
To further examine the involvement of FKBP8 on HCV replication, we established a line of Huh7 cells that stably expresses shRNA targeted to FKBP8. Huh7 was transfected with pSilencer 2.1 U6 hygro containing the cDNA of shRNA to FKBP8, and then selection was carried out with hygromycin. FKBP8 was detected in Huh7 cells harboring a control plasmid (Huh7N), whereas decreased expression of FKBP8 was clearly observed in cells expressing the shRNA to FKBP8 (Huh7FKBP8KD) (Figure 6A). In order to examine the effects of the knockdown of FKBP8 on HCV RNA replication, a chimeric HCV RNA containing the Renilla luciferase gene was transfected into these cell lines. Although the chimeric HCV RNA exhibited 5.5 times higher replication than a replication deficient GND mutant RNA in Huh7N, only a doubling of the levels of replication was observed in Huh7FKBP8KD (Figure 6B). Furthermore, HCV RNA containing a neomycin-resistant gene was transfected into the cell lines in order to examine the role played by FKBP8 in HCV RNA replication. The efficiency of colony formation in Huh7N and Huh7FKBP8KD cells with the HCV RNA were 1700 and 23 colonies/μg RNA, respectively (Figure 6C). We also examined the role of FKBP8 on the cell culture system for HCV infection. The siRNA-mediated knockdown of FKBP8 impaired both intracellular viral RNA replication and release of HCV core protein into the culture supernatants (Figure 6D). These results further confirmed that FKBP8 plays a crucial role in the efficient replication of HCV RNA.
Figure 6.
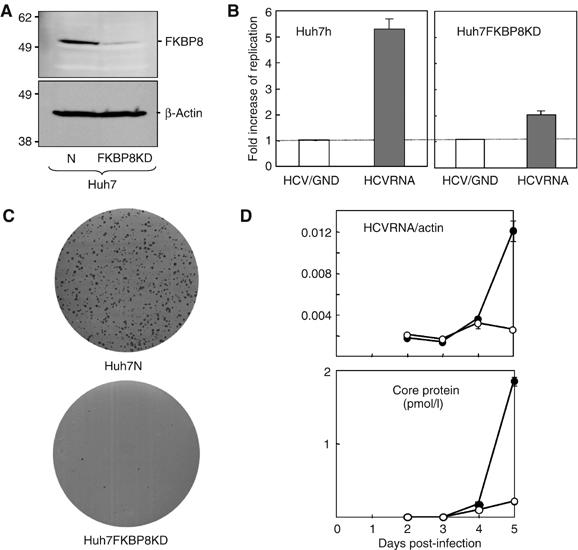
Effect of knockdown of FKBP8 on the transient replication, colony formation, and viral infection. (A) Levels of expression of FKBP8 and β-actin in Huh7N and Huh7 FKBP8KD cell lines bearing plasmids encoding shRNA for control mRNA (lane 1) and for FKBP8 mRNA (lane 2), respectively. (B) Each cell line was transfected with in vitro-transcribed HCV replicon RNA, pFK-I389 hRL/NS3-3′/NK5.1 (HCVRNA), or a replication-negative mutant, pFK-I389 hRL/NS3-3′/NK5.1GND (HCV/GND). The fold increase in replication was determined by the increase in luciferase activity at 48 h compared with that observed 4 h after standardization, as based on the activity of the replication-deficient HCV/GND replicon. (C) Huh7N and Huh7 FKBP8KD cell lines were transfected with in vitro-transcribed replicon RNA (pFK-I389 neo/NS3-3′/NK5.1) and the cells were incubated for 4 weeks. The remaining cells were fixed with 4% paraformaldehyde and then were stained. (D) Huh7.5.1 cells were transfected with either of siRNA targeted to FKBP8 (Target 1) or nontarget control at a concentration of 80 nM. The cells were inoculated with HCVcc at 24 h after transfection and cells and culture supernatants were harvested every day. Intracellular viral RNA (upper) and HCV core protein in the supernatant (lower) were determined. The data shown in each panel are representative of three independent experiments.
FKBP8 forms a multicomplex with NS5A and Hsp90
To identify the cellular proteins that associate with FKBP8, we employed a purification strategy using an MEF affinity tag composed of myc and FLAG tags fused in tandem and separated by a spacer sequence containing a TEV protease cleavage site (myc-TEV-FLAG) (Ichimura et al, 2005). The MEF expression cassette fused with FKBP8 was transfected into 293T cells and the cells were immunoprecipitated. The endogenous FKBP8-binding proteins bound to the Flag beads were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and were then visualized by silver staining. The visible protein bands were excised and determined by a nanoflow LC-MS/MS system. Major protein bands with a molecular size of 94 and 53 kDa were identified as Hsp90 and FKBP8, respectively, although it should be noted that the remaining bands detected in the samples could not be reliably identified (Figure 7A).
Figure 7.
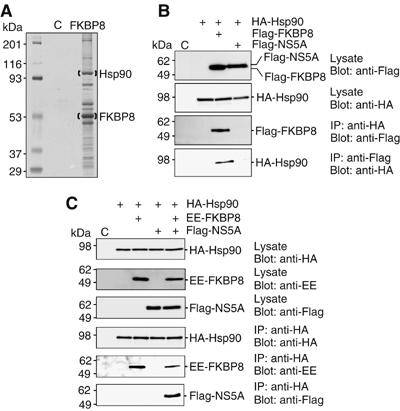
FKBP8 forms complex with NS5A and Hsp90. (A) An N-terminally myc-TEV-Flag-tagged FKBP8 was expressed in 293T cells and immunoprecipitated. The precipitated proteins were applied to SDS–PAGE and then stained with silver staining. Hsp90 and FKBP8 were identified by LC-MS/MS. (B) HA-Hsp90 was coexpressed with Flag-FKBP8 or Flag-NS5A in 293T cells, and was immunoprecipitated by anti-HA or anti-Flag antibody. Precipitates were analyzed by Western blotting. (C) HA-Hsp90 was coexpressed with EE-FKBP8 and/or Flag-NS5A in 293T cells and was immunoprecipitated with anti-HA antibody. Precipitates were analyzed by Western blotting by anti-EE, -HA or -Flag antibody.
In order to elucidate the interaction of Hsp90 with FKBP8 in mammalian cells, Flag-FKBP8 was coexpressed with HA-Hsp90 and immunoprecipitated by anti-Flag or anti-HA antibody. HA-Hsp90 and Flag-FKBP8 were co-precipitated with each other by either of the antibodies but no interaction was observed between HA-Hsp90 and Flag-NS5A (Figure 7B). To examine the interplay among NS5A, FKBP8, and Hsp90, HA-Hsp90 was coexpressed with EE-FKBP8 and/or Flag-NS5A (Figure 7C). Co-immunoprecipitation of Hsp90 and NS5A was clearly detected in the presence but not in the absence of FKBP8. The increase in NS5A expression had no effect on the interaction between FKBP8 and Hsp90 (Supplementary Figure 3). These results suggest that Hsp90 does not directly bind to NS5A but forms complex with NS5A through the interaction with FKBP8.
FKBP8 interacts with NS5A and Hsp90 via different sites in the TPR domain
Crystal structure of the TPR domain of Hop, an adaptor chaperone that binds both Hsp70 and Hsp90, revealed that C-terminal MEEVD motif of Hsp90 is held by amino-acid residues of the two-carboxylate clamp positions within the TPR domain (Scheufler et al, 2000; Brinker et al, 2002; Cliff et al, 2006). To examine the role of the C-terminal MEEVD motif of Hsp90 on the interaction with FKBP8, Hsp90 mutant lacking the MEEVD motif (HA-Hsp90ΔMEEVD) was coexpressed with Flag-FKBP8 (Figure 8A). Wild-type Hsp90 but not the mutant Hsp90 was co-precipitated with FKBP8, indicating that the FKBP8 interacts with Hsp90 via the C-terminal MEEVD motif. Lys307 and Arg311 residues in the two-carboxylate clamp positions of FKBP8 were conserved among the TPR domain of other immunophilins, such as FKBP52 and CypD (Figure 8B). To examine the role of the two-carboxylate clamp positions of FKBP8 for the interaction with Hsp90 and NS5A, FKBP8 mutant replaced Lys307 and Arg311 with Ala, designated as FKBP8TPRmut, was coexpressed with HA-Hsp90 or HA-NS5A (Figure 8C). FKBP8TPRmut exhibited no interaction with Hsp90, but still retained the capability of binding to NS5A, indicating that FKBP8 interacts with Hsp90 and NS5A through the conserved two-carboxylate clamp residues and other region in the TPR domain, respectively.
Figure 8.

FKBP8 interacts with NS5A and Hsp90 via different sites in the TPR domain and participates in HCV replication. (A) Flag-FKBP8 was coexpressed with HA-Hsp90 or HA-Hsp90ΔMEEDV lacking the C-terminal MEEDV residues and was immunoprecipitated by anti-HA or anti-Flag antibody. Precipitates were analyzed by Western blotting. (B) Sequence alignment of TPR domains of FKBP8, FKBP52, and CypD. The two bold characters (K and R) indicate amino-acid residues substituted to Ala in FKBP8TPRmut. (C) Flag-FKBP8 or Flag-FKBP8TPRmut substituted Lys307 and Arg311 to Ala was coexpressed with HA-Hsp90 (left) or HA-NS5A (right) in 293T cells, and was immunoprecipitated by anti-HA or anti-Flag antibody. Precipitates were analyzed by Western blotting. (D) Flag-FKBP8, Flag-FKBP8TPRmut, or empty plasmid was transfected into the replicon cells and HCV RNA was determined by real-time PCR after 48 h transfection. Relative replication was expressed as % increase after being normalized with the expression of β-actin mRNA. (E) The effect of geldanamycin on HCV RNA replication. HCV replicon cells (9–13 cells) were treated with 1, 3, 10, and 30 nM of geldanamycin and after 24 h treatment, HCV RNA replication was determined by real-time PCR. Relative replication was expressed as % replication after standardized by the expression of β-actin (closed circles). Cell viabilities were determined by trypan blue staining (closed triangles). (F) The effect of geldanamycin on the expression of NS5A and FKBP8. The replicon cells were examined by immunoblotting after treatment with various concentrations of geldanamycin. The data shown in each panel are representative of three independent experiments.
Hsp90 participates in the replication of HCV RNA
To examine the role of Hsp90 in the replication of HCV RNA, FKBP8TPRmut lacking the ability to bind to Hsp90 was expressed in HCV replicon cells (Figure 8D). Expression of FKBP8TPRmut resulted in 30% reduction of HCV RNA replication, suggesting that FKBP8TPRmut works as a dominant negative. Geldanamycin is well known to bind to the ATP/ADP binding site of Hsp90 and specifically inhibits the enzymatic activity of Hsp90, resulting in the promotion of the degradation of client proteins for Hsp90 (Neckers, 2002). To determine the effects of Hsp90 inhibition induced by geldanamycin on the replication of HCV RNA, HCV replicon cells were treated with various concentrations of geldanamycin. Treatment with geldanamycin clearly reduced the levels of HCV RNA replication (Figure 8E); moreover, this treatment led to the slight suppression of NS5A without reducing the levels of FKBP8 expressed in the HCV replicon cells (Figure 8F). Although the inhibition of cleavage at the NS2/NS3 junction by geldanamycin has been demonstrated in both in vitro and in vivo assays (Waxman et al, 2001), the effects of geldanamycin on the replication of HCV RNA have not yet been examined in replicon cells. The HCV replicon cell line used in the present study does not contain an NS2-coding region, and NS2 has been shown to be unnecessary for the replication of HCV subgenomic replicon (Lohmann et al, 1999). Therefore, the observed reduction in RNA replication in the HCV replicon cells by treatment with geldanamycin was not due to an inhibition of HCV polyprotein processing. In vitro pull-down assays revealed that geldanamycin inhibited the binding of FKBP8 to Hsp90 and/or NS5A domain I (Supplementary Figure 4). Thus, geldanamycin may inhibit HCV replication by disruption of NS5A/FKBP8/Hsp90 complex. These results suggest that a protein complex composed of FKBP8, Hsp90, and NS5A is involved in HCV RNA replication.
Discussion
HCV NS5A is a multifunctional protein involved in viral replication and pathogenesis (Macdonald and Harris, 2004). In this study, we demonstrated that NS5A specifically binds to FKBP8, but not to other homologous immunophilins such as FKBP52 and CypD, and that FKBP8 forms both a homomultimer and a heteromultimer with Hsp90. Mutation analyses of FKBP8 and Hsp90 suggest that FKBP8 intermediates between NS5A and Hsp90 via the different position in the TRP domain. FKBP8 has been shown to be expressed in several human tissues, including the liver (Lam et al, 1995); moreover, it has been demonstrated that FKBP8-knockout mice exhibit unusual morphological changes in brain development in the embryonic stage (Nielsen et al, 2004). However, the physiological function of FKBP8 has not been clarified to date.
Recently, the in vitro replication of the full-length HCV genome of genotype 2a (JFH1) isolated from an HCV-infected patient who developed fulminant hepatitis was reported (Lindenbach et al, 2005; Wakita et al, 2005; Zhong et al, 2005). Although binding of NS5A of the JFH1 clone to FKBP8 was weaker than that of genotypes 1a and 1b (Figure 2A), siRNA-mediated knockdown of FKBP8 impaired production of infectious HCV particles in JFH1 cell culture system (Figure 6D). In spite of a weaker interaction between FKBP8 and NS5A, these results suggest that FKBP8 is still required for HCV replication in the cell culture system of JFH1. The involvement of FKBP8 in mitochondria-mediated apoptosis remains controversial. Shirane and Nakayama (2003) reported that FKBP8 binds to Bcl-2 and that the Bcl-2/FKBP8 complex was sequestered in the mitochondria in order to suppress apoptosis. However, Edlich et al (2005) reported that FKBP8 binds to calmodulin via elevations in the calcium concentration, which in turn leads to the promotion of apoptosis in neuronal tissues. Knockdown of FKBP8 led to impaired HCV RNA replication, which was restored by the expression of an RNAi-resistant FKBP8 mutant. These results suggest that the impairment of HCV RNA replication induced by the knockdown of FKBP8 was not due to an induction of apoptosis, nor to any side effects of RNA transfection. The modulation of apoptosis by FKBP8 might be diverse in different tissue types and cell lines.
FKBP8 belongs to the FKBP family due to sequence similarity, but neither FK506 binding nor PPIase activity has been detected in the case of FKBP8 thus far (Lam et al, 1995). Apoptosis was induced in the SH-SY5Y neuroblastoma cell line by the treatment with mitochondria-mediated proapoptotic drugs, but was inhibited by the knockdown of FKBP8 and was enhanced by treatment with GPI1046, a nonimmunosuppressive FK506 derivative, whereas this result was not obtained with FK506 (Edlich et al, 2005). The inhibition constant of FKBP8 to FK506 was 50 times higher than that of FKBP12 to FK506 (Edlich et al, 2005), which suggests that the binding affinity of FKBP8 to FK506 is low. Furthermore, cyclosporin A, but not FK506, was shown to suppress HCV RNA replication via the interaction of NS5B with CypB (Watashi et al, 2003, 2005). These results support the notion that FK506 preferentially binds to FKBP members other than FKBP8 in vivo, and that it does not participate in the inhibition of HCV replication.
Cellular and viral chaperones are implicated in the processing of viral proteins and viral assembly (Maggioni and Braakman, 2005; Mayer, 2005). The NS2 protein of bovine viral diarrhea virus (BVDV), a member of the Flaviviridae family as is HCV, exhibits autoprotease activity that leads to cleavage at the NS2 and NS3 junction (Lackner et al, 2005). A noncytopathogenic strain of BVDV is unable to cleave the NS2/3 junction in the absence of the interaction of a molecular chaperone, J-domain protein interacting with viral protein (Jiv); these previous findings suggest that Jiv is necessary for the replication of a noncytopathogenic strain of BVDV and is involved in the establishment of persistent infection (Lackner et al, 2005). Furthermore, FKBP52, which shares a high homology with FKBP8, was shown to regulate replication of adeno-associated virus type 2 by interacting with viral DNA (Qing et al, 2001). In this study, we demonstrated that HCV NS5A binds to FKBP8 and forms a complex with Hsp90. FKBP8 could directly bind to NS5A domain I in vitro (Supplementary Figure 1), suggesting that Hsp90 is not required for interaction between NS5A and FKBP8. FKBP52 forms a homodimer, binds to Hsp90 through TPR domain, and regulates chaperone activity of Hsp90 (Silverstein et al, 1999; Scheufler et al, 2000; Wu et al, 2004). FKBP8 may act as cochaperone of Hsp90 to regulate HCV genome replication by interaction with NS5A. Hsp90 is a molecular chaperone that is highly expressed in most cell types in various organisms (Neckers, 2002). Here, Hsp90 was found to be able to bind to FKBP8 and form a complex with HCV NS5A. The suppression of NS5A, but not that of FKBP8, was observed in replicon cells treated with geldanamycin, thus suggesting that Hsp90 regulates the replication of HCV RNA via the interaction with FKBP8. It is well known that several host proteins such as VAPs and FBL2 interact with the HCV replication complex and regulate HCV RNA replication (Evans et al, 2004; Gao et al, 2004; Hamamoto et al, 2005; Wang et al, 2005). The TPR domain of FKBP8 is composed of 220 amino acids and is too long to determine the critical residues responsible for interaction with NS5A. Therefore, we tried to make a chimeric mutant carrying the TPR of FKBP52 to determine the critical amino-acid residues for binding to NS5A in FKBP8. However, expression of a chimeric FKBP8 possessing TPR of FKBP52 was much lower than the native form, suggesting that TPR domain is critical for stability and conformation of FKBP8. Amino-acid residues responsible for the binding to NS5A must be different from the two-carboxylate positions responsible for Hsp90 binding and locate within the TPR domain. The ternary complex consists of NS5A, FKBP8 and Hsp90 may be involved in the replication of HCV. FKBP52 possesses PPIase activity and chaperone activity in domain I (amino acids 1–148) and domain 3 (TPR domain, amino acids 264–400), respectively (Pirkl et al, 2001). Therefore, it is reasonable to speculate that the TPR domain is responsible for the chaperone activity of FKBP8, and that the FKBP8 and NS5A complex transports Hsp90 to the appropriate clients, including viral and host proteins, which in turn leads to the stabilization of the replication complex and the enhancement of HCV RNA replication.
In this study, we identified human FKBP8 as a binding partner of HCV NS5A. Our results suggest that the interaction between FKBP8 and HCV NS5A is essential for HCV replication. The NS5A protein forms a complex with FKBP8 and Hsp90, and an inhibitor of Hsp90 was shown to reduce the efficiency of HCV replication. The elucidation of the molecular mechanisms underlying the formation of the NS5A/FKBP8/Hsp90 complex may lead to the development of new therapeutics for chronic hepatitis C.
Materials and methods
Yeast two-hybrid assays
Screening for the gene-encoding host protein that interacts with HCV NS5A was performed with a yeast two-hybrid system, Matchmaker two-hybrid system 3 (Clontech, Palo Alto, CA), according to the manufacturer's protocol. Human fetal brain and liver libraries were purchased from Clontech. The cDNA of NS5A-encoding amino acids 1973–2419 of an HCV polyprotein of the J1 strain (genotype 1b) (Aizaki et al, 1998) was amplified by PCR and was cloned into the pGBKT7 vector (Clontech) (Tu et al, 1999; Hamamoto et al, 2005).
Plasmids
DNA fragments encoding NS5A were amplified from HCV genotype 1b strains J1 and Con1 (provided by Dr Bartenschlager), genotype 1a strain H77C (provided by Dr Bukh), and genotype 2a strain JFH-1 (provided by Dr Wakita) by PCR using Pfu turbo DNA polymerase (Stratagene, La Jolla, CA). The fragments were cloned into pCAGGs-PUR/N-HA, in which the sequence encoding an HA tag is inserted at the 5′-terminus of the cloning site of pCAGGs-PUR (Niwa et al, 1991). The DNA fragment encoding human FKBP8 was amplified from the total cDNA of Huh7 cells by PCR, and this fragment was introduced into pEF-FLAG pGBK puro (Huang et al, 1997), pCAGGs-PUR/NHA, pcDNA3.1-N-HA (Tu et al, 1999; Hamamoto et al, 2005), and pcDNA3.1-N-EE, in which an Glu-Glu (EE) tag is inserted in the 5′-terminus of the cloning site of pcDNA3.1 (+) (Invitrogen, Carlsbad, CA). The DNA fragments encoding human Hsp90, FKBP52, and CypD were amplified from a human fetal brain library (Clontech) by PCR, and were introduced into pcDNA3.1-N-HA. The genes encoding the deletion mutants of human FKBP8 were amplified and cloned into pCAGGs-PUR/NHA. The gene encoding an FKBP8 mutant replaced Lys307 and Arg311 with Ala, designated as FKBP8TPRmut, was generated by the method of splicing by overlap extension and introduced into pEF-Flag pGBKpuro. The gene encoding an Hsp90 mutant lacking the C-terminal MEEVD motif of Hsp90, designated as Hsp90ΔMEEVD, was amplified and cloned into pcDNA3.1-N-HA. All PCR products were confirmed by sequencing by an ABI PRSM 310 genetic analyzer (Applied Biosystems, Tokyo, Japan).
Cell lines
Human embryonic kidney 293T cells and the human hepatoma cell lines Huh7 and FLC-4 were maintained in Dulbecco's modified Eagle's medium (DMEM) (Sigma, St Louis, MO) containing 10% fetal calf serum (FCS), whereas the Huh 9–13 cell line, which possesses an HCV subgenomic replicon (Lohmann et al, 1999), was cultured in DMEM supplemented with 10% FCS and 1 mg/ml G418. All cells were cultured at 37°C in a humidified atmosphere with 5% CO2.
Antibodies
Mouse monoclonal antibodies to the HA and EE tags were purchased from Covance (Richmond, CA). Anti-Flag mouse antibody M2, horseradish peroxidase-conjugated M2 antibody, and anti-β-actin mouse monoclonal antibody were purchased from Sigma. Mouse monoclonal antibody to NS5A was from Austral Biologicals (San Ramon, CA). Mouse monoclonal antibodies to NS4B and NS5B have been described previously (Kashiwagi et al, 2002). Rabbit polyclonal antibody to NS5A was prepared as described previously (Hamamoto et al, 2005). Rabbit polyclonal antibody to thioredoxin was described previously (Moriishi et al, 1999).
Transfection, immunoblotting, and immunoprecipitation
The transfection and immunoprecipitation test were carried out by a previously described method (Hamamoto et al, 2005). The immunoprecipitates boiled in the loading buffer were subjected to 12.5% SDS–PAGE. The proteins were transferred to polyvinylidene difluoride membranes (Millipore, Bedford, MA) and were reacted with the appropriate antibodies. The immune complexes were visualized with Super Signal West Femto substrate (Pierce, Rockford, IL) and they were detected by an LAS-3000 image analyzer system (Fujifilm, Tokyo, Japan). The density of protein band was determined by using IMAGE-PRO PLUS 5.1 software (Media Cybernetics, Silver Springs, MD).
Gene silencing by siRNA
The siRNA targeted to FKBP8, Target-1: 5′-GAGUGGCUGGACAUUCUGG-3′, and negative control siRNA, that is, siCONTROL NonTargeting siRNA-2, were purchased from Dharmacon (Lafayette, CO). Target-2, 5′-UCCCAUGGAAGUGGCUGUU–3′, and Target-3, 5′-GACAACAUCAAGGCUCUCU-3′ were purchased from Qiagen (Tokyo, Japan). The Huh7 cells harboring a subgenomic HCV replicon grown on six-well plates were transfected with 80 or 160 nM of siRNA with siFECTOR (B-Bridge International, Sunnyvale, CA). The cells were grown in DMEM containing 10% FCS and were then harvested at 48 or 72 h post-transfection.
Real-time PCR
Total RNA was prepared from cell lines by using RNeasy mini kit (Qiagen). First-strand cDNA was synthesized by using a first-strand cDNA synthesis kit (Amersham Pharmacia Biotech, Franklin Lakes, NJ) and random primers. Each cDNA was estimated by Platinum SYBR Green qPCR SuperMix UDG (Invitrogen) according to the manufacturer's protocol. Fluorescent signals were analyzed by an ABI PRISM 7000 (Applied Biosystems). The HCV NS5A, human β-actin, and human FKBP8 genes were amplified using the primer pairs of 5′-AGTCAGTTGTCTGCGCTTTC-3′ and 5′-CGGGGAATTTCCTGGTCTTC-3′, 5′-TGGAGTCCTGTGGCATCCACGAAACTACCTTC AACTC-3′ and 5′-CGGACTCGTCATACTCCTGCTTGCTGATCCAC ATC-3′, and 5′-GGCTGTTGAGGAAGAAGACG-3′ and 5′-CTTGGAGTCAGCAGTGACCA-3′, respectively. The FKBP8 primers are located at different exons in order to prevent the false-positive amplification of contaminated genomic DNA. The values of the HCV genome and FKBP8 mRNA were normalized with those of β-actin mRNA. Each PCR product was detected as a single band of the correct size upon agarose gel electrophoresis (data not shown).
Establishment of cell lines expressing an siRNA-resistant FKBP8 mutant and knockdown FKBP8 expression
A, G, and T at nucleotides 273, 276, and 288 from the 5′ end of the open-reading frame of human FKBP8 were replaced with G, A, and C, respectively, according to a splicing method achieved by overlap extension; these silent mutations were then cloned into pEF-Flag pGBKpuro. The resulting plasmid encoding a mutant FKBP8 resistant to knockdown by siRNA was transfected into Huh7 cells harboring the HCV RNA replicon. The culture medium was replaced with DMEM supplemented with 10% FCS and 2 μg/ml of puromycin (Nakarai Tesque, Tokyo, Japan) at 24 h post-transfection, and the cells were cultured for 7 days. The surviving cells were used for the FKBP8 knockdown experiments. The shRNAs targeted to FKBP8, the target sequences of which were 5′-GATCCGCTGGAACCTTCCAACAAGTTCAAGAG ACTTGTTGGAAGGTTCCAGCTTA-3′, and 5′-AGCTTAAGCTGGAACCTTCCAACAAGTCTCTT GAACTTGTTGGAAGGTTCCAGCG-3′, were annealed and introduced between the BamHI and HindIII sites of pSilencer™ 2.1-U6 hygro (Ambion, Austin, TX) according to the manufacturer's protocol. An HCV replicon cell line cured with IFN-α was transfected with 5 μg of the plasmid by electroporation. The culture medium was replaced with DMEM supplemented with 10% FCS and 500 μg/ml of Hygromycin B (Wako, Tokyo, Japan) at 24 h post-transfection. The remaining cells were re-seeded in 98-well plates and cloned for the colony formation and transient replication assays.
Colony formation assay
The plasmid pFK-I389 neo/NS3-3′/NK5.1 (Pietschmann et al, 2002) was obtained from R Bartenschlager. The plasmid cleaved at the ScaI site was transcribed in vitro using the MEGAscript T7 kit (Ambion) according to the manufacturer's protocol. The linearized plasmid (10 μg) was introduced into Huh7 cells at 4 million cells/0.4 ml by electroporation at 270 V and 960 μF using a Gene Pulser™ (Bio-Rad, Hercules, CA). Electroporated cells were suspended at a final volume of 10 ml of culture medium. Three-milliliter aliquots of cell suspension were mixed with 7 ml of culture medium and then the cells were seeded on culture dishes (diameter: 10 cm). The culture medium was replaced with DMEM containing 10% FCS and 1 mg/ml of G418 (Nakarai Tesque) at 24 h post-transfection. The medium was exchanged weekly for fresh DMEM containing 10% FCS and 1 mg/ml G418. The remaining colonies were fixed with 4% paraformaldehyde at 4 weeks after electroporation, and the cells were stained with crystal violet.
Transient replication assay
The cDNA encoding Renilla luciferase was introduced between the AscI and PmeI sites of the plasmid pFK-I389 neo/NS3-3′/NK5.1, in place of the neo gene. The resulting plasmid, pFK-I389 hRL/NS3-3′/NK5.1, was cleaved with ScaI and was transcribed in vitro using a MEGAscript T7 kit (Ambion). Huh7 cells were suspended at 10 million cells/ml and the suspensions were mixed with 10 μg of in vitro-transcribed RNA at a 400-μl volume; the cells were then electroporated at 270 V and 960 μF by a Gene Pulser™ (Bio-Rad). The electroporated cells were suspended in 25 ml of culture medium and then were seeded at 1 ml/well on 12-well culture plates. Luciferase activity was measured at 4 and 48 h post-transfection using a Renilla Luciferase assay system (Promega, Madison, WI) according to the manufacturer's protocol. Luciferase activity at 4 h after electroporation was used to determine the transfection efficiency.
Generation of infectious HCV particles
The viral RNA of JFH1 was introduced into Huh7.5.1 according to the method of Wakita et al (2005). The supernatant was collected at 7 days post-transfection and used as HCV particles that are infectious in cell culture (HCVcc). The naïve Huh7.5.1 cells were transfected with siRNA of nontarget control or FKBP8-Target 1 at a concentration of 80 nM. The siRNA-treated Huh7.5.1 cells were inoculated with HCVcc at 24 h post-transfection. Infected cells and culture supernatants were harvested every day until 5 days post-infection.
Determination of FKBP8-binding proteins
MEF purification was carried out by a previously described method (Ichimura et al, 2005). The FKBP8 gene was amplified by PCR and introduced into pcDNA3.1 encoding the myc-TEV-Flag epitope tag (Ichimura et al, 2005). The resulting plasmid was transfected into 293T cells, which were then subjected to MEF purification. FKBP8-binding proteins were separated by SDS–PAGE and visualized by silver staining. The stained bands were excised, digested in gels with Lys-C, and analyzed by the direct nanoflow LC-MS/MS system (Ichimura et al, 2005).
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Material
Acknowledgments
We thank H Murase for secretarial work and H Miyamoto for discussion. We are also grateful to J Bukh, R Bartenschlager, and T Wakita for providing the HCV cDNAs and DCS Huang for the pEF-FLAG pGBK puro. This work was supported in part by grants-in-aid from the Ministry of Health, Labor, and Welfare; the Ministry of Education, Culture, Sports, Science, and Technology; the 21st Century Center of Excellence Program; and the Foundation for Biomedical Research and Innovation.
References
- Aizaki H, Aoki Y, Harada T, Ishii K, Suzuki T, Nagamori S, Toda G, Matsuura Y, Miyamura T (1998) Full-length complementary DNA of hepatitis C virus genome from an infectious blood sample. Hepatology 27: 621–627 [DOI] [PubMed] [Google Scholar]
- Appel N, Pietschmann T, Bartenschlager R (2005) Mutational analysis of hepatitis C virus nonstructural protein 5A: potential role of differential phosphorylation in RNA replication and identification of a genetically flexible domain. J Virol 79: 3187–3194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boguski MS, Sikorski RS, Hieter P, Goebl M (1990) Expanding family. Nature 346: 114. [DOI] [PubMed] [Google Scholar]
- Brinker A, Scheufler C, Von Der Mulbe F, Fleckenstein B, Herrmann C, Jung G, Moarefi I, Hartl FU (2002) Ligand discrimination by TPR domains. Relevance and selectivity of EEVD-recognition in Hsp70 × Hop × Hsp90 complexes. J Biol Chem 277: 19265–19275 [DOI] [PubMed] [Google Scholar]
- Chadli A, Bouhouche I, Sullivan W, Stensgard B, McMahon N, Catelli MG, Toft DO (2000) Dimerization and N-terminal domain proximity underlie the function of the molecular chaperone heat shock protein 90. Proc Natl Acad Sci USA 97: 12524–12529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliff MJ, Harris R, Barford D, Ladbury JE, Williams MA (2006) Conformational diversity in the TPR domain-mediated interaction of protein phosphatase 5 with Hsp90. Structure 14: 415–426 [DOI] [PubMed] [Google Scholar]
- Edlich F, Weiwad M, Erdmann F, Fanghanel J, Jarczowski F, Rahfeld JU, Fischer G (2005) Bcl-2 regulator FKBP38 is activated by Ca(2+)/calmodulin. EMBO J 24: 2688–2699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans MJ, Rice CM, Goff SP (2004) Phosphorylation of hepatitis C virus nonstructural protein 5A modulates its protein interactions and viral RNA replication. Proc Natl Acad Sci USA 101: 13038–13043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer G, Aumuller T (2003) Regulation of peptide bond cis/trans isomerization by enzyme catalysis and its implication in physiological processes. Rev Physiol Biochem Pharmacol 148: 105–150 [DOI] [PubMed] [Google Scholar]
- Gao L, Aizaki H, He JW, Lai MM (2004) Interactions between viral nonstructural proteins and host protein hVAP-33 mediate the formation of hepatitis C virus RNA replication complex on lipid raft. J Virol 78: 3480–3488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamamoto I, Nishimune Y, Okamoto T, Aizaki H, Lee K, Mori Y, Abe T, Lai MC, Miyamura T, Moriishi K, Matsuura Y (2005) Human VAP-B is involved in HCV replication through interaction with NS5A and NS5B. J Virol 79: 13473–13482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano T, Kinoshita N, Morikawa K, Yanagida M (1990) Snap helix with knob and hole: essential repeats in S. pombe nuclear protein nuc2+. Cell 60: 319–328 [DOI] [PubMed] [Google Scholar]
- Huang DC, Cory S, Strasser A (1997) Bcl-2, Bcl-XL and adenovirus protein E1B19kD are functionally equivalent in their ability to inhibit cell death. Oncogene 14: 405–414 [DOI] [PubMed] [Google Scholar]
- Ichimura T, Yamamura H, Sasamoto K, Tominaga Y, Taoka M, Kakiuchi K, Shinkawa T, Takahashi N, Shimada S, Isobe T (2005) 14-3-3 proteins modulate the expression of epithelial Na+ channels by phosphorylation-dependent interaction with Nedd4–2 ubiquitin ligase. J Biol Chem 280: 13187–13194 [DOI] [PubMed] [Google Scholar]
- Inoue K, Sekiyama K, Yamada M, Watanabe T, Yasuda H, Yoshiba M (2003) Combined interferon alpha2b and cyclosporin A in the treatment of chronic hepatitis C: controlled trial. J Gastroenterol 38: 567–572 [DOI] [PubMed] [Google Scholar]
- Kapadia SB, Chisari FV (2005) Hepatitis C virus RNA replication is regulated by host geranylgeranylation and fatty acids. Proc Natl Acad Sci USA 102: 2561–2566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashiwagi T, Hara K, Kohara M, Iwahashi J, Hamada N, Honda-Yoshino H, Toyoda T (2002) Promoter/origin structure of the complementary strand of hepatitis C virus genome. J Biol Chem 277: 28700–28705 [DOI] [PubMed] [Google Scholar]
- Koch JO, Bartenschlager R (1999) Modulation of hepatitis C virus NS5A hyperphosphorylation by nonstructural proteins NS3, NS4A, and NS4B. J Virol 73: 7138–7146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackner T, Muller A, Konig M, Thiel HJ, Tautz N (2005) Persistence of bovine viral diarrhea virus is determined by a cellular cofactor of a viral autoprotease. J Virol 79: 9746–9755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam E, Martin M, Wiederrecht G (1995) Isolation of a cDNA encoding a novel human FK506-binding protein homolog containing leucine zipper and tetratricopeptide repeat motifs. Gene 160: 297–302 [DOI] [PubMed] [Google Scholar]
- Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM (2005) Complete replication of hepatitis C virus in cell culture. Science 309: 623–626 [DOI] [PubMed] [Google Scholar]
- Lohmann V, Korner F, Koch J, Herian U, Theilmann L, Bartenschlager R (1999) Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285: 110–113 [DOI] [PubMed] [Google Scholar]
- Macdonald A, Harris M (2004) Hepatitis C virus NS5A: tales of a promiscuous protein. J Gen Virol 85: 2485–2502 [DOI] [PubMed] [Google Scholar]
- Maggioni C, Braakman I (2005) Synthesis and quality control of viral membrane proteins. Curr Top Microbiol Immunol 285: 175–198 [DOI] [PubMed] [Google Scholar]
- Manns MP, McHutchison JG, Gordon SC, Rustgi VK, Shiffman M, Reindollar R, Goodman ZD, Koury K, Ling M, Albrecht JK (2001) Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet 358: 958–965 [DOI] [PubMed] [Google Scholar]
- Mayer MP (2005) Recruitment of Hsp70 chaperones: a crucial part of viral survival strategies. Rev Physiol Biochem Pharmacol 153: 1–46 [DOI] [PubMed] [Google Scholar]
- Moriishi K, Inoue S, Koura M, Amano F (1999) Inhibition of listeriolysin O-induced hemolysis by bovine lactoferrin. Biol Pharm Bull 22: 1167–1172 [DOI] [PubMed] [Google Scholar]
- Moriishi K, Matsuura Y (2003) Mechanisms of hepatitis C virus infection. Antivir Chem Chemother 14: 285–297 [DOI] [PubMed] [Google Scholar]
- Nakagawa M, Sakamoto N, Tanabe Y, Koyama T, Itsui Y, Takeda Y, Chen CH, Kakinuma S, Oooka S, Maekawa S, Enomoto N, Watanabe M (2005) Suppression of hepatitis C virus replication by cyclosporin a is mediated by blockade of cyclophilins. Gastroenterology 129: 1031–1041 [DOI] [PubMed] [Google Scholar]
- Neckers L (2002) Hsp90 inhibitors as novel cancer chemotherapeutic agents. Trends Mol Med 8: S55–S61 [DOI] [PubMed] [Google Scholar]
- Neddermann P, Clementi A, De Francesco R (1999) Hyperphosphorylation of the hepatitis C virus NS5A protein requires an active NS3 protease, NS4A, NS4B, and NS5A encoded on the same polyprotein. J Virol 73: 9984–9991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen JV, Mitchelmore C, Pedersen KM, Kjaerulff KM, Finsen B, Jensen NA (2004) Fkbp8: novel isoforms, genomic organization, and characterization of a forebrain promoter in transgenic mice. Genomics 83: 181–192 [DOI] [PubMed] [Google Scholar]
- Niwa H, Yamamura K, Miyazaki J (1991) Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108: 193–199 [DOI] [PubMed] [Google Scholar]
- Pietschmann T, Lohmann V, Kaul A, Krieger N, Rinck G, Rutter G, Strand D, Bartenschlager R (2002) Persistent and transient replication of full-length hepatitis C virus genomes in cell culture. J Virol 76: 4008–4021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietschmann T, Lohmann V, Rutter G, Kurpanek K, Bartenschlager R (2001) Characterization of cell lines carrying self-replicating hepatitis C virus RNAs. J Virol 75: 1252–1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirkl F, Fischer E, Modrow S, Buchner J (2001) Localization of the chaperone domain of FKBP52. J Biol Chem 276: 37034–37041 [DOI] [PubMed] [Google Scholar]
- Qing K, Hansen J, Weigel-Kelley KA, Tan M, Zhou S, Srivastava A (2001) Adeno-associated virus type 2-mediated gene transfer: role of cellular FKBP52 protein in transgene expression. J Virol 75: 8968–8976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheufler C, Brinker A, Bourenkov G, Pegoraro S, Moroder L, Bartunik H, Hartl FU, Moarefi I (2000) Structure of TPR domain-peptide complexes: critical elements in the assembly of the Hsp70-Hsp90 multichaperone machine. Cell 101: 199–210 [DOI] [PubMed] [Google Scholar]
- Shirane M, Nakayama KI (2003) Inherent calcineurin inhibitor FKBP38 targets Bcl-2 to mitochondria and inhibits apoptosis. Nat Cell Biol 5: 28–37 [DOI] [PubMed] [Google Scholar]
- Silverstein AM, Galigniana MD, Kanelakis KC, Radanyi C, Renoir JM, Pratt WB (1999) Different regions of the immunophilin FKBP52 determine its association with the glucocorticoid receptor, hsp90, and cytoplasmic dynein. J Biol Chem 274: 36980–36986 [DOI] [PubMed] [Google Scholar]
- Tellinghuisen TL, Marcotrigiano J, Gorbalenya AE, Rice CM (2004) The NS5A protein of hepatitis C virus is a zinc metalloprotein. J Biol Chem 279: 48576–48587 [DOI] [PubMed] [Google Scholar]
- Tellinghuisen TL, Marcotrigiano J, Rice CM (2005) Structure of the zinc-binding domain of an essential component of the hepatitis C virus replicase. Nature 435: 374–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu H, Gao L, Shi ST, Taylor DR, Yang T, Mircheff AK, Wen Y, Gorbalenya AE, Hwang SB, Lai MM (1999) Hepatitis C virus RNA polymerase and NS5A complex with a SNARE-like protein. Virology 263: 30–41 [DOI] [PubMed] [Google Scholar]
- Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Krausslich HG, Mizokami M, Bartenschlager R, Liang TJ (2005) Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med 11: 791–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Gale M Jr, Keller BC, Huang H, Brown MS, Goldstein JL, Ye J (2005) Identification of FBL2 as a geranylgeranylated cellular protein required for hepatitis C Virus RNA replication. Mol Cell 18: 425–434 [DOI] [PubMed] [Google Scholar]
- Wasley A, Alter MJ (2000) Epidemiology of hepatitis C: geographic differences and temporal trends. Semin Liver Dis 20: 1–16 [DOI] [PubMed] [Google Scholar]
- Watashi K, Hijikata M, Hosaka M, Yamaji M, Shimotohno K (2003) Cyclosporin A suppresses replication of hepatitis C virus genome in cultured hepatocytes. Hepatology 38: 1282–1288 [DOI] [PubMed] [Google Scholar]
- Watashi K, Ishii N, Hijikata M, Inoue D, Murata T, Miyanari Y, Shimotohno K (2005) Cyclophilin B is a functional regulator of hepatitis C virus RNA polymerase. Mol Cell 19: 111–122 [DOI] [PubMed] [Google Scholar]
- Waxman L, Whitney M, Pollok BA, Kuo LC, Darke PL (2001) Host cell factor requirement for hepatitis C virus enzyme maturation. Proc Natl Acad Sci USA 98: 13931–13935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Li P, Liu Y, Lou Z, Ding Y, Shu C, Ye S, Bartlam M, Shen B, Rao Z (2004) 3D structure of human FK506-binding protein 52: implications for the assembly of the glucocorticoid receptor/Hsp90/immunophilin heterocomplex. Proc Natl Acad Sci USA 101: 8348–8353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Wang C, Sumpter R Jr, Brown MS, Goldstein JL, Gale M Jr (2003) Disruption of hepatitis C virus RNA replication through inhibition of host protein geranylgeranylation. Proc Natl Acad Sci USA 100: 15865–15870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi M, Lemon SM (2004) Adaptive mutations producing efficient replication of genotype 1a hepatitis C virus RNA in normal Huh7 cells. J Virol 78: 7904–7915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV (2005) Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci USA 102: 9294–9299 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Material