

Abstract
Regulation of E2F1-mediated apoptosis is essential for proper cellular growth. This control requires TopBP1, a BRCT (BRCA1 carboxyl-terminal) domain-containing protein, which interacts with E2F1 but not other E2Fs and represses its proapoptotic activity. We now show that the regulation of E2F1 by TopBP1 involves the phosphoinositide 3-kinase (PI3K)–Akt signaling pathway, and is independent of pocket proteins. Akt phosphorylates TopBP1 in vitro and in vivo. Phosphorylation by Akt induces oligomerization of TopBP1 through its seventh and eighth BRCT domains. The Akt-dependent oligomerization is crucial for TopBP1 to interact with and repress E2F1. Akt phosphorylation is also required for interaction between TopBP1 and Miz1 or HPV16 E2, and repression of Miz1 transcriptional activity, suggesting a general role for TopBP1 oligomerization in the control of transcription factors. Together, this study defines a novel pathway involving PI3K–Akt–TopBP1 for specific control of E2F1 apoptosis, in parallel with cyclin–Cdk–Rb for general control of E2F activities.
Keywords: Akt, apoptosis, E2F1, Miz1, oligomerization, TopBP1
Introduction
The balance between proliferation and cell death is delicately controlled throughout growth and development. Central to control of proliferation is the Rb/E2F pathway. The Rb tumor suppressor protein controls cellular proliferation through its ability to bind and repress E2F activity. E2F is a family of transcription factors, including eight members, E2F1–E2F8 (Attwooll et al, 2004; Dimova and Dyson, 2005). E2Fs activate a large array of genes that encode proteins important for DNA replication and cell cycle progression. Although E2Fs are important for cell growth, de-regulated E2F1 is able to induce apoptosis. Among the E2F family members, E2F1 has the unique ability to trigger apoptosis when expressed in the absence of growth factors (Hallstrom and Nevins, 2003). Although E2F2 and E2F3 also induce apoptosis to a lesser extent than E2F1 in some experiments, their proapoptotic activity is mediated through transactivation of E2F1 (Lazzerini Denchi and Helin, 2005). Therefore, E2F1 possesses the unique ability to promote both proliferation and death, and these activities must be under tight control for proper cellular proliferation.
As the level of E2F1 surges during G1/S transition and S phase of each cell cycle, the proapoptotic activity of E2F1 must be blocked to allow cell cycle progression. The control of E2F1 apoptotic activity requires the action of the Ras–phosphoinositide 3-kinase (PI3K)–Akt pathway, because constitutively active Akt/protein kinase B (PKB) (referred hereafter as Akt) attenuates E2F1-induced apoptosis and a PI3K inhibitor abrogates serum-mediated suppression of E2F1 apoptosis (Hallstrom and Nevins, 2003). Nevertheless, the mechanism of this Akt-mediated control of E2F1 remains unclear. Recently, we have identified an E2F1-interacting protein, TopBP1 (topoisomerase II β-binding protein), that inhibits E2F1-dependent apoptosis during normal growth and DNA damage (Liu et al, 2003, 2004). Through its sixth BRCT domain, TopBP1 interacts with and represses E2F1 but not other E2F factors. The repression is mediated through recruitment of the Brg1/Brm chromatin-remodeling complex by TopBP1 to E2F1-responsive promoters, thereby repressing the transcriptional activity of E2F1. The interaction occurs not only after DNA damage but also during G1/S transition, and this regulation is crucial for the control of E2F1-dependent apoptosis during normal proliferation and DNA damage. The selectivity of TopBP1 toward E2F1, but not E2F2 or E2F3, would allow E2F2 and E2F3 to function and induce S-phase entry while E2F1 apoptosis is inhibited. Therefore, TopBP1 appears to be the key regulator for E2F1. Whether the PI3K–Akt signaling pathway controls E2F1 through TopBP1 is the subject of investigation in this study.
TopBP1 is an evolutionally conserved BRCT domain-rich protein that appears to be involved in DNA replication, DNA damage checkpoint response and transcriptional regulation (Garcia et al, 2005). Human TopBP1 contains eight BRCT domains, whereas its yeast homologs, Cut5/Rad4 (Schizosaccharomyces pombe) and Dpb11 (Saccharomyces cerevisiae), contain only four. The homology between TopBP1 and Cut5/Rad4 or Dpb11 lies at its BRCT domains 1–2, and 4–5. The interaction between Rad9 and the BRCT domains 4–5 of TopBP1 is believed to recruit TopBP1 to DNA damage sites and activate the Chk1 checkpoint response (Greer et al, 2003; Furuya et al, 2004). The additional carboxyl BRCT domains of metazoan TopBP1 are responsible for interactions with transcription factors, such as E2F1, Miz1 (Herold et al, 2002) and HPV16 E2 (Boner et al, 2002), suggesting that new functions of transcriptional regulation were acquired by metazoan TopBP1 during evolution. Miz1 mediates cell cycle arrest by transactivating p15INK4b (Seoane et al, 2001; Staller et al, 2001) and p21Cip1 (Herold et al, 2002; Seoane et al, 2002). TopBP1 interacts with and represses the transcriptional activities of Miz1. Thus, TopBP1 may have a more general role in transcriptional regulation.
Here, we show that TopBP1 is regulated by the PI3K–Akt pathway. Phosphorylation of TopBP1 by Akt kinase induces TopBP1 oligomerization and binding to E2F1. Phosphorylation by Akt is also required for TopBP1 to interact with other transcription factors, such as HPV16 E2 and Miz1, and repress Miz1 activity. These results link PI3K–Akt survival signaling with the control of E2F1-induced apoptosis and Miz1-mediated cell cycle arrest through regulation of TopBP1 oligomerization.
Results
Akt phosphorylates TopBP1 in vitro and in vivo, and induces the interaction between TopBP1 and E2F1
As E2F1-induced apoptosis is inhibited by the Akt pathway (Hallstrom and Nevins, 2003), we speculated that TopBP1-mediated E2F1 regulation might be regulated by Akt. Upon examination of the TopBP1 sequence, we identified a single Ser1159 residue lying within an optimal surrounding sequence for Akt phosphorylation (RXRXXS/T). Moreover, this site and its surrounding sequences (RARLAS1159) are completely conserved among frog, mouse, rat and human TopBP1 (Figure 1A). Thus, we hypothesized that Akt might phosphorylate TopBP1 at this site. Indeed, purified Akt effectively phosphorylated GST-TopBP1, which was produced and purified from Escherichia coli, and the phosphorylation was completely abrogated by mutation of Ser1159 to Ala (S1159A) (Figure 1B). This result demonstrates that Akt can directly phosphorylate TopBP1 at Ser1159 in vitro. To test whether endogenous TopBP1 was phosphorylated in vivo, we utilized a monoclonal antibody (110B7) specifically recognizing phospho-(Ser/Thr) Akt substrates. This antibody can specifically recognize the phosphorylated Ser1159 peptide, but not its unphosphorylated counterpart (Supplementary Figure 1A). It also specifically recognized pSer1159-TopBP1 on immunoblotting (Figures 2E, 5C and 6H). The endogenous TopBP1 was immunoprecipitated from HEK293 cells with a TopBP1-specific antibody and immunoblotted with this phospho-Akt substrate-specific antibody. As shown in Figure 1C, TopBP1 was recognized by this phospho-specific antibody in an Akt-dependent manner. Endogenous Akt was at least partially active in growing HEK293 cells, as it could be recognized by a phospho-Ser473-specific Akt antibody, a marker of Akt activity (Figure 1C, lower panels). When transfected with a dominant-negative Akt (dn-Akt) or treated with a PI-3 kinase inhibitor LY294002, phosphorylation of TopBP1 was inhibited (Figure 1C). This result strongly suggests that TopBP1 is phosphorylated by Akt in vivo. We further tested the response to physiological Akt activators. Phosphorylation of TopBP1 was greatly induced by insulin and serum (Figure 1D and E). Both insulin and serum also stimulated the interaction between endogenous TopBP1 and E2F1. To confirm that Ser1159 was the phosphorylation site in vivo, we expressed TopBP1 or S1159A mutant in the presence of dn-Akt or constitutively active Akt (ca-Akt) in HEK293 cells and performed metabolic labeling with 32P-orthophosphate. Phosphorylation of wild-type TopBP1 was much stronger in the presence of ca-Akt compared with that in dn-Akt, and importantly, mutation of Ser1159 of TopBP1 to Ala blocked Akt-dependent phosphorylation (Figure 1F). Consistent with the interaction between TopBP1 and E2F1 during G1/S (Liu et al, 2004), phosphorylation of TopBP1 by Akt also peaks at G1/S transition (Supplementary Figure 1B). Together, these results indicate that Akt phosphorylates TopBP1 at Ser1159 in vitro and in vivo, and induces the interaction between TopBP1 and E2F1.
Figure 1.
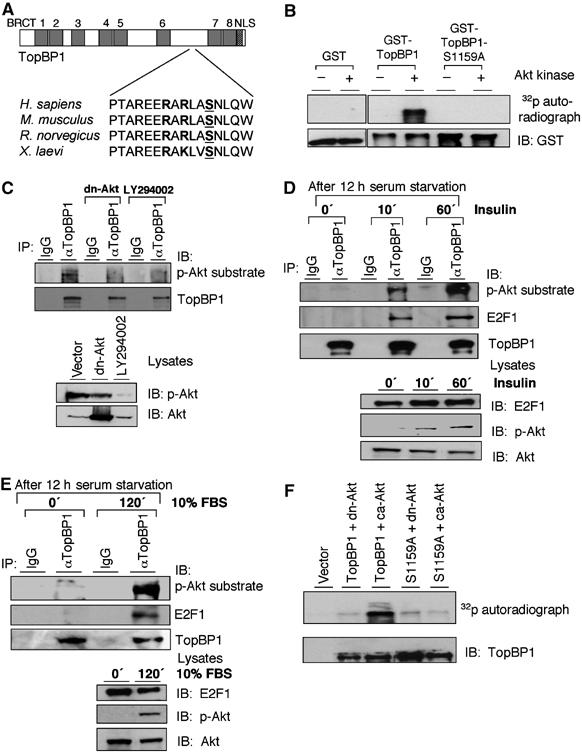
Akt phosphorylates TopBP1 in vitro and in vivo, and induces the interaction between endogenous TopBP1 and E2F1. (A) The RXRXXS surrounding Ser1159 is conserved among human, mouse, rat and frog TopBP1. (B) Purified GST, GST-TopBP1 or GST-TopBP1(S1159A) protein was incubated with recombinant Akt kinase in the presence of [γ-32P]ATP. GST proteins were then pulled down with glutathione-Sepharose followed by SDS–PAGE. The upper panel depicts the 32P autoradiograph and the lower panel shows an immunoblot with GST antibody. (C) HEK293 cells were either transfected with dn-Akt or treated with LY294002 (25 μM for 24 h) and lysates were immunoprecipitated (IP) with TopBP1 antibody (αTopBP1) or normal mouse IgG and immunoblotted (IB). An aliquot of input lysates was subject to SDS–PAGE and immunoblotted with antibodies for Akt or phospho-Akt (Ser473) (lower panel). (D) HEK293 cells were starved in serum-free medium for 12 h and then treated with insulin (500 nM) for 10 and 60 min. The cell lysates were immunoprecipitated with TopBP1 antibody or normal mouse IgG and immunoblotted. An aliquot of input lysates was subjected to SDS–PAGE and immunoblotted with antibodies for E2F1, Akt or phospho-Akt (Ser473) (lower panel). (E) HEK293 cells were starved in serum-free medium for 12 h and then stimulated by 10% FBS for 120 min. Immunoprecipitation and immunoblotting were performed as panel D. (F) HEK293 cells were trasnfected with FLAG-tagged TopBP1 or S1159A mutant, and dn-Akt or ca-Akt, as indicated. The cells were then metabolically labeled with 32P-orthophosphate followed by anti-FLAG beads immunoprecipitation and autoradiograph (upper panel) or immunoblotting (lower panel).
Figure 2.
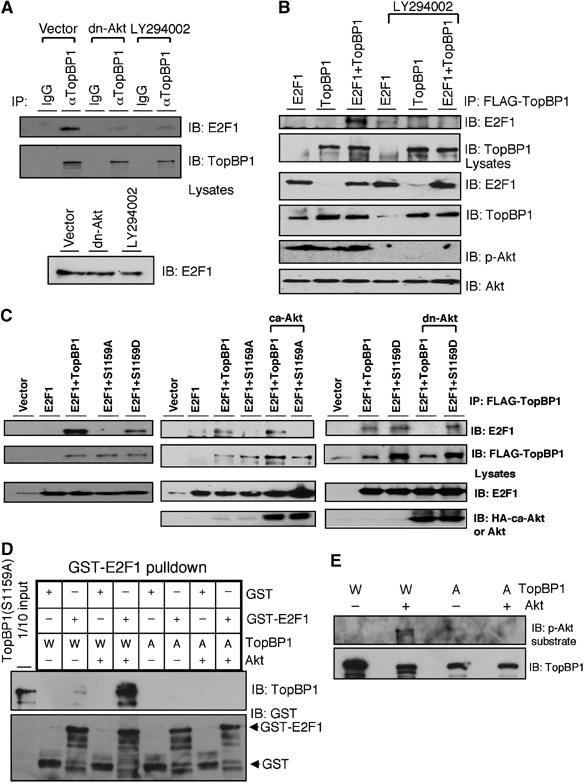
Akt phosphorylation is required for TopBP1 to interact with E2F1. (A) The membrane for endogenous TopBP1 immunoprecipitates shown in Figure 1C was striped and probed with an E2F1 antibody to detect the interaction between endogenous E2F1 and TopBP1. (B) HEK293 cells were transfected with E2F1 and FLAG-TopBP1 and treated with LY294002. The lysates were then immunoprecipitated with anti-FLAG beads followed by immunoblotting. The endogenous E2F1 was barely detectable in these films due to a relatively short exposure in detecting overexpressed proteins. (C) HEK293 cells were transfected with E2F1, FLAG-TopBP1 (either wild type, or S1159A or S1159D) and ca-Akt or dn-Akt, and the lysates were subjected to immunoprecipitation and immunoblotting as in panel A. (D) Akt phosphorylation induces the interaction between TopBP1 and E2F1 in vitro. Purified TopBP1(W: wild type) or TopBP1(A: S1159A) protein was first phosphorylated with Akt kinase and then incubated with purified GST or GST-E2F1. GST pulldown with glutathione-Sepharose was performed and probed with indicated antibodies. (E) The input TopBP1 protein after phosphorylation with Akt kinase in panel D was immunoblotted with phospho-Akt substrate antibody (110B7) or TopBP1 antibody.
Figure 5.
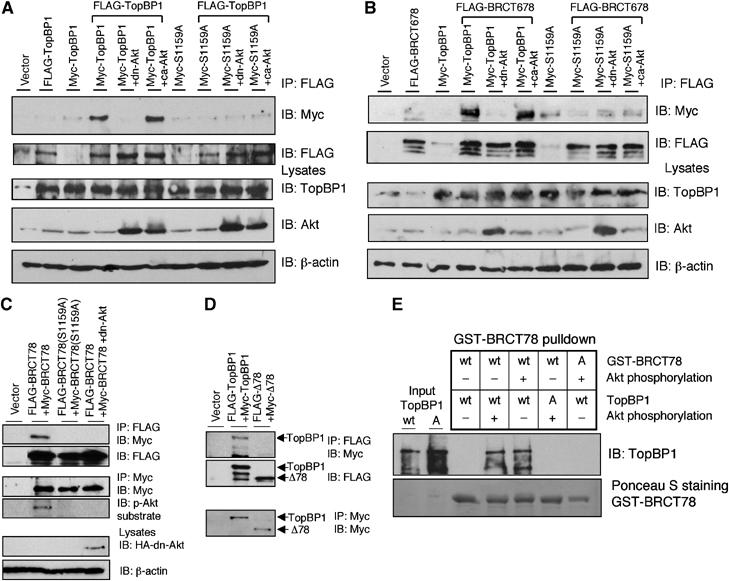
Akt-dependent oligomerization of TopBP1. (A) HEK293 cells were transfected with FLAG-TopBP1 along with Myc-TopBP1 or Myc-TopBP1(S1159A) (Myc-S1159A) as well as Akt plasmids. The lysates were then immunoprecipitated with anti-FLAG beads followed by immunoblotting as indicated. The overexpression of TopBP1 and dn-Akt in the input lysates is shown in lower panels. The ca-Akt, HA-myr(Δ4–129) Akt, cannot be detected by this Akt antibody, which recognizes the N-terminus of Akt, but can be detected by HA antibody (Figure 2C and data not shown). Transfection of ca-Akt may lead to increasing expression of endogenous Akt in some experiments. (B) HEK293 cells were transfected with FLAG-BRCT678 along with Myc-TopBP1 or Myc-TopBP1(S1159A) (Myc-S1159A) as well as Akt plasmids. The immunoprecipitation by anti-FLAG beads was then carried out as in panel A. (C) HEK293 cells were transfected with FLAG-BRCT78 and Myc-BRCT78 or their S1159A mutant counterparts, or with dn-Akt. The lysates were then immunoprecipitated with anti-FLAG beads followed by immunoblotting. The expression of Myc-BRCT78 or S1159A mutant was confirmed by immunoprecipitation and immunoblotting with anti-Myc (middle panels). Phosphorylation of Ser1159 in Myc-BRCT78 was detected by phospho-Akt substrate antibody. Total lysates were also immunoblotted directly to detect the expression of HA-Akt (lower panel). (D) HEK293 cells were transfected with FLAG-TopBP1 and Myc-TopBP1 or FLAG-TopBP1Δ78 (FLAG-Δ78) and Myc-TopBP1Δ78 (Myc-Δ78). The lysates were then immunoprecipitated with anti-FLAG beads followed by immunoblotting with Myc antibody (upper panel) and FLAG antibody (middle panel) or immunoprecipitated with anti-Myc beads followed by immunoblotting with Myc antibody (lower panel). (E) Akt phosphorylation induces self-association of TopBP1 in vitro. Purified TopBP1 was incubated with GST-BRCT78. Some TopBP1 or GST-BRCT78 was phosphorylated with recombinant Akt kniase as indicated before incubation. S1159A mutant (A) of TopBP1 or BRCT78 serves as a control for its wild-type (wt) counterpart. GST pulldown with glutathione-Sepharose was then performed and probed with TopBP1 antibody to detect the interaction between full-length TopBP1 and BRCT78. GST-BRCT78 was visualized by Ponceau S staining.
Figure 6.
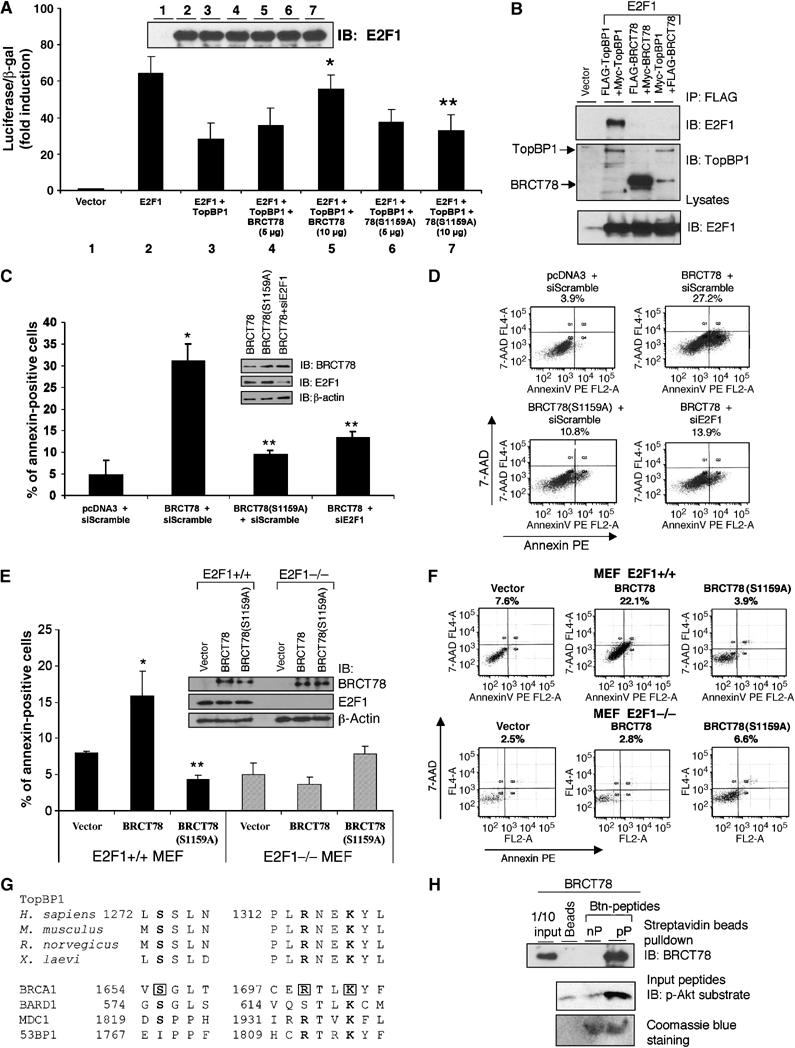
E2F1 binding and regulation require oligomerization of TopBP1. A TopBP1 dominant-negative mutant induces E2F1-dependent apoptosis. (A) E2F1 transcriptional activity was determined by p14ARF promoter-driven luciferase assay in the presence of TopBP1 and BRCT78 or BRCT78(S1159A) at different ratios. A TopBP1-expressing plasmid (10 μg) was cotransfected with plasmids expressing BRCT78 or BRCT78(S1159A) at 5 or 10 μg. E2F1 immunoblot from lysates of each transfection is shown on the top. (*) P<0.05 (t-test) compared with E2F1+TopBP1; (**) P<0.05 (t-test) compared with E2F1+TopBP1+BRCT78 (10 μg). (B) HEK293 cells were transfected with E2F1 plasmid along with TopBP1 or BRCT78 or TopBP1 and BRCT78 plasmids. The lysates were immunoprecipitated with anti-FLAG beads and immunoblotted with E2F1 antibody (upper panel) or TopBP1 antibody against the carboxyl terminus of TopBP1, which recognizes both TopBP1 and BRCT78 (middle panel). The input lysates were also immunoblotted with E2F1 antibody (lower panel). (C) HEK293 cells were transfected with the plasmid expressing TopBP1-BRCT78, or TopBP1-BRCT78(S1159A), and GFP along with pSUPER-siE2F1 or a control siRNA pSUPER-siScramble. Two days later, cells were stained with annexin V-PE and 7-AAD, and GFP-positive cells were gated and analyzed by flow cytometry. The data shown are the mean±s.e. of three independent experiments. An aliquot of cell lysates was analyzed by Western blot with BRCT78, E2F1 and β-actin antibodies. (*) P<0.001 (t-test) compared with pcDNA3+siScramble; (**) P<0.05 (t-test) compared with BRCT78+siScramble. (D) A representative profile of apoptosis as described in panel C. The percentage of annexin-positive cells is shown on top of each profile. (E) Wild-type and E2F1−/− MEFs were transfected with the plasmid expressing TopBP1-BRCT78, or TopBP1-BRCT78(S1159A), and GFP. Apoptosis was analyzed as described in panel C. The data shown are the mean±s.e. of three independent transfections. An aliquot of cell lysates was analyzed by Western blot with BRCT78, E2F1 and β-actin antibodies (upper panels). (*) P<0.05 (t-test) compared with pcDNA3 vector control; (**) P<0.05 (t-test) compared with BRCT78. (F) A representative profile of apoptosis as described in panel E. The percentage of annexin-positive cells is shown on top of each profile. (G) Sequence alignment of TopBP1 orthologs from human, mouse, rat and frog, and other BRCT repeats-containing proteins. The boxed sequences of BRCA1 represent the residues that form hydrogen bonds with pSer990 of BACH1 (Shiozaki et al, 2004). (H) GST-BRCT78 was produced in E. coli and purified with glutathione-Sepharose. The GST portion was removed by PreScission protease. Purified BRCT78 was incubated with Btn-pS1159 peptide (pP) or Btn-nonphosophorylated peptide (nP) followed by streptavidin Sepharose pulldown and immunoblotting with anti-TopBP1 carboxyl-terminus antibody. To ensure the loading of peptides, the input peptides were separated in 20% SDS–PAGE and immunoblotted with p-Akt substrate antibody or visualized by Coomassie blue staining.
Akt phosphorylation is required for TopBP1 to bind E2F1 and regulate E2F1 activity
Next we investigated the role of Akt phosphorylation in the interaction between TopBP1 and E2F1. The interaction between endogenous E2F1 and TopBP1 was inhibited by LY294002 or dn-Akt, as assayed by immunoprecipitation (Figure 2A). The interaction between transfected E2F1 and TopBP1 was also inhibited by LY294002 treatment or dn-Akt (Figure 2B and C). The S1159A mutant failed to interact with E2F1 even in the presence of ca-Akt (Figure 2C). The S1159D mutant of TopBP1 that mimics the phosphorylated form was able to interact with E2F1, although to a lesser extent when compared with wild type. However, its binding was no longer inhibited by the expression of dn-Akt (Figure 2C). The interaction of purified proteins was also tested in a cell-free system. Phosphorylation of TopBP1 at Ser1159 by a recombinant Akt kinase greatly induced its interaction with E2F1 in vitro (Figure 2D and E). Based on these results, we conclude that phosphorylation of TopBP1 by Akt at Ser1159 is required for TopBP1 to bind E2F1.
We then investigated whether Akt phosphorylation was required for TopBP1 to repress E2F1 activity by examining the endogenous E2F1 target genes. Previously, we showed that knockdown of TopBP1 by small interfering RNAs (siRNAs) de-repressed E2F1 target genes in T98G cells (Liu et al, 2004). TopBP1 knockdown in NIH3T3 cells by siRNA against murine TopBP1 also de-repressed E2F1 target genes such as p73, Apaf-1, caspase 3, cyclin E, thymidine kinase 1 (TK) and p107 (Figure 3A). The expression of TopBP1 was then reconstituted with wild-type or S1159A TopBP1 by recombinant adenoviruses that express human TopBP1 transcripts resistant to the siRNA against murine TopBP1 due to codon degeneracy. Importantly, reconstitution of wild-type TopBP1 but not S1159A mutant to physiological levels re-suppressed the expression of E2F1 target genes (Figure 3A). To further test the model that Akt controls E2F1 through TopBP1, we examined whether activated Akt could suppress the expression of E2F1 target genes after genotoxic stress and whether this effect depended on TopBP1. As shown in Figure 3B, the expressions of p73, Apaf-1, caspase 3, cyclin E, TK and p107 were induced by adriamycin, and expression of ca-Akt suppressed these gene expressions in a TopBP1-dependent manner. We cannot rule out the possibility that some of the effect from expression of ca-Akt may be p53-dependent; however, given the established role of E2F1 in controlling the expression of these target genes, particularly cyclin E, TK and p107, the observed effect most likely reflects E2F activity. Interestingly, the levels of cyclin D1 (Figure 3A and B) transcripts were not significantly changed by TopBP1 knockdown or Akt activation. As the cyclin D1 promoter is also directly regulated by E2F1 (Ohtani et al, 1995; Lee et al, 2000), this result suggests that TopBP1 may specifically regulate a subset of E2F1 target genes.
Figure 3.
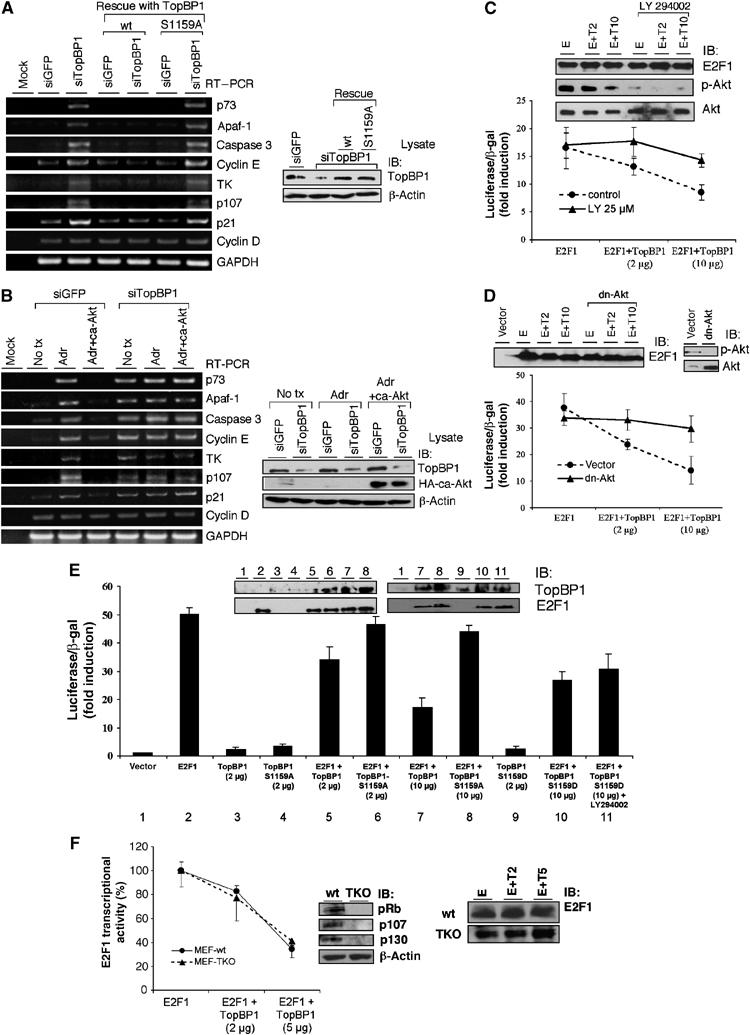
A physiological role of Akt phosphorylation of TopBP1 in the control of endogenous E2F1 target gene expression and E2F1 transcriptional activity. (A) NIH3T3 cells were transfected with a control siRNA vector (pSUPER-siGFP) or pSUPER-siTopBP1. Twenty-four hours later, the cells were infected with adenoviruses either harboring an empty vector or expressing TopBP1 or S1159A mutant TopBP1 at an m.o.i. of 400. Two days later, RNA was extracted and RT–PCR analysis was performed. Right panel: An aliquot of cell lysates was analyzed by Western blot with TopBP1 or β-actin antibody. (B) NIH3T3 cells were transfected with a control vector (pSUPER-siGFP) or pSUPER-siTopBP1, with or without coexpression of ca-Akt. Two days later, the cells were left untreated (no tx) or treated with adriamycin (Adr, 1 μM) for 16 h, and RT–PCR and Western blot analysis were performed as in panel A. (C) E2F1 activity was measured in HEK293 cells by p14ARF promoter-luciferase activity assay (Liu et al, 2003). The cells were either left untreated or treated with LY294002 for 18 h. Luciferase activity of transfected E2F1 was determined as fold induction relative to that of empty vector control. Each sample was performed in triplicate. The experiments were repeated multiple times with consistent results. One-tenth of each sample was subjected to Western blot analysis (upper panels, E: E2F1, T2: TopBP1 2 μg, T10: TopBP1 10 μg). (D) E2F1 activity was assayed in HEK293 cells with or without cotransfection of dn-Akt plasmid. (E) TopBP1, either wild type, S1159A or S1159D mutant, was cotransfected with E2F1 to REF52 fibroblasts. Some transfected cells were treated with LY294002 as in panel C. The E2F1 activity was then assayed. The expression levels of E2F1 and TopBP1 were determined by immunoblotting (upper panels). (F) The regulation of E2F1 by TopBP1 is independent of all pocket proteins. E2F1 transcriptional activity was assayed in wild-type (wt) MEFs or Rb−/−;p107−/−;p130−/− TKO MEFs. The activity of E2F1 in the presence of TopBP1 was determined as the percentage relative to that in the absence of TopBP1. An aliquot of each sample was subjected to E2F1 Western blot analysis (right panels, E: E2F1, T2: TopBP1 2 μg, T5: TopBP1 5 μg). The experiments were performed in two independent TKO MEFs with consistent results.
The regulation of E2F1 transcriptional activity by TopBP1 was also demonstrated by an E2F1 reporter assay. The ability of TopBP1 to repress E2F1 activity was blocked by LY294002 (Figure 3C) and dn-Akt (Figure 3D). Mutation of Ser1159 to Ala also blocked TopBP1's ability to repress E2F1 in both REF52 fibroblasts (Figure 3E) and HEK293 cells (Supplementary Figure 2). Consistent with the findings in co-immunoprecipitation experiments (Figure 2C), S1159D mutant also repressed E2F1, although to a lesser extent; nevertheless, its repressive effect was not significantly inhibited by LY294002 (Figure 3E).
Taken together, we conclude that Akt phosphorylates TopBP1 at Ser1159 to repress E2F1 transcriptional activity.
Regulation of E2F1 by TopBP1 is independent of all pocket proteins
Previous work showed that TopBP1 could repress E2F1 in Rb-deficient cells (Saos-2 and Rb−/− mouse embryonic fibroblasts (MEFs)) (Liu et al, 2004). To rule out the possibility that the other pocket proteins p107 and p130 may be involved, we examined the effect of TopBP1 on E2F1 transcriptional activity in Rb−/−;p107−/−;p130−/− triple knockout (TKO) MEFs (Sage et al, 2000). TopBP1 effectively repressed E2F1 activity both in wild-type and TKO MEFs (Figure 3F). Thus, the regulation of E2F1 by TopBP1 is distinct from the classic Rb family/E2F control.
Akt phosphorylation of TopBP1 is required for TopBP1 to repress E2F1-mediated apoptosis
We also examined the ability of TopBP1 mutants in repressing E2F1-induced apoptosis. Prior studies have shown that TopBP1 siRNA induced E2F1-dependent apoptosis in HEK293 cells and MEFs (Liu et al, 2004). TopBP1 knockdown in NIH3T3 cells also induced apoptosis, which was suppressed by reconstitution of wild-type TopBP1 expression (Figure 4A and B). Importantly, reconstitution with S1159A-TopBP1 failed to repress the apoptosis. This result is consistent with the examination of E2F1 target genes (Figure 3A), and strongly argues for a physiological role of Akt phosphorylation in the control of E2F1 proapoptotic function.
Figure 4.
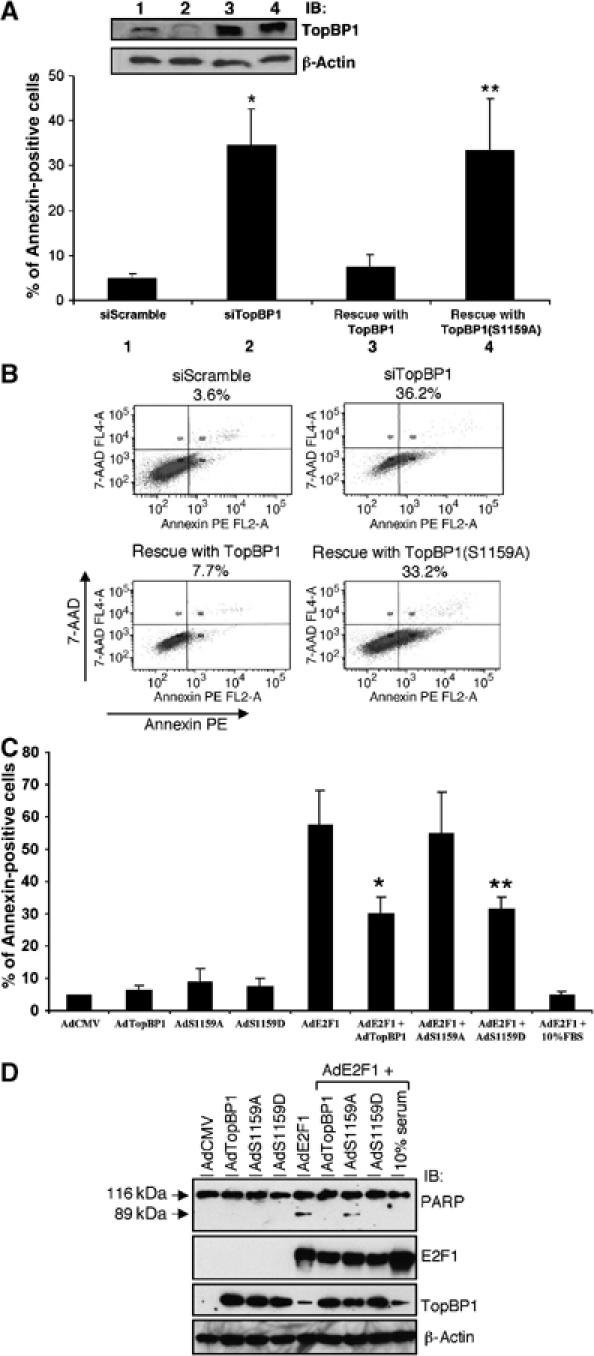
Akt phosphorylation of TopBP1 is required for TopBP1 to repress E2F1-mediated apoptosis under physiological conditions. (A) NIH3T3 cells were transfected with a control siRNA vector (pSUPER-siScramble) or pSUPER-siTopBP1 along with GFP-expressing plasmid. Twenty-four hours later, the cells were infected with adenoviruses either harboring an empty vector or expressing TopBP1 or S1159A mutant TopBP1 at an m.o.i. of 400. Two days later, cells were stained with annexin V-PE and 7-AAD, and GFP-positive cells were gated and analyzed by flow cytometry. The data shown are the mean±s.e. of three independent experiments. An aliquot of cell lysates was analyzed by Western blot with TopBP1 and β-actin antibodies (upper panels). (*) P<0.001 (t-test) compared with siScramble; (**) P<0.01 (t-test) compared with wild-type TopBP1 rescue. (B) A representative profile of apoptosis as described in panel A. The percentage of annexin-positive cells is shown on top of each profile. (C) REF52 cells were serum-starved and infected with AdE2F1 with/without AdTopBP1 or its mutants at an m.o.i. of 100. The cells were then grown in 0.25% serum for 3 days and apoptosis was scored by annexin V-PE/7-AAD staining. Some AdE2F1-infected cells were grown in 10% serum after infection. The data shown are the mean±s.e. of four independent experiments. (*) P<0.05 (t-test) compared with AdE2F1; (**) P<0.05 (t-test) compared with AdE2F1. (D) A parallel experiment as in panel C was performed except that the cellular lysates were harvested 2 days after infection. Immunoblottings were performed to ensure equal expression of E2F1 and TopBP1 among different samples. Presence of the 89 kDa proteolytic fragment of PARP is indicative of apoptosis.
The requirement of Akt phosphorylation for TopBP1 to control E2F1-induced apoptosis was further demonstrated by an independent assay. Infection of recombinant adenoviruses expressing E2F1 in serum-starved REF52 cells induces apoptosis, which is inhibited by coexpression of TopBP1 (Liu et al, 2003). We infected serum-starved REF52 cells with recombinant adenoviruses expressing E2F1 (AdE2F1) and either TopBP1 (AdTopBP1) or its mutants, and assessed apoptosis by annexin staining (Figure 4C and Supplementary Figure 3). Expression of E2F1 induced apoptosis in the absence of serum, which was largely abrogated by addition of 10% fetal bovine serum (FBS). Whereas AdTopBP1 (wild type) and S1159D mutant inhibited E2F1-induced apoptosis, AdTopBP1 (S1159A) failed to do so. Although the phosphorylation status of TopBP1 was not assayed in these cells, Akt is at least partially activated by AdE2F1 infection in these serum-starved REF52 cells according to phospho-Akt antibody immunoblotting (data not shown), a result consistent with a prior report that E2F1 can upregulate Akt activity (Chaussepied and Ginsberg, 2004). Similar results were obtained by measuring the proteolytic cleavage of poly(ADP-ribose) polymerase (PARP) (Figure 4D), an independent assay for apoptosis. Together, these results strongly support a requirement of Akt phosphorylation for TopBP1 to bind E2F1 and repress E2F1 activity.
Phosphorylation of TopBP1 by Akt is required for oligomerization of TopBP1
Akt phosphorylation can regulate the intracellular localization of certain proteins. Phosphorylation of p21, p27, CDC25B, Forkhead transcriptional factors, AHNAK and YAP by Akt results in retention or relocalization of these proteins in the cytosol (Franke et al, 2003). On the other hand, Akt phosphorylation can promote nuclear localization of Mdm2 (Mayo and Donner, 2001). To test whether Akt regulates TopBP1 localization, we examined the localization of TopBP1 in the presence of LY294002 or dn-Akt, or TopBP1(S1159A) mutant by fluorescence microscopy. We found that the nuclear localization of TopBP1 was not affected by LY294002 treatment, coexpression of dn-Akt or S1159A mutation (data not shown). Therefore, we conclude that Akt phosphorylation does not regulate the nuclear localization of TopBP1. We speculated that phosphorylation at Ser1159 by Akt might alter the structure of TopBP1 and stabilize the interaction between TopBP1 and E2F1. Serendipitously, we observed an interaction between full-length TopBP1 and the carboxyl terminus of TopBP1 containing the sixth, seventh and eighth BRCT domains (BRCT678) in a co-immunoprecipitation experiment. This result suggested an inter-molecular interaction between TopBP1 proteins. Structural analysis revealed that the interaction between BRCA1-BRCT domain and BACH1 phosphopeptide occurred at the junction of two BRCT repeats (Shiozaki et al, 2004). Therefore, we hypothesized that Akt phosphorylation might induce oligomerization of TopBP1 and bring two BRCT6 domains to proximity to stabilize E2F1 binding. Indeed, an interaction between two different TopBP1 molecules was observed and the interaction required Akt phosphorylation. As shown in Figure 5A, FLAG-tagged TopBP1 interacted with Myc-tagged TopBP1 and the interaction was blocked by dn-Akt or S1159A mutation. This result demonstrates that phosphorylation of TopBP1 by Akt at Ser1159 induces oligomerization. As Ser1159 is located between BRCT6 and BRCT7 domains, we speculated that the carboxyl BRCT6, BRCT7 and BRCT8 domains might be responsible for Akt-dependent self-association. TopBP1-BRCT678 indeed interacted with full-length TopBP1 in an Akt-dependent manner (Figure 5B). The domains responsible for self-association were further narrowed down to the carboxyl BRCT7 and BRCT8. FLAG-BRCT78 interacted with Myc-BRCT78 in an Akt-dependent manner (Figure 5C), whereas deletion of BRCT7 and BRCT8 abrogated their self-association (Figure 5D). To investigate whether Akt phosphorylation directly induced self-association of TopBP1, we examined the interaction between purified TopBP1 and BRCT78 proteins in vitro. The interaction between TopBP1 and BRCT78 was not seen in the absence of Akt phosphorylation, but was greatly induced upon phosphorylation by Akt (Figure 5E). Based on these results, we conclude that TopBP1 interacts with itself in an Akt-dependent manner through its BRCT7 and BRCT8 domains.
A truncated TopBP1-BRCT78 mutant serves as a dominant-negative inhibitor in preventing TopBP1 to interact with E2F1, and induces E2F1-dependent apoptosis
The observation that Akt regulated TopBP1 self-association and E2F1 interaction suggested that oligomerization of TopBP1 might be required for E2F1 binding and repression. To test this idea, we made use of a TopBP1 truncation mutant. TopBP1-BRCT78 lacks the E2F1-interacting BRCT6 domain, but is able to interact with full-length TopBP1; therefore, once TopBP1-BRCT78 binds full-length TopBP1, it may form nonfunctional complexes. As expected, TopBP1-BRCT78 inhibited full-length TopBP1's ability to repress E2F1 activity, whereas BRCT78(S1159A) did not (Figure 6A). We further provide evidence that it is a result of the formation of nonfunctional complexes. As shown in Figure 6B, full-length TopBP1 bound to E2F1, but TopBP1-BRCT78 failed to interact with E2F1. More importantly, formation of TopBP1/BRCT78 complexes prevented TopBP1 interaction with E2F1. Thus, we conclude that oligomerization of TopBP1 is essential for TopBP1 to bind and repress E2F1.
To further demonstrate the physiological role of Akt/TopBP1 in the control of E2F1-dependent apoptosis, we made use of the dominant-negative function of the BRCT78 fragment, which perturbs the interaction of TopBP1 and E2F1. Indeed, expression of BRCT78, but not the BRCT78 S1159A mutant, induced E2F1-dependent apoptosis in HEK293 cells (Figure 6C and D). We noticed a partial rescue effect obtained by the E2F1 siRNA. This could be a result of incomplete knockdown of E2F1 level by the E2F1 siRNA (Figure 6C, upper panels). Alternatively, BRCT78 could also induce apoptosis in an E2F1-independent manner. To investigate these possibilities further, we performed similar experiments in a pair of E2F1+/+ and E2F1−/− MEFs. Indeed, expression of BRCT78 induced apoptosis only in E2F1+/+, but not in E2F1−/− MEFs (Figure 6E and F); thus, we conclude that the apoptosis induced by BRCT78 is exclusively dependent on E2F1. The degree of apoptosis induced by BRCT78 in MEFs is less than that in HEK293. We observed a similar difference in the apoptosis induced by TopBP1 siRNA (Liu et al, 2004). This is likely to be a consequence of higher E2F1 activity residing in HEK293 cells.
Together with the experiments of TopBP1 reconstitution in NIH3T3 cells (Figures 3 and 4), these results strongly support the critical role of Akt/TopBP1 in controlling E2F1-mediated apoptosis under physiological conditions.
TopBP1-BRCT78 binds to a phosphoserine-1159-containing peptide
The BRCT repeats can bind phosphopeptides (Manke et al, 2003; Yu et al, 2003). A sequence alignment analysis between the BRCT motif of BRCA1 and TopBP1 revealed 34% similarity and 25% identity between BRCA1-BRCT and TopBP1-BRCT78. More importantly, the residues in the BRCT repeats of BRCA1 that make critical contact to phosphorylated Ser990 of BACH1 (Shiozaki et al, 2004) are conserved among TopBP1 of human, mouse, rat and frog origin (Figure 6G). Given the sequence homology between TopBP1-BRCT78 and the BRCT motif of BRCA1, as well as the phosphopeptide-binding activity of BRCA1 tandem BRCT domains, we postulated that phosphorylation of Ser1159 by Akt might create a docking site for BRCT78 and stimulate self-association. To test this, we synthesized a biotinylated peptide flanking Ser1159 (REERARLAS1159NLQWPS) and a biotinylated phosphopeptide (REERARLApS1159NLQWPS). We incubated purified BRCT78 proteins with biotinylated peptide or biotinylated phosphopeptide and pulled down the peptides with streptavidin-Sepharose followed by TopBP1 immunoblotting. As shown in Figure 6H, BRCT78 bound to a pS1159-containing phosphopeptide, but not nonphosphorylated S1159 peptide. This result supports a model that phosphorylation of Ser1159 creates a binding site for BRCT78 domains and induces TopBP1 oligomerization.
Akt phosphorylation is also required for TopBP1 to interact with HPV16 E2 and Miz1, but not Rad9 or topoisomerase IIβ
In addition to E2F1, TopBP1 also interacts with several other proteins, such as HPV16 E2, Miz1, topoisomerase IIβ (Yamane et al, 1997) and Rad9 (Makiniemi et al, 2001; Greer et al, 2003). As Akt phosphorylation regulates the structure of TopBP1, it may also regulate the interactions with other proteins and therefore represent a more general control of TopBP1 function. To investigate this hypothesis, we performed co-immunoprecipitation assays to investigate the interaction between TopBP1 and these proteins. Consistent with other reports, we were able to observe the interaction between TopBP1 and HPV16 E2, Miz1, topoisomerase IIβ or Rad9 by co-immunoprecipitation (Figure 7A–C and Supplementary Figures 4 and 5). TopBP1(S1159A) mutant also interacted with topoisomerase IIβ and Rad9 equally well as wild-type TopBP1 (Supplementary Figures 4 and 5), suggesting that these interactions do not require oligomerization or phosphorylation by Akt. In contrast, mutation of S1159A diminished the interaction between TopBP1 and HPV16 E2 or Miz1 (Figure 7A–C). These results suggest that Akt phosphorylation also regulates the function of TopBP1 in controlling HPV16 E2 and Miz1. We further tested whether S1159A mutation blocked the ability of TopBP1 to control Miz1 activity. Miz1 has been shown to bind directly to the p21Cip1 promoter and activate its transcription, whereas the binding of TopBP1 to Miz1 inhibits this function (Herold et al, 2002; Wanzel et al, 2005). We utilized a p21 promoter-driven luciferase assay to measure the transcriptional activity of Miz1. Consistent with previous reports (Herold et al, 2002; Wanzel et al, 2005), we observed the transcriptional activity of Miz1 on the p21 promoter construct and repression of Miz1 activity by TopBP1. Importantly, S1159A mutation blocked the ability of TopBP1 to repress Miz1 transcriptional activity (Figure 7D). We also examined the expression of endogenous p21 in the TopBP1- or S1159A-reconstituted NIH3T3 cells described in Figure 3A and B. TopBP1 siRNA de-repressed the expression of p21, and repression of p21 by TopBP1 required Ser1159 (Figure 3A). Akt also inhibited p21 expression in a TopBP1-dependent manner (Figure 3B). These results demonstrate the role of Akt/TopBP1 in the regulation of p21. The regulation of p21 could be mediated through either Miz1 or p53 (Herold et al, 2002). The role of Miz1 in this control is further supported by the fact that TopBP1 siRNA also de-represses p21 expression in a p53-mutant cell line T98G, either during normal growth or upon DNA damage (Figure 7E). These data suggest a role for Akt to regulate TopBP1 in the control of Miz1 activity.
Figure 7.
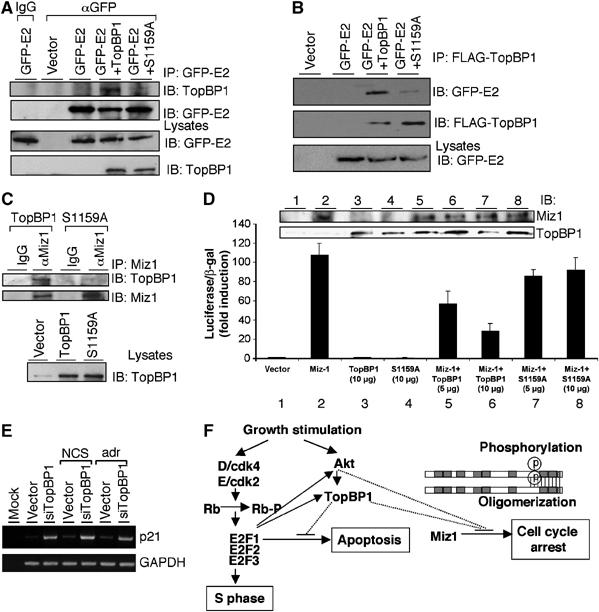
Akt phosphorylation is also required for TopBP1 to interact with HPV16 E2 and Miz1, and regulate Miz1 function. (A) HEK293 cells were transfected with expression plasmids for GFP-E2 and/or TopBP1 and the lysates were immunoprecipitated with either a GFP monoclonal antibody (αGFP) or normal mouse IgG, followed by immunoblotting. An aliquot of input lysates was immunoblotted directly (lower panels). (B) HEK293 cells were transfected with expression plasmids for GFP-E2 and/or FLAG-TopBP1, and lysates were immunoprecipitated with anti-FLAG beads followed by immunoblotting with indicated antibodies. (C) HEK293 cells were transfected with expression plasmids for TopBP1 or S1159A mutant and the lysates were immunoprecipitated with a goat polyclonal anti-Miz1 antibody (αMiz1) or normal goat IgG followed by immunoblotting. An aliquot of input lysates was immunoblotted with TopBP1 antibody to demonstrate equal expression of wild-type and S1159A TopBP1 (lower panel). (D) HEK293 cells were transfected with a p21 promoter-luciferase plasmid and pCMV-β-gal for Miz1 activity reporter assay. Miz1 was coexpressed with TopBP1 or S1159A mutant at different ratios. Luciferase activity of transfected Miz1 was determined as fold induction relative to that of empty vector control. Each sample was performed in triplicate. (E) The RNA extraction from TopBP1 siRNA-transfected T98G cells was described previously (Liu et al, 2004). Briefly, T98G cells were transfected with an empty vector or pSUPER-siTopBP1. Two days later, cells were either left untreated or treated with neocarzinostatin (NCS) at 300 ng/ml or adriamycin at 1 μM for 5 h. RNA was then extracted and RT–PCR analysis for p21 and GAPDH was performed. The RT–PCR analysis for E2F target genes from these samples has been presented (Liu et al, 2004). (F) A model for the role of Akt–TopBP1 in the control of apoptosis through regulation of E2F1 and in the control of cell cycle arrest through regulation of Miz1.
Discussion
The PI3K–Akt is a key regulator in mediating cell survival (Franke et al, 2003). We have now delineated a novel pathway linking PI3K–Akt with a BRCT domain protein TopBP1 for the control of E2F1 and Miz1 activities. Through the Akt–TopBP1 pathway, both E2F1-induced apoptosis and Miz1-induced cell cycle arrest can be controlled to ensure progression of normal proliferation. Moreover, a new mechanism of regulation of TopBP1 function through Akt-dependent oligomerization has also been elucidated.
Akt–TopBP1 pathway in the control of E2F1-mediated apoptosis
We provide compelling evidence that Akt phosphorylates TopBP1 at a single residue Ser1159. This phosphorylation event is necessary for TopBP1 to repress the transcriptional and proapoptotic activities of E2F1 under physiological conditions. We also demonstrate that regulation of E2F1 by TopBP1 is independent of all pocket proteins. Taken together, we now propose a pathway involving Akt–TopBP1 in parallel to the classical Cdk–Rb pathway (Figure 7F) for the control of E2F1-dependent apoptosis. In response to growth factor stimulation, activation of Cdk leads to pRb phosphorylation and release of free E2F to induce S-phase entry. Activation of PI3K–Akt is important to hold in check the proapoptotic activity of E2F1 through regulation of TopBP1 during G1/S transition. To ensure proper control of apoptosis, there exist several feedback loops. TopBP1 in fact is an E2F target and its level peaks at G1/S and S phases (Liu et al, 2004; Yoshida and Inoue, 2004). E2F also activates Gab2 and upregulates Akt activity (Chaussepied and Ginsberg, 2004). During S phase, E2F1 activity is inhibited by cyclin A/Cdk2 (Krek et al, 1994; Xu et al, 1994). Together, these mechanisms provide a multi-layered regulation to control E2F1-mediated apoptosis during cellular proliferation.
The Akt–TopBP1 pathway may control E2F1 during DNA damage as well. The interaction between TopBP1 and E2F1 is induced upon phosphorylation of E2F1 at Ser31 by ATM (Liu et al, 2003). TopBP1 can be phosphorylated by ATM (Yamane et al, 2002) and the level of TopBP1 protein is induced by the radiomimetic agent neocarzinostain (NCS) (Liu et al, 2003). Ionizing radiation activates Akt (Contessa et al, 2002; Viniegra et al, 2005), which was confirmed in our hands by NCS treatment (data not shown). ATM mediates the phosphorylation of Akt at Ser473 and contributes to activation of Akt in response to ionizing radiation (Viniegra et al, 2005). Activation of Akt during adriamycin treatment inhibits the expression of E2F1 target genes through TopBP1 action (Figure 3B). Hence, in response to ionizing radiation, ATM kinase may target the Akt–TopBP1 pathway to control E2F1-mediated apoptosis and allow DNA repair to complete.
Akt–TopBP1 pathway in the control of HPV 16 E2 and Miz1
The interaction between TopBP1 and HPV16 E2 or Miz1 requires the Akt phosphorylation site as well. These results suggest that Akt–TopBP1 also regulates other biological processes. HPV16 E2 is essential for the life cycle of HPVs. It regulates transcription of the viral genome. It also interacts with viral replication factor E1 and recruits E1 to replication origins for viral replication. TopBP1 interacts with HPV16 E2 and enhances its ability to activate transcription and replication (Boner et al, 2002). During HPV infection, HPV E7 targets members of the pocket protein family such as pRb to release E2Fs. It also activates Akt through an interaction with PP2A, preventing dephosphorylation of activated Akt (Pim et al, 2005). Thus, the activation of Akt by E7 could regulate TopBP1 and enhance E2-mediated activation of viral transcription and replication.
Miz1 is a Myc-associated zinc-finger transcription factor. Miz1 is involved in cell cycle arrest through its activity to transactivate p15INK4b and p21Cip1 (Wanzel et al, 2003). Myc interacts with Miz1 and represses transcriptional activation by Miz1, at least in part through competition with p300 for binding to Miz1 (Staller et al, 2001). The interaction with Miz1 plays a crucial role in mediating transcriptional repression by Myc. TopBP1 interacts with Miz1 through its BRCT7 and BRCT8 domains, and represses Miz1 transcriptional activity (Herold et al, 2002). Our data further suggest that phosphorylation by Akt is required for TopBP1 to bind and repress Miz1. In addition to the control of Miz1 by Akt–TopBP1, Akt also phosphorylates Miz1 directly; this phosphorylation leads to binding of 14-3-3η to the Miz1 DNA binding domain and inhibits Miz1 function (Wanzel et al, 2005). Therefore, Akt regulates Miz1 to control cell cycle arrest directly through phosphorylation of Miz1 as well as indirectly through phosphorylation of TopBP1 (Figure 7F). The 14-3-3η-mediated and TopBP1-mediated inhibition of Miz1 are likely two alternative processes, as there is no detectable interaction between TopBP1 and 14-3-3 to suggest a trimeric Miz1/TopBP1/14-3-3 complex formation (Supplementary Figure 6). The inhibition of p21 expression during adriamycin treatment by ca-Akt is abolished by TopBP1 siRNA (Figure 3B), suggesting that the regulation of Miz1 by Akt, at least in the control of p21 during DNA damage, is mainly mediated by TopBP1. A role of Miz1 for G1 arrest has been demonstrated in the context of DNA damage (Seoane et al, 2002; Wanzel et al, 2005) and TGFβ signaling (Seoane et al, 2001). The regulation of Miz1 activity by Akt–TopBP1 suggests that Miz1 may be under constant repression by Akt–TopBP1 during normal proliferation, similar to the control for E2F1-mediated apoptosis. The hypothesis is supported by the fact that Akt–TopBP1 is required to control the expression of p21, one of the Miz1 target genes, during normal growth (Figures 3A and 7E). This may explain the lack of measurable effect on gene expression in unstressed cells by depletion of Miz1 (Wanzel et al, 2005).
Akt phosphorylation regulates oligomerization of TopBP1 protein
Akt generally regulates its substrates by altering their enzymatic activity or increasing the affinity of the substrates for 14-3-3 proteins, thereby retaining phosphorylated substrates in the cytosol. The nuclear localization of TopBP1 is not regulated by Akt, nor can we detect the interaction between 14-3-3 and TopBP1 in growing HEK293 cells (Supplementary Figure 6). We present evidence that phosphorylation of TopBP1 by Akt leads to direct self-association of TopBP1 BRCT domains. To our knowledge, this represents the first example that phosphorylation by Akt induces protein oligomerization as a regulatory mechanism. Considering that BACH1 phosphopeptide binds to the BRCT repeats of BRCA1 through specific interactions with a surface cleft at the junction of the two BRCT repeats (Shiozaki et al, 2004), the TopBP1 BRCT6 domain alone may not be sufficient to foster stable interaction between TopBP1 and E2F1. The oligomerization of TopBP1 could bring two BRCT6 to proximity to stabilize its interaction with E2F1. It is not clear at this moment whether TopBP1 forms as dimers, trimers, tetramers or higher-order multimers within the cells. It would be very informative to analyze the multimeric structure of the TopBP1 complex. The structural basis for the role of phosphorylated Ser1159 in TopBP1 self-association is also a very interesting question. The phosphopeptide-binding activity of BRCT78 toward a pSer1159-peptide suggests a model that phosphorylation of Ser1159 creates a binding site for the BRCT78 of other TopBP1 molecules and induces oligomerization.
Although TopBP1 is involved in several aspects of DNA metabolism, protein oligomerization appears to be specifically required for its interaction with transcription factors. Given the conservation of Ser1159 and its surrounding sequence in metazoan TopBP1, the Akt-dependent oligomerization is likely conserved as well. The carboxyl oligomerization domain of TopBP1 is not found in yeast homologs. Thus, it appears that both oligomerization and the function of transcriptional regulation were acquired in metazoan TopBP1 during evolution. It is tempting to speculate that Akt might selectively regulate the transcriptional function of TopBP1. Although the Akt phosphorylation site is not required for its binding to Rad9 or topoisomerase IIβ, it would be very interesting to test whether oligomerization of TopBP1 is involved in checkpoint activation or DNA replication. It is worth noting that the interaction between TopBP1 and Rad9 depends on the fourth and fifth BRCT motifs, and does not require the carboxyl oligomerization domain. TopBP1 has also been shown to be an ATR activator (Kumagai et al, 2006). Although the ATR-activating domain (residues 978–1286 of human TopBP1) contains Ser1159 residue, it only contains a portion of the seventh BRCT motif. Thus, it is unlikely that oligomerization is involved in this process. Whether Akt phosphorylation is involved in ATR activation warrants further investigation. The identification of Akt phosphorylation and TopBP1 oligomerization will greatly facilitate future studies in TopBP1 functions.
The role of TopBP1 self-association in E2F1 control may have therapeutic implications. Most cancer cells harbor excessive E2F1 activities due to de-regulation of the Rb pathway. Downregulation of TopBP1 induces E2F1-dependent apoptosis (Liu et al, 2004); therefore, cells containing higher E2F1 levels would be more susceptible to TopBP1 inhibitors. The oligomerization domain of TopBP1 could be considered as a therapeutic target to de-repress E2F1 activity and potentiate E2F1-mediated apoptosis during chemotherapy.
Materials and methods
Cell culture and transfection
HEK293, NIH3T3, REF52 and MEF cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS, penicillin (50 IU/ml) and streptomycin (50 μg/ml) in a humidified incubator of 5% CO2 and 95% air at 37°C. HEK293 cells were transfected with appropriate vectors using a standard calcium phosphate method. NIH3T3 cells were transfected with the Gene Pulser Xcell electroporation system. REF52 and MEF cells were transfected using Lipofectamine 2000 (Invitrogen). After transfection, cells were incubated at 37°C for 2 days before analysis. Wild-type, E2F1−/− MEFs (passage 3) and two independent Rb−/−;p107−/−;p130−/−TKO MEFs (passage 6) (Sage et al, 2000; Liu et al, 2003) were used.
Akt in vitro phosphorylation and GST pulldown assay
GST, GST-TopBP1 and GST-TopBP1(S1159A) proteins were induced by 0.1 mM IPTG in E. coli strain BL21 and purified as described (Lin et al, 2001). A 1 μg portion of GST-TopBP1, GST-TopBP1(S1159A) or GST was incubated with 1 μg Akt kinase (Akt1/PKB, active; Upstate) and [γ-32P]ATP at 30°C for 30 min. GST-TopBP1 was pulled down with glutathione Sepharose and the beads were washed six times with PBS buffer, and then subjected to 10% SDS–PAGE and analyzed by 32P autoradiograph and Western blot. For GST-E2F1 and GST-BRCT78 pulldown assays, GST-E2F1, GST-BRCT78 and GST-TopBP1 proteins were induced and purified from E. coli, and the GST portion of GST-TopBP1 was excised by PreScission Protease (Pharmacia).
In vivo labeling
One day after transfection, HEK293 cells were starved for 30 min at 37°C in 3 ml of phosphate-free DMEM, and 32P-orthophosphate (2 mCi; Perkin-Elmer) was added for 2 h of incubation. Cells were washed and lysed in TNN buffer supplemented with protease inhibitors. Lysates were clarified by centrifugation and TopBP1 was immunoprecipitated with anti-FLAG beads (Sigma). The samples were subjected to 10% SDS–PAGE, and 32P autoradiograph and Western blot were carried out.
Adenoviral recombinants construction and infection and flow cytometry
AdE2F1 and AdTopBP1 were described before (Liu et al, 2003). The cDNAs of TopBP1(S1159A) and TopBP1(S1159D) were constructed into adenovirus with AdEasy system as described (He et al, 1998). Viruses were purified by CsCl banding. REF52 cells were starved in 0.25% FBS for 2 days followed by adenovirus infection. To assay apoptosis, cells were stained with annexin V-PE (Pharmingen). At least 5000 cells were gated for each sample, and annexin profile of these cells was analyzed by flow cytometry. Annexin-positive cells were scored as apoptotic.
Plasmid construction, immunoprecipitation and Western blot analysis, RT–PCR, in vitro peptide binding assay and luciferase assay
See Supplementary data.
Supplementary Material
Supplementary Research Data
Acknowledgments
We thank Drs Julien Sage and Tyler Jacks for Rb−/−;p107−/−;p130−/− TKO MEFs, Dr Louise Chow for providing GFP-E2 expressing plasmid and Dr Xinbin Chen for pGL2-p21 promoter-luciferase plasmid. We also appreciate critical reading of the manuscript by Julien Sage. This work was supported by grants from NIH (CA 100857) (WCL) and Department of Defense Breast Cancer Research Program (W81XWH-04-1-0442) (WCL). WCL is a Leukemia & Lymphoma Society Scholar. All authors declare that they have no financial interests that will pose a conflict of interest regarding the submitted article.
References
- Attwooll C, Denchi EL, Helin K (2004) The E2F family: specific functions and overlapping interests. EMBO J 23: 4709–4716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boner W, Taylor ER, Tsirimonaki E, Yamane K, Campo MS, Morgan IM (2002) A functional interaction between the HPV16 transcription/replication factor E2 and the DNA damage response protein TopBP1. J Biol Chem 277: 22297–22303 [DOI] [PubMed] [Google Scholar]
- Chaussepied M, Ginsberg D (2004) Transcriptional regulation of AKT activation by E2F. Mol Cell 16: 831–837 [DOI] [PubMed] [Google Scholar]
- Contessa JN, Hampton J, Lammering G, Mikkelsen RB, Dent P, Valerie K, Schmidt-Ullrich RK (2002) Ionizing radiation activates Erb-B receptor dependent Akt and p70 S6 kinase signaling in carcinoma cells. Oncogene 21: 4032–4041 [DOI] [PubMed] [Google Scholar]
- Dimova DK, Dyson NJ (2005) The E2F transcriptional network: old acquaintances with new faces. Oncogene 24: 2810–2826 [DOI] [PubMed] [Google Scholar]
- Franke TF, Hornik CP, Segev L, Shostak GA, Sugimoto C (2003) PI3K/Akt and apoptosis: size matters. Oncogene 22: 8983–8998 [DOI] [PubMed] [Google Scholar]
- Furuya K, Poitelea M, Guo L, Caspari T, Carr AM (2004) Chk1 activation requires Rad9 S/TQ-site phosphorylation to promote association with C-terminal BRCT domains of Rad4TOPBP1. Genes Dev 18: 1154–1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia V, Furuya K, Carr AM (2005) Identification and functional analysis of TopBP1 and its homologs. DNA Repair (Amst) 4: 1227–1239 [DOI] [PubMed] [Google Scholar]
- Greer DA, Besley BD, Kennedy KB, Davey S (2003) hRad9 rapidly binds DNA containing double-strand breaks and is required for damage-dependent topoisomerase II beta binding protein 1 focus formation. Cancer Res 63: 4829–4835 [PubMed] [Google Scholar]
- Hallstrom TC, Nevins JR (2003) Specificity in the activation and control of transcription factor E2F-dependent apoptosis. Proc Natl Acad Sci USA 100: 10848–10853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B (1998) A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci USA 95: 2509–2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herold S, Wanzel M, Beuger V, Frohme C, Beul D, Hillukkala T, Syvaoja J, Saluz HP, Haenel F, Eilers M (2002) Negative regulation of the mammalian UV rby Myc through association with Miz-1. Mol Cell 10: 509–521 [DOI] [PubMed] [Google Scholar]
- Krek W, Ewen ME, Shirodkar S, Arany Z, Kaelin WG Jr, Livingston DM (1994) Negative regulation of the growth-promoting transcription factor E2F-1 by a stably bound cyclin A-dependent protein kinase. Cell 78: 161–172 [DOI] [PubMed] [Google Scholar]
- Kumagai A, Lee J, Yoo HY, Dunphy WG (2006) TopBP1 activates the ATR–ATRIP complex. Cell 124: 943–955 [DOI] [PubMed] [Google Scholar]
- Lazzerini Denchi E, Helin K (2005) E2F1 is crucial for E2F-dependent apoptosis. EMBO Rep 6: 661–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RJ, Albanese C, Fu M, D'Amico M, Lin B, Watanabe G, Haines GK III, Siegel PM, Hung MC, Yarden Y, Horowitz JM, Muller WJ, Pestell RG (2000) Cyclin D1 is required for transformation by activated Neu and is induced through an E2F-dependent signaling pathway. Mol Cell Biol 20: 672–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin WC, Lin FT, Nevins JR (2001) Selective induction of E2F1 in response to DNA damage, mediated by ATM-dependent phosphorylation. Genes Dev 15: 1833–1844 [PMC free article] [PubMed] [Google Scholar]
- Liu K, Lin FT, Ruppert JM, Lin WC (2003) Regulation of E2F1 by BRCT-domain containing protein TopBP1. Mol Cell Biol 23: 3287–3304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Luo Y, Lin FT, Lin WC (2004) TopBP1 recruits Brg1/Brm to repress E2F1-induced apoptosis, a novel pRb-independent and E2F1-specific control for cell survival. Genes Dev 18: 673–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makiniemi M, Hillukkala T, Tuusa J, Reini K, Vaara M, Huang D, Pospiech H, Majuri I, Westerling T, Makela TP, Syvaoja JE (2001) BRCT domain-containing protein TopBP1 functions in DNA replication and damage response. J Biol Chem 276: 30399–30406 [DOI] [PubMed] [Google Scholar]
- Manke IA, Lowery DM, Nguyen A, Yaffe MB (2003) BRCT repeats as phosphopeptide-binding modules involved in protein targeting. Science 302: 636–639 [DOI] [PubMed] [Google Scholar]
- Mayo LD, Donner DB (2001) A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc Natl Acad Sci USA 98: 11598–11603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtani K, DeGregori J, Nevins JR (1995) Regulation of the cyclin E gene by transcription factor E2F1. Proc Natl Acad Sci USA 92: 12146–12150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pim D, Massimi P, Dilworth SM, Banks L (2005) Activation of the protein kinase B pathway by the HPV-16 E7 oncoprotein occurs through a mechanism involving interaction with PP2A. Oncogene 24: 7830–7838 [DOI] [PubMed] [Google Scholar]
- Sage J, Mulligan GJ, Attardi LD, Miller A, Chen S, Williams B, Theodorou E, Jacks T (2000) Targeted disruption of the three Rb-related genes leads to loss of G(1) control and immortalization. Genes Dev 14: 3037–3050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seoane J, Le HV, Massague J (2002) Myc suppression of the p21(Cip1) Cdk inhibitor influences the outcome of the p53 response to DNA damage. Nature 419: 729–734 [DOI] [PubMed] [Google Scholar]
- Seoane J, Pouponnot C, Staller P, Schader M, Eilers M, Massague J (2001) TGFbeta influences Myc, Miz-1 and Smad to control the CDK inhibitor p15INK4b. Nat Cell Biol 3: 400–408 [DOI] [PubMed] [Google Scholar]
- Shiozaki EN, Gu L, Yan N, Shi Y (2004) Structure of the BRCT repeats of BRCA1 bound to a BACH1 phosphopeptide: implications for signaling. Mol Cell 14: 405–412 [DOI] [PubMed] [Google Scholar]
- Staller P, Peukert K, Kiermaier A, Seoane J, Lukas J, Karsunky H, Moroy T, Bartek J, Massague J, Hanel F, Eilers M (2001) Repression of p15INK4b expression by Myc through association with Miz-1. Nat Cell Biol 3: 392–399 [DOI] [PubMed] [Google Scholar]
- Viniegra JG, Martinez N, Modirassari P, Losa JH, Parada Cobo C, Lobo VJ, Luquero CI, Alvarez-Vallina L, Ramon y Cajal S, Rojas JM, Sanchez-Prieto R (2005) Full activation of PKB/Akt in response to insulin or ionizing radiation is mediated through ATM. J Biol Chem 280: 4029–4036 [DOI] [PubMed] [Google Scholar]
- Wanzel M, Herold S, Eilers M (2003) Transcriptional repression by Myc. Trends Cell Biol 13: 146–150 [DOI] [PubMed] [Google Scholar]
- Wanzel M, Kleine-Kohlbrecher D, Herold S, Hock A, Berns K, Park J, Hemmings B, Eilers M (2005) Akt and 14-3-3eta regulate Miz1 to control cell-cycle arrest after DNA damage. Nat Cell Biol 7: 30–41 [DOI] [PubMed] [Google Scholar]
- Xu M, Sheppard KA, Peng CY, Yee AS, Piwnica-Worms H (1994) Cyclin A/CDK2 binds directly to E2F-1 and inhibits the DNA-binding activity of E2F-1/DP-1 by phosphorylation. Mol Cell Biol 14: 8420–8431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamane K, Kawabata M, Tsuruo T (1997) A DNA-topoisomerase-II-binding protein with eight repeating regions similar to DNA-repair enzymes and to a cell-cycle regulator. Eur J Biochem 250: 794–799 [DOI] [PubMed] [Google Scholar]
- Yamane K, Wu X, Chen J (2002) A DNA damage-regulated BRCT-containing protein, TopBP1, is required for cell survival. Mol Cell Biol 22: 555–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Inoue I (2004) Expression of MCM10 and TopBP1 is regulated by cell proliferation and UV irradiation via the E2F transcription factor. Oncogene 23: 6250–6260 [DOI] [PubMed] [Google Scholar]
- Yu X, Chini CC, He M, Mer G, Chen J (2003) The BRCT domain is a phospho-protein binding domain. Science 302: 639–642 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Research Data