

Abstract
Hypoxia promotes genetic instability for tumor progression. Recent evidence indicates that the transcription factor HIF-1α impairs DNA mismatch repair, yet the role of HIF-1α isoform, HIF-2α, in tumor progression remains obscure. In pursuit of the involvement of HIF-α in chromosomal instability, we report here that HIF-1α, specifically its PAS-B, induces DNA double-strand breaks at least in part by repressing the expression of NBS1, a crucial DNA repair gene constituting the MRE11A–RAD50–NBS1 complex. Despite strong similarities between the two isoforms, HIF-2α fails to do so. We demonstrate that this functional distinction stems from phosphorylation of HIF-2α Thr-324 by protein kinase D1, which discriminates between subtle differences of the two PAS-B in amino-acid sequence, thereby precluding NBS1 repression. Hence, our findings delineate a molecular pathway that functionally distinguishes HIF-1α from HIF-2α, and arguing a unique role for HIF-1α in tumor progression by promoting genomic instability.
Keywords: double-strand break, hypoxia, NBS1, PAS-B, PKD1
Introduction
Cancer is essentially a genetic disease (Vogelstein and Kinzler, 2004). As a result of gene mutations, activation of oncogenes and inactivation of tumor-suppressor genes collaborate on the neoplastic process by stimulating cell proliferation, or inhibiting cell death or cell-cycle arrest. Furthermore, hereditary inactivation of stability genes or caretakers, mainly involved in DNA repair, gives rise to genetic alternations leading to tumorigenesis. Genetic instability is a hallmark of most human cancers, arising from changes at the nucleotide and the chromosomal levels (Lengauer et al, 1998).
The primary structure of DNA is constantly subjected to alteration by cellular metabolites and exogenous DNA-damaging agents. DNA repair is essential to safeguard the genetic integrity by correcting mutations arising from myriad types of damage (Friedberg, 2003). Multiple distinct mechanisms of excision repair have been identified, including nucleotide excision repair, base excision repair, and mismatch repair. Apart from coping with damaged bases or replication errors, cells frequently suffer breakage of the DNA duplex. DNA double-strand breaks (DSBs) arise primarily from stalled replication forks, and genetically programmed processes in developing lymphocytes, and exogenous factors such as ionizing radiation. Proper DSB repair is fundamental to the prevention of chromosome loss, translocations, and truncations.
NBS1/nibrin is part of the evolutionarily conserved MRE11A–RAD50–NBS1 (MRN) complex, which interacts with DSBs early in the DNA damage response (Carney et al, 1998; D'Amours and Jackson, 2002). The genetic defect in NBS1 gene, stemming predominantly from a 5-base-pair deletion in exon 6, is responsible for the Nijmegan breakage syndrome, a hereditary disorder characterized by chromosomal instability and a predisposition to malignancies (Varon et al, 1998). Cells derived from NBS patients show high sensitivity to ionizing radiation, chromosome fragility, accelerated shortening of telomeres, and deficiency in cell-cycle checkpoints (D'Amours and Jackson, 2002). It has been shown that Nbs1 knockout mice manifested increased chromosomal breaks, owing to reduced gene conversion and sister chromatid exchanges (Tauchi et al, 2002; Frappart et al, 2005). At the molecular level, NBS1 also interacts with γ-H2AX, a phosphorylated histone H2AX detected at the sites of nascent DSBs (Rogakou et al, 1998; Paull et al, 2000), for relocating MRE11A–RAD50 to the vicinity of DNA damage (Kobayashi et al, 2002).
Although deficiency in DNA repair arising from germline mutations has been linked to various hereditary cancers, no somatic mutation in DNA repair genes has been observed in majority of sporadic cancers, suggesting the possibility of functional impairment of DNA repair in these cancers. Numerous studies have indicated that the tumor microenvironment, characterized by hypoxia, low pH, and nutrient deprivation, promotes genetic instability and tumor progression (Bindra and Glazer, 2005). Hypoxia has been shown to induce chromosomal fragility and polyploidy in cell-culture and animal models (Coquelle et al, 1998; Nelson et al, 2004). Recent investigations indicated that hypoxia inhibits DNA repair by down-regulating genes involved in mismatch repair and homologous recombination (Mihaylova et al, 2003; Bindra et al, 2004; Bindra et al, 2005; Koshiji et al, 2005).
The transcription factor HIF-1α serves as a master regulator of oxygen homeostasis by activating expression of various hypoxia-responsive genes, such as those for angiogenesis and glycolysis (Wenger, 2002; Giaccia et al, 2003; Huang and Bunn, 2003; Pugh and Ratcliffe, 2003; Semenza, 2003; Poellinger and Johnson, 2004; Kaelin, 2005). Interestingly, HIF-2α, a close member of the HIF-α family, seems to exert different biological functions in vivo (Tian et al, 1998; Compernolle et al, 2002; Scortegagna et al, 2003), even though the underlying mechanisms remain elusive. Apart from binding to the hypoxia-responsive element for gene activation, HIF-1α also functions via the HIF-1α–Myc pathway, by which HIF-1α competes with the transcription activator Myc for Sp1 binding in the target gene promoter, resulting in transcriptional downregulation of the DNA mismatch repair genes (Koshiji et al, 2005). To explore the differential role of HIF-1α and HIF-2α in DNA repair, we embarked on an investigation into the molecular basis that accounts for their distinct functions in genetic instability. Our data demonstrated that the phosphorylation status of a highly conserved threonine in the PAS-B domain distinguishes HIF-1α from HIF-2α in DNA repair gene expression.
Results
HIF-1α, distinct from HIF-2α, is critical for NBS1 downregulation by hypoxia
In pursuit of the involvement of HIF-α in chromosomal instability, we focused our investigation on the expression of the MRN complex. Results from real-time PCR showed that NBS1 mRNA levels in HCT116 cells were reduced by 50% after a 16-h hypoxic treatment (1% O2) (Figure 1A), similar to the MSH2 repression. However, MRE11A and RAD50 expression were unaffected by the treatment (Figure S1). Interestingly, similar NBS1 downregulation was observed in HCT116 TP53−/− cells, in stark contrast to the strict p53-dependence for MSH2 inhibition (Koshiji et al, 2005). Again, neither MRE11A nor RAD50 was inhibited in these cells, indicating specific NBS1 inhibition by a p53-independent mechanism. Furthermore, NBS1 protein levels were markedly decreased after 8- and 16-h hypoxic treatment (Figure S2).
Figure 1.
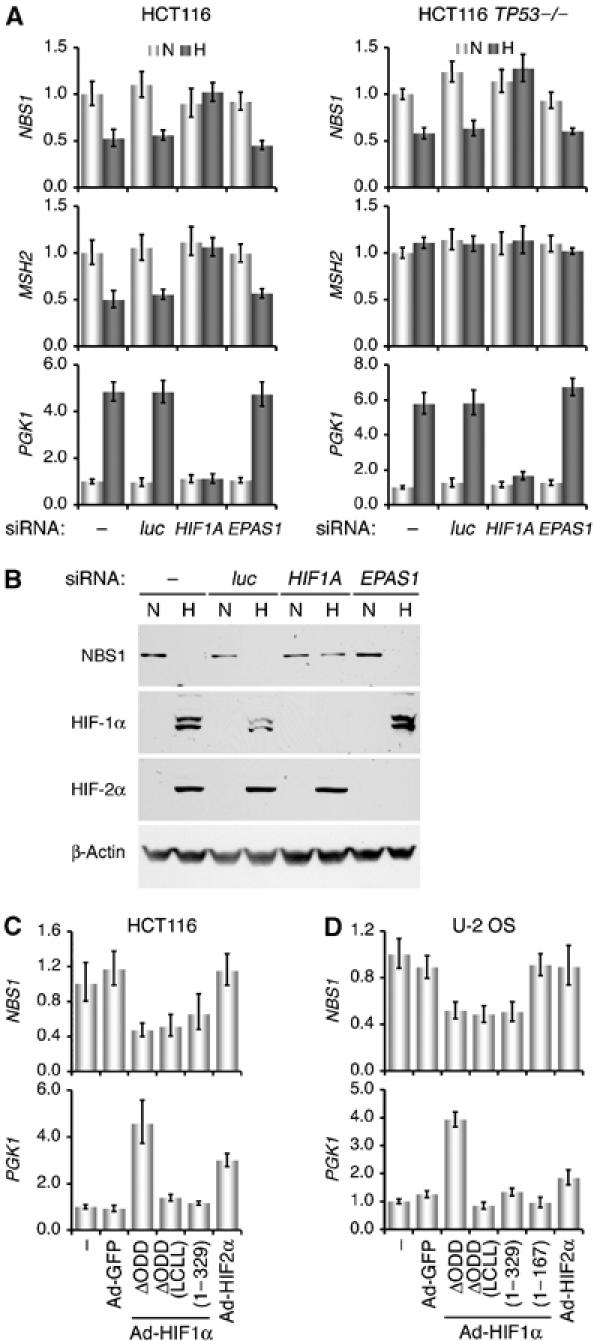
Distinct roles of HIF-1α and HIF-2α in mediating NBS1 repression by hypoxia. (A) HCT116 and HCT116 TP53−/− cells, transfected with siRNA targeting HIF1A or EPAS1 (encoding HIF-2α) were subjected to normoxic (N) or hypoxic (H) treatment (1% O2, 16 h). Mock transfection (−) and luciferase siRNA (luc) were used as negative controls. NBS1, MSH2, and PGK1 mRNA levels were determined with quantitative real-time PCR. Representative results from three independent experiments in triplicate are presented as means+standard errors. (B) U-2 OS cells were treated as in (A) and assayed for specific protein levels by sequential probing of the same blot with the corresponding antibodies. (C) HCT116 cells were infected with adenoviruses expressing HIF-1αΔODD, HIF-1αΔODD (LCLL), HIF-1α (1–329), and HIF-2α for 16 h. Cells without treatment (−) or infected with Ad-GFP served as controls. NBS1 and PGK1 mRNA levels were determined with real-time PCR. (D) U-2 OS cells were infected with the adenoviruses as above and examined with real-time PCR. An adenovirus expressing an N-terminal HIF-1α lacking the PAS-B domain, HIF-1α (1–167), was also included.
To test the requirement of HIF-α for NBS1 inhibition, we utilized small interfering RNA (siRNA) targeting HIF1A and EPAS1 (encoding HIF-2α). In Figure 1A, HIF1A siRNA abrogated the NBS1 and MSH2 downregulation, as well as the PGK1 upregulation in hypoxic cells, whereas EPAS1 siRNA showed no obvious effects. Likewise, NBS1 protein levels remained equivalent in hypoxic U-2 OS cells when HIF-1α protein expression was abolished (Figure 1B). Collectively, these findings suggest that HIF-1α, but not HIF-2α, is required for NBS1 repression by hypoxia.
Next, we asked whether HIF-1α is sufficient to inhibit NBS1 expression by infecting HCT116 cells with recombinant adenoviruses expressing a stable HIF-1α (Ad-HIF1αΔODD) (Huang et al, 1998; Koshiji et al, 2004). Results in Figure 1C show that forced expression of HIF1αΔODD reduced NBS1 mRNA levels by ∼60%. More importantly, two HIF-1α variants lacking functional transactivation domains, Ad-HIF1αΔODD (LCLL) (Gu et al, 2001; Koshiji et al, 2004) and Ad-HIF1α (1–329) (see Figure 4A), were also effective in NBS1 repression. By contrast, no inhibition of NBS1 was observed with the forced expression of HIF-2α, even though PGK1 was upregulated. Likewise, overexpression of the three HIF-1α variants in U-2 OS cells also lowered NBS1 mRNA levels by ∼50% (Figure 1D), but not with Ad-HIF1α (1–167) devoid of PAS-B (see Figure 4A). Similarly, Ad-HIF2α showed no obvious effect on NBS1 expression, but markedly stimulated the expression of HIF-2α specific target CITED2 (data not shown). It should be noted that although during viral replication adenoviral E4 inactivates the MRN complex (Stracker et al, 2002), the recombinant adenoviruses used here are replication-defective, and our results show specific inhibition of NBS1 by HIF-1α. Therefore, we conclude that HIF-1α, especially its N-terminal portion, is sufficient to mediate NBS1 repression by hypoxia.
Figure 4.
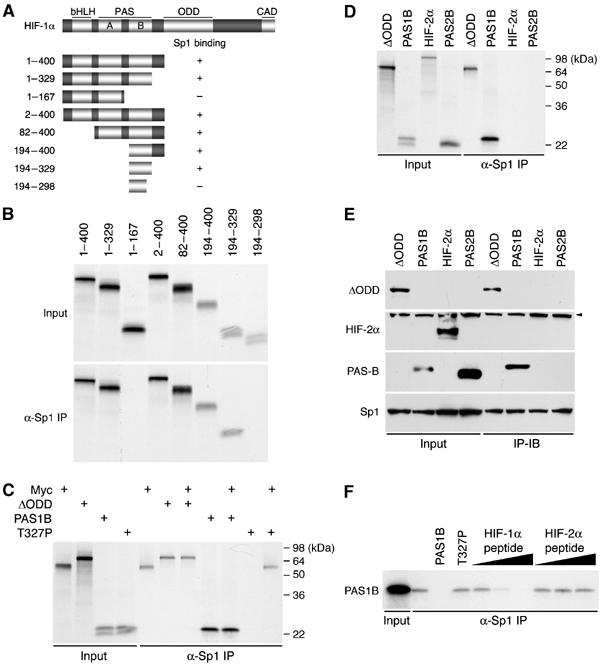
HIF-1α PAS-B differs from HIF-2α PAS-B in Sp1 binding. (A) A schematic representation of HIF-1α and its deletion mutants. Structural domains of HIF-1α (Huang and Bunn, 2003) are indicated at the top, and the corresponding residues on the left. Sp1-binding activity of each mutant is summarized. (B) The deletion mutants of N-terminal HIF-1α, as indicated, were translated in rabbit reticulocyte lysate with [35S]methionine and subjected to anti-Sp1 immunoprecipitations (α-Sp1). Input, 10% of lysates before immunoprecipitations. (C) HIF-1αΔODD (ΔODD), PAS1B, PAS1B T327P (T327P), and Myc were translated in vitro as above. Equal amount of the translated products were mixed as denoted and subjected to anti-Sp1 immunoprecipitations to determine their competitiveness for Sp1 binding. Molecular weight markers are indicated. (D, E) HIF-1αΔODD, PAS1B, HIF-2α, and PAS2B were translated in vitro as above and subjected to anti-Sp1 immunoprecipitations (D), or transfected into HeLa cells and immunoprecipitated with an anti-Sp1 antibody, followed by immunoblotting (IP-IB) with respective antibodies against specific proteins as indicated (E). To block PAS-B proteolysis, cells were treated with Cbz-LLL for 4 h before harvest. Input, 10% of total whole-cell extract subjected to direct immunoblotting. Arrowhead denotes nonspecific detection. (F) In vitro translated PAS1B was immunoprecipitated with anti-Sp1 antibody in the presence of unlabeled PAS1B or its T327P mutant. Addition of increasing amounts (10, 100, and 1000 pmol) of synthetic peptide corresponding to HIF-1α residues 299–329 competed with PAS1B for Sp1 binding.
Hypoxic repression of NBS1 is associated with the induction of DNA DSBs
To ascertain the functional relevance of NBS1 repression by hypoxia, we assessed the extent of DNA damage by immunofluorescent staining of γ-H2AX foci, a most sensitive method for quantifying DNA DSBs (Rogakou et al, 1998, 1999). In normoxia, U-2 OS cells displayed an average of two γ-H2AX foci per cell, whereas 72-h hypoxic treatment markedly increased the number by ∼3-fold (Figure S3; Table S1). In particular, some of the hypoxic nuclei exhibited numerous, intensified γ-H2AX foci. Cells treated with desferrioxamine, a hypoxia mimic agent, displayed a much greater increase of these intensified foci in a time-dependent manner (data not shown). Furthermore, results also show strikingly colocalized foci of γ-H2AX and 53BP1, the latter of which is known to interact with various DNA repair proteins including γ-H2AX in response to DSBs (Schultz et al, 2000). Thus, hypoxic cells experience DNA DSBs, presumably resulting from the NBS1 repression.
Given its ability to repress NBS1 expression, HIF-1α was tested for the induction of DNA DSBs. Similar to the effects of Ad-HIF-1αΔODD and Ad-HIF-1αΔODD (LCLL), Ad-HIF-1α (1–329) infection also gave rise to a significant increase in γ-H2AX foci, which were well colocalized with the 53BP1 foci (Figure 2; Figure S4; Table S1). Moreover, in accordance with the result above, further removal of PAS-B domain abolished the induction of these foci. Taken together, these results indicate that NBS1 repression by HIF-1α, HIF-1α (1–329) in particular, is associated with increased DNA DSBs.
Figure 2.
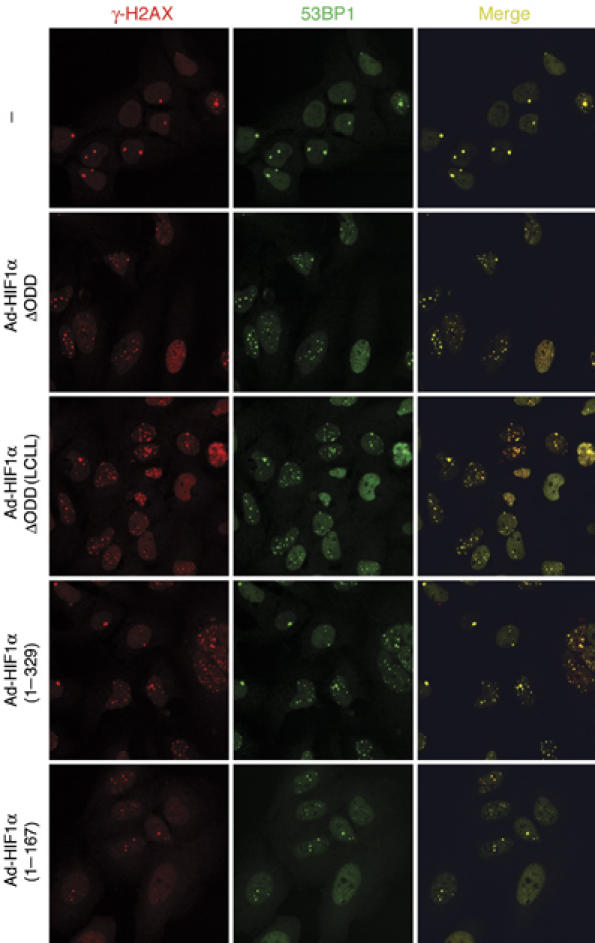
The N-terminal HIF-1α is sufficient to induce DNA DSB. U-2 OS cells were infected with adenoviruses expressing HIF-1α variants as indicated, and stained by immunofluorescence with antibodies against γ-H2AX (red) and 53BP1 (green). Representative fields are presented together with merged images. All of the HIF-1α variants, with the exception of Ad-HIF-1α (1–167), significantly increased the number of colocalized γ-H2AX and 53BP1 foci.
The HIF-1α–Myc pathway is responsible for NBS1 repression
Previously, we demonstrated that HIF-1α functionally counteracts the transcription factor Myc by altering Myc promoter occupancy (Koshiji et al, 2004, 2005). To test the relevance of Myc displacement in NBS1 repression, we performed chromatin immunoprecipitations to demonstrate the change in occupancy of the NBS1 locus (Matsuura et al, 1998) by the relevant transcription factors under hypoxia. Four sets of PCR primers were used, as illustrated in Figure 3A, to cover the NBS1 promoter from 1.3 kb upstream of the 5′ untranslated region to 75 base-pair downstream of the ATG start codon, as well as intron 1 harboring an E-box required for Myc-activated transcription (Chiang et al, 2003). Results in Figure 3B show that in addition to direct binding to the intron, Myc also bound under normoxia to a region clustered with five putative Sp1-binding sites spanned by the primer set P2. In stark contrast to the intron however, the P2 region showed a marked decrease in Myc binding under hypoxia, in concomitance with a gain of HIF-1α binding. It is noteworthy that no Sp1 and HIF-1α binding was detected in intron 1. Furthermore, unlike Myc displacement from the MSH2 promoter requiring wild-type p53 (Koshiji et al, 2005), no p53 binding was present in the scanned regions of NBS1. Thus, we conclude that HIF-1α selectively displaces Myc from the NBS1 promoter without affecting those bound to the E-box in intron 1.
Figure 3.
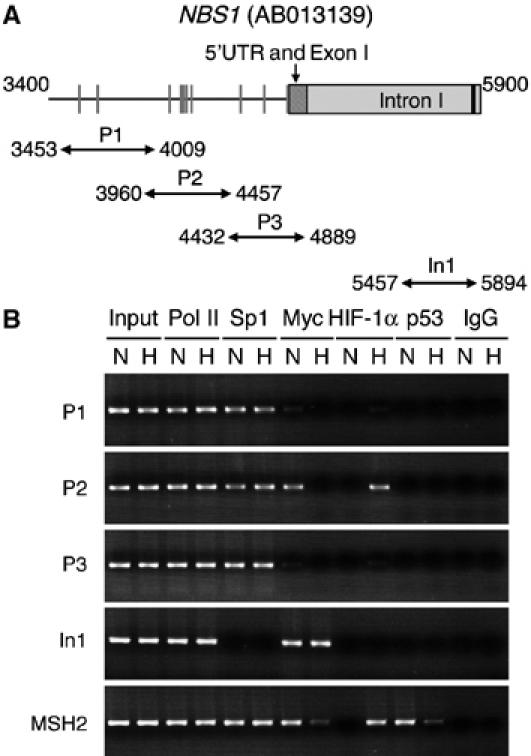
Selective Myc displacement in NBS1 gene under hypoxia. (A) A schematic representation of part of the human NBS1 is shown from nucleotides 3400–5900 of gene locus AB013139. The hatched box depicts 5′UTR and exon 1, and the gray box part of intron 1. The black bar specifies an E-box in the intron. Predicted Sp1 sites are in gray bars in the promoter. Four sets of PCR primers, designated as P1, P2, P3, and In1, were used for amplification of the NBS1 regulatory regions, as marked with double-arrow lines. (B) Chromatin immunoprecipitations were performed with normoxic (N) and hypoxic (H) U-2 OS cells. Antibodies used are against RNA polymerase II (Pol II), Sp1, Myc, HIF-1α, and p53. Normal immunoglobulin (IgG) served as a negative control, whereas the Pol II antibody served as a positive control. Sheared genomic DNA before immunoprecipitations was included as control of input DNA. The same immunoprecipitates were subjected to PCR amplification with four sets of NBS1 primers and one set of MSH2 primers (Koshiji et al, 2005). Myc displacement occurred in P2 but not in intron 1.
The PAS-B of HIF-1α, but not of HIF-2α, is essential for Sp1 binding
Previously, we showed that Myc displacement is mediated by the N-terminal HIF-1α (1–329), which competes with Myc for Sp1 binding in the MSH2 promoter (Koshiji et al, 2005). To identify the molecular basis that distinguishes HIF-1α from HIF-2α in DNA repair, we constructed a series of deletion mutants (Figure 4A) to narrow down the region responsible for Sp1 binding. These HIF-1α fragments were translated in vitro in a rabbit reticulocyte lysate system and tested for their binding to the rabbit reticulocyte Sp1. In accordance with our previous finding, deletion of PAS-B resulted in loss of Sp1 binding (Figure 4B). By contrast, deletion of the bHLH and PAS-A failed to do so, suggesting that PAS-B (codon 194–329) is sufficient for Sp1 interaction. Of note, removal of part of the PAS-B from the C terminus (codon 299–329) destroyed Sp1 binding.
To provide evidence that HIF-1α PAS-B is sufficient to compete with Myc for Sp1 binding, PAS-B and Myc were translated separately in vitro. As expected, in the absence of one another, Sp1 captured HIF-1α fragments or Myc (Figure 4C). However, when Myc was mixed with PAS-B or ΔODD, only the HIF-1α fragments, but not Myc, bound Sp1. Intriguingly, mutation of HIF-1α Thr-327 (T327P, see below) disabled PAS-B. Collectively, these results suggest that HIF-1α PAS-B is crucial for the HIF-1α–Myc pathway.
HIF-1α PAS-B shares 67% identity in amino-acid sequence with its HIF-2α counterpart, yet functionally HIF-2α differs strikingly in DNA repair. To that end, we tested whether HIF-2α PAS-B binds Sp1. For simplicity, we refer to the HIF-1α PAS-B hereinafter as PAS1B, and the HIF-2α PAS-B (codons 195–331) as PAS2B.
Interestingly, neither full-length HIF-2α nor PAS2B exhibited Sp1-binding activity when expressed in the rabbit reticulocyte lysate (Figure 4D). Likewise, when they were expressed ectopically in HeLa cells, no Sp1 binding was detected (Figure 4E). Moreover, neither PAS2B (data not shown) nor the PAS1B T327P mutant was able to compete with PAS1B for Sp1 binding (Figure 4F). Furthermore, addition of a synthetic peptide corresponding to HIF-1α residues 299–329 (required for Sp1 binding (Figure 4A)), but not of its HIF-2α counterpart, prevented Sp1 binding in a dose-dependent manner. Thus, we speculated that the functional divergence between HIF-1α and HIF-2α arises from limited differences in amino-acid sequence spanning from HIF-1α residues 299–329.
HIF-2α Pro-329 is responsible for abrogating Sp1 binding
Sequence analysis of HIF-1α residues 299–329 revealed that HIF-1α harbors two unique residues: Thr-301 and Thr-327, corresponding to Val-303 and Pro-329 in HIF-2α (Figure 5A). Hence, these two residues were replaced with those of HIF-2α. Interestingly, only the PAS1B (T327P) mutant lost Sp1 binding when expressed in the in vitro translation system (Figure 5B). Similar results were obtained when these substitutions were made in the context of HIF-1αΔODD (Figure 5C).
Figure 5.
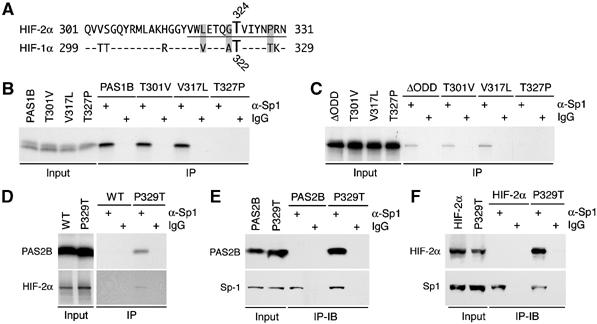
HIF-2α Pro-329 precludes Sp1 binding. (A) Sequence alignment of HIF-2α residues 301–331 with those of HIF-1α. A predicted phosphorylation motif at Thr-324 by PKD1 is underlined. Both HIF-2α Thr-324 and HIF-1α Thr-322 are specified. Shaded residues are unique in HIF-2α and critical for the phosphorylation motif. (B, C) T301V, V317L, and T327P mutants in the context of PAS1B (B) and HIF-1αΔODD (C) were translated in vitro as above and subjected to anti-Sp1 immunoprecipitations. (D) Wild-type (WT) HIF-2α and PAS2B, and their P329T mutants were analyzed as in (B). (E, F) P329T mutant in the context of PAS2B (E) and HIF-2α (F) were transfected into HeLa cells. Anti-Sp1 immunoprecipitations were performed, followed by immunoblotting to detect the co-immunoprecipitates as indicated.
Conversely, we substituted HIF-2α Pro-329 with threonine in the context of PAS2B and full-length HIF-2α. Results in Figure 5D–F show that in contrast to their original forms, these mutants gained Sp1 binding in both cell-free and cell-culture systems. Therefore, HIF-2α Pro-329 is responsible for abrogating Sp1 binding.
Phosphorylation of HIF-2α Thr-324 by PKD1 controls Sp1 binding
The gain of Sp1 binding by HIF-2α might be a result of substituted threonine, which is presumably subjected to modification required for Sp1 binding. To test this possibility, we compared PAS1B produced in rabbit reticulocyte lysate (modification-proficient) with the one from a wheat germ extract (modification-deficient). Unexpectedly, Sp1 captured PAS1B produced in both systems (Figure 6A), suggesting the irrelevance of threonine modification in Sp1 binding. In keeping with this, the PAS1B T327P mutant, disabled for Sp1 binding in rabbit reticulocyte, regained binding when produced from the wheat germ, and so did PAS2B (Figure 6B). These unexpected results indicted (i) no requirement of post-translational modification of HIF-1α for Sp1 binding; (ii) the nonessential role of HIF-1α Thr-327 for the binding; and most importantly (iii) the involvement of post-translational modification of HIF-2α that requires Pro-329 for abrogating Sp1 binding.
Figure 6.
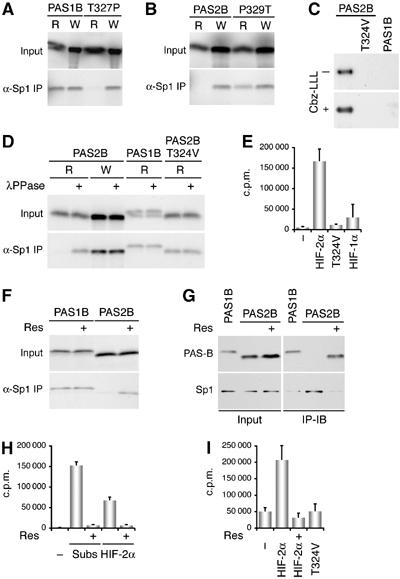
HIF-2α Thr-324 is phosphorylated by PKD1, thereby abrogating Sp1 binding. (A) PAS1B and its T327P mutant were translated in vitro in rabbit reticulocyte lysate (R) or in wheat germ extract (W). Their interactions with Sp1 were determined by anti-Sp1 immunoprecipitations. (B) PAS2B and its P329T mutant were analyzed as in (A). (C) PAS2B and its T324V mutant, as well as PAS1B were transfected into U-2 OS cells, and labeled with 32P-orthophosphate for 4 h. The expressed proteins were immunoprecipitated with anti-FLAG antibodies. The proteasome inhibitor Cbz-LLL was added (+) to ensure adequate PAS1B expression. (D) PAS-B variants, produced from rabbit reticulocyte lysate or wheat germ extract as indicated, were subjected to treatment with λ protein phosphatase (λPPase, 100 U for 30 min) before anti-Sp1 immunoprecipitations. (E) A synthetic peptide of HIF-2α containing the predicted PKD1 motif was used as a substrate of recombinant PKD1 in an in vitro kinase assay. Two additional synthetic peptides, one harboring mutation at HIF-2α Thr-324 and the other a HIF-1α equivalent, were included as controls. PKD1-mediated phosphorylation was presented in counts per minute (c.p.m.). (F) PAS1B and PAS2B were translated in rabbit reticulocyte lysate in the presence of resveratrol (Res, 100 μM). Their Sp1-binding activity was analyzed as above. (G) HeLa cells expressing PAS1B and PAS2B were treated with 5 nM of resveratrol. Anti-Sp1 immunoprecipitations were performed, followed by immunoblotting (IP-IB) as indicated. Cells were treated with Cbz-LLL for 4 h before harvest to increase the PAS-B protein levels. (H) Synthetic peptides containing a PKD1 consensus sequence (Subs) and the predicted PKD1 site in HIF-2α were tested, respectively, for phosphorylation by recombinant PKD1 in the presence or absence of resveratrol in an in vitro kinase assay. Recombinant PKD1 was preincubated with resveratrol (100 μM) for 10 min. (I) Immunoprecipitated endogenous PKD1 was used for in vitro kinase assays with synthetic peptides as indicated in the absence or presence of resveratrol.
To that end, the PAS2B sequence from codon 301–331 (Figure 5A) was analyzed by a motif-based searching algorithm (http://scansite.mit.edu/motifscan_seq.phtml) (Yaffe et al, 2001). Result from a high-stringency search suggested that HIF-2α Thr-324 is harbored within a phosphorylation motif for protein kinase D1 (PKD1), originally classified within the PKC family as PKCμ (Rykx et al, 2003). Although HIF-1α possesses an equivalent threonine (Thr-322), the algorithm indicates that the lack of a proline at Thr-327 plus the variation of Val-317 and Ala-321 precludes PKD1-mediated phosphorylation (Table S2). In essence, HIF-2α Pro-329 is obligatory for phosphorylation at Thr-324, in support of its role in abrogating Sp1 binding.
To ascertain phosphorylation of HIF-2α Thr-324, we performed in vivo 32P-orthophosphate labeling of ectopically expressed PAS1B and PAS2B. Results in Figure 6C show that PAS2B, but not PAS1B, was phosphorylated, and furthermore mutation of Thr-324 eliminated the phosphorylation. To demonstrate the effect of phosphorylation on Sp1 binding, we first treated PAS2B produced in the rabbit reticulocyte with λ protein phosphatase. Dephosphorylation rendered PAS2B able to bind Sp1 (Figure 6D). Furthermore, mutation of PAS2B Thr-324 to valine also established binding, arguing that dephosphorylation of HIF-2α Thr-324 is required for Sp1 binding. Hence, HIF-2α Thr-324 is phosphorylated in vivo, which functionally distinguishes PAS2B from PAS1B in Sp1 binding.
To determine that it is PKD1 that phosphorylates HIF-2α, we utilized recombinant PKD1 in an in vitro kinase assay. Figure 6E shows that a HIF-2α peptide, but not its HIF-1α counterpart, was robustly phosphorylated, whereas replacement of Thr-324 with valine abolished phosphorylation. Moreover, incubation with 100-μM resveratrol, a PKD1 inhibitor, during the synthesis of PAS2B in the rabbit reticulocyte established Sp1 binding (Figure 6F). Likewise, treatment of cells with resveratrol yielded PAS2B–Sp1 immunoprecipitated complexes (Figure 6G). Furthermore, resveratrol abrogated PKD1-mediated phosphorylation of its consensus substrate peptide as well as the HIF-2α peptide (Figure 6H), confirming the inhibitory effect of resveratrol on PKD1 kinase activity. Finally, endogenous PKD1 immunoprecipitated from 786-O cells specifically phosphorylated HIF-2α at Thr-324 (Figure 6I). In aggregate, these results argue that PKD1 phosphorylates HIF-2α Thr-324, thereby precluding Sp1 binding.
Nonphosphorylated PAS-B represses NBS1 expression and induces DNA DSBs
To determine the effect of PAS-B phosphorylation on NBS1 repression, we first created two mutants in PAS1B that mimic phosphorylation at Thr-322: one with glutamic acid substitution (T322E), and the other (VAT) undergoing simultaneous replacement of Val-317, Ala-321, and Thr-327 with the corresponding HIF-2α residues (Figure 5A), yielding an identical score for PKD1 phosphorylation as HIF-2α (Table S2). As expected, both T322E and VAT mutants lost Sp1 binding (Figure 7A). In contrast, a T322V mutation maintained PAS1B activity, indicating the nonessential role of Thr-322 per se for Sp1 binding. These findings further argue that maintaining HIF-1α Thr-322 or HIF-2α Thr-324 in a nonphosphorylated state is obligatory for Sp1 binding.
Figure 7.
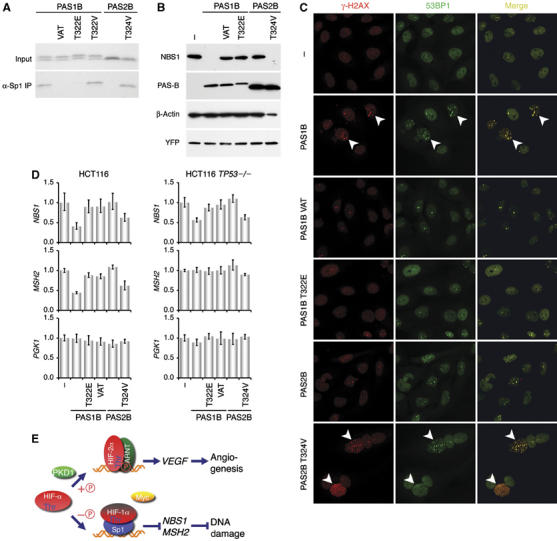
Nonphosphorylated PAS-B induces NBS1 repression and DNA DSBs. (A) PAS1B, PAS2B, and their mutants, as indicated, were translated in rabbit reticulocyte lysate and subjected to anti-Sp1 immunoprecipitations. (B) The PAS-B variants were expressed transiently in U-2 OS cells. Endogenous NBS1 and the transfected PAS-B were determined by sequential probing of the same blot with the corresponding antibodies, as indicated. (C) γ-H2AX and 53BP1 foci were determined by immunofluorescence as in Figure 2 in U-2 OS expressing the PAS-B variants. PAS1B and PAS2B T324V expression significantly increased γ-H2AX foci, as pointed by arrowheads. (D) The PAS-B variants were transfected into HCT116 and HCT116 TP53−/− cells. NBS1, MSH2 and PGK1 mRNA levels were determined with real-time PCR. Representative results are shown as in Figure 1. (E) A model depicts PKD1-mediated threonine (Thr) phosphorylation ( ) that differentiates the major role of HIF-2α from that of HIF-1α in the hypoxic response. Whereas nonphosphorylated HIF-1α competes with Myc for Sp1 binding, resulting in NBS1 and MSH2 repression and consequently DNA damage, phosphorylated HIF-2α primarily engages in the canonical hypoxia-responsive pathway.
) that differentiates the major role of HIF-2α from that of HIF-1α in the hypoxic response. Whereas nonphosphorylated HIF-1α competes with Myc for Sp1 binding, resulting in NBS1 and MSH2 repression and consequently DNA damage, phosphorylated HIF-2α primarily engages in the canonical hypoxia-responsive pathway.
Next, we asked how these PAS-B mutants relate to their abilities in NBS1 repression. Figure 7B shows that forced expression of PAS1B in U-2 OS cells was sufficient to eliminate NBS1 protein levels, whereas the PAS1B VAT and T322E mutants failed to do so. Conversely, the PAS2B T324V mutant, unlike PAS2B, gained its ability to inhibit NBS1 protein levels. Similar effects were observed at NBS1 mRNA levels with these PAS-B variants in both U-2 OS (Figure S5) and HCT116 cells (Figure 7D). Again, neither MRE11A nor RAD50 was affected (Figures S5 and S6). Interestingly, in TP53−/− HCT116 cells, although MSH2 expression was unaffected by PAS1B and PAS2B T324V, NBS1 mRNA levels were downregulated. This distinct difference is consistent with a p53-independent mechanism for NBS1 repression. Hence, in accordance with the requirement for Sp1 binding, these results underscore the fundamental difference between HIF-1α and HIF-2α in NBS1 repression, resulting from a unique threonine modification in the PAS-B.
To substantiate the role of threonine phosphorylation controlling NBS1 repression, we asked whether these phosphorylation mutants induce the formation of γ-H2AX foci. The results in Figure 7C show that the PAS-B fragments capable to repress NBS1 expression (PAS1B and PAS2B T324V mutant) significantly increased γ-H2AX foci that were colocalized with those of 53BP1. In contrast, those unable to affect NBS1 expression failed to do so. Taken together, these results strengthen the hypothesis that threonine phosphorylation in PAS-B is the molecular determinant that distinguishes HIF-1α from HIF-2α in DNA repair gene regulation.
Discussion
For all the remarkable advancement in recent years of knowledge of HIF-1α and HIF-2α in oxygen homeostasis, little is known about how these transcription factors, if indeed, are involved in genetic instability. Despite its similarities to HIF-1α in amino-acid sequence and protein structure, HIF-2α apparently has separate functions, presumably due to its distinct spatiotemporal expression pattern in vivo (Tian et al, 1998; Compernolle et al, 2002; Scortegagna et al, 2003). However, other studies indicate widespread expression of HIF-2α, along with HIF-1α, in various cell types (Wiesener et al, 1998; Talks et al, 2000). Furthermore, whether HIF-2α generally promotes or suppresses tumor growth remains a contentious issue (Blancher et al, 2000; Kondo et al, 2002; Maranchie et al, 2002; Acker et al, 2005; Covello et al, 2005; Raval et al, 2005). Recently, distinct HIF-1α and HIF-2α target genes have been reported (Hu et al, 2003; Covello et al, 2006), yet how the two isoforms selectively bind to their specific target genes for transcriptional activation remains obscure.
In pursuit of an in-depth understanding of hypoxic effects on DNA repair pathways, we show here that HIF-2α, in contrast to HIF-1α, is unable to participate in the HIF-1α–Myc pathway for repressing DNA repair genes, and that this functional difference stems from PKD1-mediated phosphorylation of HIF-2α Thr-324, thereby precluding HIF-2α from competing with Myc for Sp1 binding. Our findings argue PAS-B phosphorylation serves as a molecular determinant that governs the ability of HIF-α to impair DNA repair and distinguishes between HIF-1α and HIF-2α functionally (Figure 7E). Of note, PAS-B defined in this study lacks the last β-strand of the structural domain (Erbel et al, 2003), implying a nonessential role for a folded PAS-B in the HIF-1α–Myc pathway. Furthermore, we observed the induction of apoptosis by HIF-2α overexpression, as reported recently (Acker et al, 2005), along with increased γ-H2AX foci (Rogakou et al, 2000). Although the mechanism for HIF-2α triggered apoptosis and DNA fragmentation remains unknown, it is evident that no NBS1 downregulation is involved. Finally, our results show that the NBS1 inhibition by hypoxia and HIF-1α was apparently more effective at protein levels than at mRNA levels, and therefore alternative post-transcriptional mechanisms may also regulate NBS1 expression.
The identification of the PKD1–HIF-2α signaling pathway raises several unanswered questions. Although HIF-1α Thr-322 is not phosphorylated by PKD1, whether other kinases modify this threonine cannot be excluded thus far. Conversely, how HIF-2α phosphorylation is regulated warrants further investigation. PKD1 can be activated thorough PKC-dependent and -independent pathways by various agents including phorbol esters, oxidative stress, tumor necrosis factor α, and ATP, and has been associated with cell proliferation and survival (Rykx et al, 2003). It is particularly interesting to note that HIF-1α plays an essential role in maintaining intracellular ATP levels by stimulating glycolysis and curtailing ATP consumption (To et al, 2005). Furthermore, HIF-1 represses mitochondrial function and O2 consumption by inducing pyruvate dehydrogenase kinase 1 (Kim et al, 2006; Papandreou et al, 2006). Therefore, the maintenance of ATP concentration by HIF-1α might be a contributory factor for maintaining PKD1 activity and thereby HIF-2α phosphorylation. Consequently, HIF-2α tends to function in the canonical hypoxia-responsive pathway for cell proliferation and survival (Figure 7E).
We show here that HIF-1α, the PAS-B in particular, induces DNA DSBs, at least in part, by repressing NBS1 expression. Although previous studies by relying the less-sensitive comet assay indicated no DNA damage under hypoxia (Hammond et al, 2002, 2003), isolated hypoxic S-phase cells exhibited DNA damage when ATR activity was inactivated (Hammond et al, 2004), suggesting the existence of cellular mechanism in response to hypoxia-induced DNA damage. In keeping with this view, apoptosis-defective cells manifested salient genomic alterations including aberrant metaphases and polyploidy changes under hypoxia (Nelson et al, 2004), implicating that apoptosis is part of the hypoxic response to eliminate cells suffering an intolerable amount of DNA damage. Interestingly, the DNA damage response (e.g. by ionizing radiation) in mammalian cells comprises cell-cycle checkpoint activation, transcriptional response, and apoptosis, in addition to DNA repair to maintain genomic stability (Zhou and Elledge, 2000). Previously, we and others reported that HIF-1α induces cell-cycle arrest (Carmeliet et al, 1998; Goda et al, 2003; Koshiji et al, 2004; Mack et al, 2005). Given the involvement of NBS1 in cell-cycle checkpoints (D'Amours and Jackson, 2002), it is also interesting to ascertain whether the hypoxic effect on cell cycle is related to the DNA damage arising from hypoxic stress. To that end, the hypoxic response particularly during tumor development comprises activation of cell-cycle checkpoints, transcriptional activation of hypoxia-responsive genes, induction/avoidance of apoptosis, and impairment of DNA repair pathways in order to acquire genetic alterations necessary for tumor survival and progression (To et al, 2005).
In the name of cell survival, the hypoxic response presumably provides opportunities for genetic change. However, when and what type of genetic changes take place during tumor development is still an open question. Based on the observation that wild-type p53 is required for the hypoxic impairment of mismatch repair pathway, we surmised that hypoxic impairment of mismatch repair occurs during incipient tumorigenesis because majority of the developed cancers harbor mutated p53 (Koshiji et al, 2005). The current study however indicates that hypoxia also participates in tumor progression by inducing chromosomal instability. Although hypoxia-induced DNA DSBs may occur irrespective of the p53 status, whether cells can tolerate the resulting chromosomal aberrations largely depend on the biological integrity of the cells. Cells defective in p53 and apoptosis have a greater propensity to acquire genomic instability during tumor progression (Nelson et al, 2004).
Targeting HIF-α for therapeutics has generated enormous interests in recent years for both cancers and ischemic diseases by primarily focusing on inhibiting endogenous HIF-α expression and delivering stable HIF-α for overexpression (Giaccia et al, 2003; Semenza, 2003). Given the complex roles of HIF-α in adaptation to the hypoxic stress, understanding of the mechanisms of action—the bull's eye—is key to the successful development of specific drugs (Huang, 2004). Our studies have suggested that HIF-α PAS-B may represent a novel drug target, not only because of the identification of the molecular basis that functionally differentiates HIF-1α from HIF-2α but also the revelation of the PKD1–HIF-2α signaling pathway, which may provide a target of manipulation for functionally converting the HIF-α isoforms to one another for various purposes. Thus, these findings may spur further investigations into a better understanding of the mechanisms underlying HIF-α differential roles.
Materials and methods
Plasmids
A series of FLAG-tagged expression plasmids encoding various N-terminal fragments of HIF-1α and HIF-2α were constructed by PCR amplification. The PCR fragments were inserted in-frame into NotI- and XbaI-digested p3XFLAG-CMV10 (Sigma), and were subcloned into BamH1 and XbaI sites in pcDNA3 (Invitrogen). Site-directed mutagenesis was performed as described previously (Huang et al, 2002).
Immunofluorescence
Immunofluorescence staining was performed essentially as described previously (Rogakou et al, 1999) with mouse anti-γ-H2AX antibody (Upstate) and rabbit anti-53BP1 (Novus Biologicals). Secondary antibodies were anti-mouse Alexa-546 and anti-rabbit Alexa-488 (Molecular Probes). Samples were mounted in antifade media, and were examined by fluorescent and laser-scanning confocal microscopy with a Nikon PCM 2000.
In vivo 32P-orthophosphate labeling
U2OS cells were transfected with FLAG-tagged PAS1B, PAS2B, or PAS2B T324V mutant. At 48 h after transfection, cells were washed in phosphate-free DMEM, followed by incubation in phosphate-free DMEM for 10 min. Cells were then incubated at 37°C for 20 min in phosphate-free DMEM containing 100 μCi 32Pi (phosphorus-32 as orthophosphate in aqueous solution (HCl-free, carrier-free; 10 mCi/ml; Amersham). Subsequently, cells were washed three times with PBS and subjected to anti-FLAG immunoprecipitation. Ectopically expressed PASB was resolved by SDS–PAGE and subjected to autoradiography.
In vitro kinase assay
The procedure for the in vitro kinase assay was modified from a previous report (Storz et al, 2003). Briefly, recombinant PKD1 (Upstate Biotechnology) was used as the source of enzyme in 20 μl of kinase buffer (50 mM Tris/HCl, pH 7.4, 10 mM MgCl2, 2 mM dithiothreitol). The kinase reaction was carried out for 30 min at room temperature after addition of 10 μl of kinase substrate mixture (150 μM substrate peptide, 50 μM ATP, 10 μCi of [γ-32P]ATP in kinase buffer). A PKD1-specific substrate (Santa Cruz Biotechnology) was employed as a positive control. A peptide corresponding to HIF-1α residues 299–329 (Ac-GQVTTGQYRMLAKRGGYVWVETQATVIYN TKN-NH2), one including HIF-2α residues 314–331 (Ac-YGRKKRRQRRRGGGVWLETQGTVIYNPRN L-NH2), and another harboring HIF-2α T324V mutation (Ac-YGRKKRRQRRRGGGVWLETQGVVIYNPRN L-NH2) were synthesized by Quality Control Biochemicals. The reactions were terminated by adding 100 μl of 0.75% H3PO4, and 30 μl of the mixed supernatant was spotted to P-81 phosphocellulose paper (Whatman) in triplicates. Papers were washed thoroughly three times with 0.75% phosphoric acid, once with acetone, dried, and radioactivity incorporated into the synthetic peptide was determined in a scintillation counter. Endogenous PKD1 was immunoprecipitated with an anti-PKD1 antibody (SC-935; Santa Cruz) from 786-O cells lysed in a buffer containing 137 mM NaCl, 20 mM Tris (pH 7.5), 1 mM EGTA, 1 mM EDTA, 10% (v/v) glycerol, 1% (v/v) Nonidet P-40, 1 mM vanadate, and protease inhibitor cocktail (Roche). The immune complexes were washed three times with TBS (50 mM Tris/HCl, pH 7.4, 150 mM NaCl) and subjected to in vitro kinase assays.
Supplementary Material
Supplementary Information
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. We thank Shu-Chun Teng and Kou-Juey Wu for providing assistance in the NBS1 promoter analysis. LEH was an NCI Scholar of the National Institutes of Health.
References
- Acker T, Diez-Juan A, Aragones J, Tjwa M, Brusselmans K, Moons L, Fukumura D, Moreno-Murciano MP, Herbert JM, Burger A, Riedel J, Elvert G, Flamme I, Maxwell PH, Collen D, Dewerchin M, Jain RK, Plate KH, Carmeliet P (2005) Genetic evidence for a tumor suppressor role of HIF-2alpha. Cancer Cell 8: 131–141 [DOI] [PubMed] [Google Scholar]
- Bindra RS, Gibson SL, Meng A, Westermark U, Jasin M, Pierce AJ, Bristow RG, Classon MK, Glazer PM (2005) Hypoxia-induced down-regulation of BRCA1 expression by E2Fs. Cancer Res 65: 11597–11604 [DOI] [PubMed] [Google Scholar]
- Bindra RS, Glazer PM (2005) Genetic instability and the tumor microenvironment: towards the concept of microenvironment-induced mutagenesis. Mutat Res 569: 75–85 [DOI] [PubMed] [Google Scholar]
- Bindra RS, Schaffer PJ, Meng A, Woo J, Maseide K, Roth ME, Lizardi P, Hedley DW, Bristow RG, Glazer PM (2004) Down-regulation of Rad51 and decreased homologous recombination in hypoxic cancer cells. Mol Cell Biol 24: 8504–8518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blancher C, Moore JW, Talks KL, Houlbrook S, Harris AL (2000) Relationship of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha expression to vascular endothelial growth factor induction and hypoxia survival in human breast cancer cell lines. Cancer Res 60: 7106–7113 [PubMed] [Google Scholar]
- Carmeliet P, Dor Y, Herbert JM, Fukumura D, Brusselmans K, Dewerchin M, Neeman M, Bono F, Abramovitch R, Maxwell P, Koch CJ, Ratcliffe P, Moons L, Jain RK, Collen D, Keshet E (1998) Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature 394: 485–490 [DOI] [PubMed] [Google Scholar]
- Carney JP, Maser RS, Olivares H, Davis EM, Le Beau M, Yates JR III, Hays L, Morgan WF, Petrini JH (1998) The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: linkage of double-strand break repair to the cellular DNA damage response. Cell 93: 477–486 [DOI] [PubMed] [Google Scholar]
- Chiang YC, Teng SC, Su YN, Hsieh FJ, Wu KJ (2003) c-Myc directly regulates the transcription of the NBS1 gene involved in DNA double-strand break repair. J Biol Chem 278: 19286–19291 [DOI] [PubMed] [Google Scholar]
- Compernolle V, Brusselmans K, Acker T, Hoet P, Tjwa M, Beck H, Plaisance S, Dor Y, Keshet E, Lupu F, Nemery B, Dewerchin M, Van Veldhoven P, Plate K, Moons L, Collen D, Carmeliet P (2002) Loss of HIF-2alpha and inhibition of VEGF impair fetal lung maturation, whereas treatment with VEGF prevents fatal respiratory distress in premature mice. Nat Med 8: 702–710 [DOI] [PubMed] [Google Scholar]
- Coquelle A, Toledo F, Stern S, Bieth A, Debatisse M (1998) A new role for hypoxia in tumor progression: induction of fragile site triggering genomic rearrangements and formation of complex DMs and HSRs. Mol Cell 2: 259–265 [DOI] [PubMed] [Google Scholar]
- Covello KL, Kehler J, Yu H, Gordan JD, Arsham AM, Hu CJ, Labosky PA, Simon MC, Keith B (2006) HIF-2alpha regulates Oct-4: effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev 20: 557–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covello KL, Simon MC, Keith B (2005) Targeted replacement of hypoxia-inducible factor-1alpha by a hypoxia-inducible factor-2alpha knock-in allele promotes tumor growth. Cancer Res 65: 2277–2286 [DOI] [PubMed] [Google Scholar]
- D'Amours D, Jackson SP (2002) The Mre11 complex: at the crossroads of DNA repair and checkpoint signalling. Nat Rev Mol Cell Biol 3: 317–327 [DOI] [PubMed] [Google Scholar]
- Erbel PJ, Card PB, Karakuzu O, Bruick RK, Gardner KH (2003) Structural basis for PAS domain heterodimerization in the basic helix--loop--helix-PAS transcription factor hypoxia-inducible factor. Proc Natl Acad Sci USA 100: 15504–15509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frappart PO, Tong WM, Demuth I, Radovanovic I, Herceg Z, Aguzzi A, Digweed M, Wang ZQ (2005) An essential function for NBS1 in the prevention of ataxia and cerebellar defects. Nat Med 11: 538–544 [DOI] [PubMed] [Google Scholar]
- Friedberg EC (2003) DNA damage and repair. Nature 421: 436–440 [DOI] [PubMed] [Google Scholar]
- Giaccia A, Siim BG, Johnson RS (2003) HIF-1 as a target for drug development. Nat Rev Drug Discov 2: 803–811 [DOI] [PubMed] [Google Scholar]
- Goda N, Ryan HE, Khadivi B, McNulty W, Rickert RC, Johnson RS (2003) Hypoxia-inducible factor 1alpha is essential for cell cycle arrest during hypoxia. Mol Cell Biol 23: 359–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Milligan J, Huang LE (2001) Molecular mechanism of hypoxia-inducible factor 1alpha–p300 interaction. A leucine-rich interface regulated by a single cysteine. J Biol Chem 276: 3550–3554 [DOI] [PubMed] [Google Scholar]
- Hammond EM, Denko NC, Dorie MJ, Abraham RT, Giaccia AJ (2002) Hypoxia links ATR and p53 through replication arrest. Mol Cell Biol 22: 1834–1843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond EM, Dorie MJ, Giaccia AJ (2003) ATR/ATM targets are phosphorylated by ATR in response to hypoxia and ATM in response to reoxygenation. J Biol Chem 278: 12207–12213 [DOI] [PubMed] [Google Scholar]
- Hammond EM, Dorie MJ, Giaccia AJ (2004) Inhibition of ATR leads to increased sensitivity to hypoxia/reoxygenation. Cancer Res 64: 6556–6562 [DOI] [PubMed] [Google Scholar]
- Hu CJ, Wang LY, Chodosh LA, Keith B, Simon MC (2003) Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol Cell Biol 23: 9361–9374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang LE (2004) Targeting HIF-alpha: when a magic arrow hits the bull's eye. Drug Discov Today 9: 869. [DOI] [PubMed] [Google Scholar]
- Huang LE, Bunn HF (2003) Hypoxia-inducible factor and its biomedical relevance. J Biol Chem 278: 19575–19578 [DOI] [PubMed] [Google Scholar]
- Huang LE, Gu J, Schau M, Bunn HF (1998) Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin–proteasome pathway. Proc Natl Acad Sci USA 95: 7987–7992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang LE, Pete EA, Schau M, Milligan J, Gu J (2002) Leu-574 of HIF-1alpha is essential for the von Hippel–Lindau (VHL)-mediated degradation pathway. J Biol Chem 277: 41750–41755 [DOI] [PubMed] [Google Scholar]
- Kaelin WG (2005) Proline hydroxylation and gene expression. Annu Rev Biochem 74: 115–128 [DOI] [PubMed] [Google Scholar]
- Kim JW, Tchernyshyov I, Semenza GL, Dang CV (2006) HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 3: 177–185 [DOI] [PubMed] [Google Scholar]
- Kobayashi J, Tauchi H, Sakamoto S, Nakamura A, Morishima K, Matsuura S, Kobayashi T, Tamai K, Tanimoto K, Komatsu K (2002) NBS1 localizes to gamma-H2AX foci through interaction with the FHA/BRCT domain. Curr Biol 12: 1846–1851 [DOI] [PubMed] [Google Scholar]
- Kondo K, Klco J, Nakamura E, Lechpammer M, Kaelin WG (2002) Inhibition of HIF is necessary for tumor suppression by the von Hippel–Lindau protein. Cancer Cell 1: 237–246 [DOI] [PubMed] [Google Scholar]
- Koshiji M, Kageyama Y, Pete EA, Horikawa I, Barrett JC, Huang LE (2004) HIF-1alpha induces cell cycle arrest by functionally counteracting Myc. EMBO J 23: 1949–1956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshiji M, To KK, Hammer S, Kumamoto K, Harris AL, Modrich P, Huang LE (2005) HIF-1alpha induces genetic instability by transcriptionally downregulating MutSalpha expression. Mol Cell 17: 793–803 [DOI] [PubMed] [Google Scholar]
- Lengauer C, Kinzler KW, Vogelstein B (1998) Genetic instabilities in human cancers. Nature 396: 643–649 [DOI] [PubMed] [Google Scholar]
- Mack FA, Patel JH, Biju MP, Haase VH, Simon MC (2005) Decreased growth of Vhl−/− fibrosarcomas is associated with elevated levels of cyclin kinase inhibitors p21 and p27. Mol Cell Biol 25: 4565–4578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maranchie JK, Vasselli JR, Riss J, Bonifacino JS, Linehan WM, Klausner RD (2002) The contribution of VHL substrate binding and HIF1-alpha to the phenotype of VHL loss in renal cell carcinoma. Cancer Cell 1: 247–255 [DOI] [PubMed] [Google Scholar]
- Matsuura S, Tauchi H, Nakamura A, Kondo N, Sakamoto S, Endo S, Smeets D, Solder B, Belohradsky BH, Der Kaloustian VM, Oshimura M, Isomura M, Nakamura Y, Komatsu K (1998) Positional cloning of the gene for Nijmegen breakage syndrome. Nat Genet 19: 179–181 [DOI] [PubMed] [Google Scholar]
- Mihaylova VT, Bindra RS, Yuan J, Campisi D, Narayanan L, Jensen R, Giordano F, Johnson RS, Rockwell S, Glazer PM (2003) Decreased expression of the DNA mismatch repair gene Mlh1 under hypoxic stress in mammalian cells. Mol Cell Biol 23: 3265–3273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson DA, Tan TT, Rabson AB, Anderson D, Degenhardt K, White E (2004) Hypoxia and defective apoptosis drive genomic instability and tumorigenesis. Genes Dev 18: 2095–2107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC (2006) HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab 3: 187–197 [DOI] [PubMed] [Google Scholar]
- Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM (2000) A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol 10: 886–895 [DOI] [PubMed] [Google Scholar]
- Poellinger L, Johnson RS (2004) HIF-1 and hypoxic response: the plot thickens. Curr Opin Genet Dev 14: 81–85 [DOI] [PubMed] [Google Scholar]
- Pugh CW, Ratcliffe PJ (2003) Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med 9: 677–684 [DOI] [PubMed] [Google Scholar]
- Raval RR, Lau KW, Tran MG, Sowter HM, Mandriota SJ, Li JL, Pugh CW, Maxwell PH, Harris AL, Ratcliffe PJ (2005) Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel–Lindau-associated renal cell carcinoma. Mol Cell Biol 25: 5675–5686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou EP, Boon C, Redon C, Bonner WM (1999) Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol 146: 905–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou EP, Nieves-Neira W, Boon C, Pommier Y, Bonner WM (2000) Initiation of DNA fragmentation during apoptosis induces phosphorylation of H2AX histone at serine 139. J Biol Chem 275: 9390–9395 [DOI] [PubMed] [Google Scholar]
- Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM (1998) DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 273: 5858–5868 [DOI] [PubMed] [Google Scholar]
- Rykx A, De Kimpe L, Mikhalap S, Vantus T, Seufferlein T, Vandenheede JR, Van Lint J (2003) Protein kinase D: a family affair. FEBS Lett 546: 81–86 [DOI] [PubMed] [Google Scholar]
- Schultz LB, Chehab NH, Malikzay A, Halazonetis TD (2000) p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J Cell Biol 151: 1381–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scortegagna M, Ding K, Oktay Y, Gaur A, Thurmond F, Yan LJ, Marck BT, Matsumoto AM, Shelton JM, Richardson JA, Bennett MJ, Garcia JA (2003) Multiple organ pathology, metabolic abnormalities and impaired homeostasis of reactive oxygen species in Epas1−/− mice. Nat Genet 35: 331–340 [DOI] [PubMed] [Google Scholar]
- Semenza GL (2003) Targeting HIF-1 for cancer therapy. Nat Rev Cancer 3: 721–732 [DOI] [PubMed] [Google Scholar]
- Storz P, Doppler H, Johannes FJ, Toker A (2003) Tyrosine phosphorylation of protein kinase D in the pleckstrin homology domain leads to activation. J Biol Chem 278: 17969–17976 [DOI] [PubMed] [Google Scholar]
- Stracker TH, Carson CT, Weitzman MD (2002) Adenovirus oncoproteins inactivate the Mre11–Rad50–NBS1 DNA repair complex. Nature 418: 348–352 [DOI] [PubMed] [Google Scholar]
- Talks KL, Turley H, Gatter KC, Maxwell PH, Pugh CW, Ratcliffe PJ, Harris AL (2000) The expression and distribution of the hypoxia-inducible factors HIF-1alpha and HIF-2alpha in normal human tissues, cancers, and tumor- associated macrophages. Am J Pathol 157: 411–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauchi H, Kobayashi J, Morishima K, van Gent DC, Shiraishi T, Verkaik NS, vanHeems D, Ito E, Nakamura A, Sonoda E, Takata M, Takeda S, Matsuura S, Komatsu K (2002) Nbs1 is essential for DNA repair by homologous recombination in higher vertebrate cells. Nature 420: 93–98 [DOI] [PubMed] [Google Scholar]
- Tian H, Hammer RE, Matsumoto AM, Russell DW, McKnight SL (1998) The hypoxia-responsive transcription factor EPAS1 is essential for catecholamine homeostasis and protection against heart failure during embryonic development. Genes Dev 12: 3320–3324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- To KK, Koshiji M, Hammer S, Huang LE (2005) Genetic instability: the dark side of the hypoxic response. Cell Cycle 4: 881–882 [DOI] [PubMed] [Google Scholar]
- Varon R, Vissinga C, Platzer M, Cerosaletti KM, Chrzanowska KH, Saar K, Beckmann G, Seemanova E, Cooper PR, Nowak NJ, Stumm M, Weemaes CM, Gatti RA, Wilson RK, Digweed M, Rosenthal A, Sperling K, Concannon P, Reis A (1998) Nibrin, a novel DNA double-strand break repair protein, is mutated in Nijmegen breakage syndrome. Cell 93: 467–476 [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Kinzler KW (2004) Cancer genes and the pathways they control. Nat Med 10: 789–799 [DOI] [PubMed] [Google Scholar]
- Wenger RH (2002) Cellular adaptation to hypoxia: O2-sensing protein hydroxylases, hypoxia-inducible transcription factors, and O2-regulated gene expression. FASEB J 16: 1151–1162 [DOI] [PubMed] [Google Scholar]
- Wiesener MS, Turley H, Allen WE, Willam C, Eckardt KU, Talks KL, Wood SM, Gatter KC, Harris AL, Pugh CW, Ratcliffe PJ, Maxwell PH (1998) Induction of endothelial PAS domain protein-1 by hypoxia: characterization and comparison with hypoxia-inducible factor-1alpha. Blood 92: 2260–2268 [PubMed] [Google Scholar]
- Yaffe MB, Leparc GG, Lai J, Obata T, Volinia S, Cantley LC (2001) A motif-based profile scanning approach for genome-wide prediction of signaling pathways. Nat Biotechnol 19: 348–353 [DOI] [PubMed] [Google Scholar]
- Zhou BB, Elledge SJ (2000) The DNA damage response: putting checkpoints in perspective. Nature 408: 433–439 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information