

Abstract
Transcriptional activation of the interleukin-8 (IL-8) gene is restricted to distinct cell types, although the transcriptional regulatory proteins controlling IL-8 gene expression are ubiquitous. We show that cell-specific transcription of IL-8 is due to the distinct chromatin architecture on the enhancer/promoter before the arrival of the inducing signal. In expressing epithelial cells the enhancer/promoter is nucleosome-free, whereas in non-expressing B cells a nucleosome masks the entire regulatory region. The B-cell-specific nucleosome contains the histone variant macroH2A, which is responsible for preventing transcription factor binding. Recruitment of the repressive macroH2A nucleosome requires direct interactions between ATF-2 bound to the nearby AP1 site and macroH2A and it is regulated by DNA-induced protein allostery. siRNA against ATF-2 or macroH2A rescues IL-8 transcription in B cells. Thus, a transcription factor can work as a transcriptional repressor by orchestrating and maintaining the assembly of specialized local chromatin architectures.
Keywords: transcription factors, chromatin structure, gene expression, histone variants, protein allostery
Introduction
Cell-specific gene expression programs are hardwired in the genome in the form of gene-specific unique regulatory DNA sequences to build enhancers and promoters. However, it is the chromatin structure, the epigenome, which determines whether DNA templates are accessible to the numerous protein complexes required for DNA transcription, replication, recombination, etc. (Luger, 2003). Thus, layered upon the DNA code is the epigenetic code that provides part of the regulatory infrastructure determining when, how and where the genetic information will be expressed in developing and/or adult organisms (Turner, 2002). Epigenetic information is encoded through direct DNA modification (e.g. methylation), chromatin structure and modification, and it can also be heritably transmitted through cell division to maintain cellular identity (Jaenisch and Bird, 2003; Morgan et al, 2005). In case of transcription, the precise position of nucleosomes on regulatory DNA can interfere with the binding of several regulatory proteins ranging from transcription factors to components of the general transcriptional machinery (Lomvardas and Thanos, 2002; Ercan et al, 2004). To bypass this barrier, cells have evolved a complex array of enzymes that alter chromatin. These modifications can lead either to nucleosome sliding, nucleosome loss (eviction) or to modified nucleosomes (Flaus and Owen-Hughes, 2004). In every case, previously masked critical DNA sequences are now exposed to the transcriptional machinery to allow initiation of transcription. Our current view postulates that the combinatorial nature of covalent histone modifications generates unique signatures used to stabilize the recruitment of chromatin remodeling complexes to the promoters of selected genes. The precise combination of gene-specific histone modifications is read by a small number of chromatin-associated proteins, which are targeted to promoters in a spatially and temporally specific manner (Agalioti et al, 2002).
An additional way to mark genes for activation or repression is the replacement of canonical histones with other variants, which can induce local chromosomal changes functioning in multiple cellular pathways, with their precise role, however, remaining largely unknown (Henikoff et al, 2004; Sarma and Reinberg, 2005; Kamakaka and Biggins, 2005). The low amounts of the histone variants in chromatin suggests that they may have regulatory roles, especially during transcription. MacroH2A is a vertebrate-specific H2A variant, which in addition to the H2A homology region bears a carboxyl-terminal extension with no similarities to histones (Pehrson and Fried, 1992). MacroH2A is enriched on the inactive X chromosome but its presence is not essential for maintenance of the transcriptionally inactive state (Costanzi and Pehrson, 1998; Mermoud et al, 1999). However, biochemical studies using synthetic templates have indicated that macroH2A-containing nucleosomes are structurally different in the vicinity of the dyad axis, and this correlates with the inability of transcription factors to bind to DNA sites inserted nearby (Angelov et al, 2003; Abbott et al, 2004; Chakravarthy et al 2005). These observations suggest that incorporation of macroH2A into nucleosomes could confer an epigenetic mark for gene repression. However, there are no known gene targets for macroH2A-dependent transcriptional repression, and no evidence for the mechanisms by which macroH2A could be recruited to specific genes and repress transcription in vivo.
Here, we show that the cell-lineage-specific gene expression program of human IL-8 depends on the presence of macroH2A. Ordinarily, this gene is activated in response to proinflammatory cytokines, viral infection and stress (Hoffmann et al, 2002). One of the most remarkable properties of IL-8 is the variation in expression levels between different cell types and this is important for mounting an efficient immune response. Transcriptional activation depends on a relatively simple enhancer located immediately upstream from the start site of transcription. This enhancer is bound cooperatively by the transcription factors NF-κB and C/EBPβ, which form an enhanceosome-like structure whose transcriptional potential is facilitated, at least in some cell types and/or signals, by ATF/AP1 proteins bound 30 bp upstream (Hoffmann et al, 2002). However, the ubiquitous expression and/or activation of the AP1, C/EBP and NF-κB proteins cannot explain the wide variation of expression levels and the cell-type-specific transcription of the IL-8 gene. We showed that the constitutive DNA binding of the ATF2/JunD heterodimer in B cells leads to the recruitment of a macroH2A-containing nucleosome on the IL-8 enhancer/promoter, thus inhibiting transcriptional activation.
Results
Cell-type-specific chromatin architecture at the IL-8 promoter regulates gene transcription
Figures 1A and B show that virus infection activates transcription of the IL-8 gene in epithelial HeLa and not in Namalwa B cells. However, IFN-β gene expression is induced in both cells types.
Figure 1.
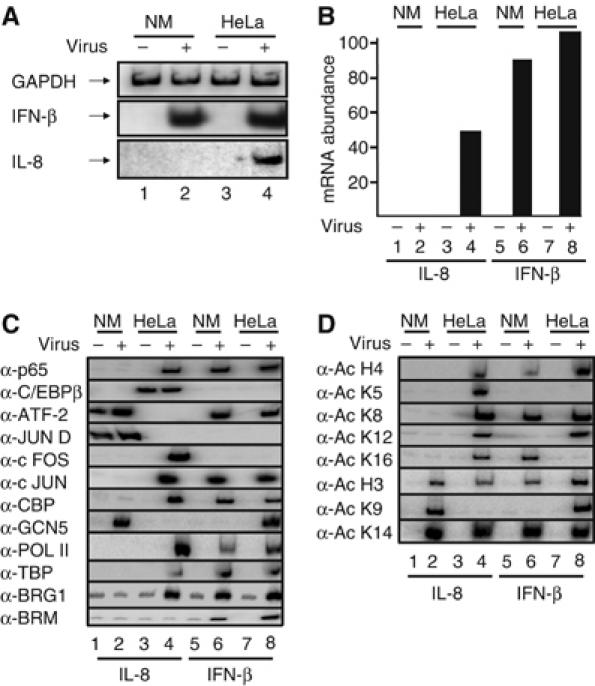
Cell-type-specific recruitment of transcriptional regulatory proteins to the IL-8 enhancer/promoter. (A) Total RNA was isolated from mock- or virus-infected HeLa and Namalwa (NM) cells for 6 h, and used in RT–PCR reactions as a template with primers detecting the transcripts shown on the left part of the gel. (B) Real-time RT–PCR analysis illustrating cell-type-specific transcriptional activation of the IL-8 gene. (C) Crosslinked chromatin prepared from mock or 6 h virus-infected HeLa and NM cells was immunoprecipitated with the indicated antibodies against transcriptional regulatory proteins required for IL-8 or IFN-β gene expression. The precipitated IL-8 and IFN-β promoters were detected by PCR using 32P-dCTP in the reaction. (D) Same as in (C) except the antibodies used reacted with the indicated acetylated histone lysine residues.
The inability of the IL-8 gene to be activated upon viral infection in Namalwa B cells was unexpected, as previous studies have shown that transcriptional activation of IL-8 is mediated via a simple enhancer bound by the transcription factors NF-κB, C/EBP and ATF/AP1, all of which are present in B cells (Hoffmann et al, 2002). To investigate transcription factor occupancy on the IL-8 promoter, we carried out chromatin immunoprecipitation (ChIP) experiments using chromatin prepared from either mock- or virus-infected HeLa and Namalwa cells, precipitated with a specific set of antibodies recognizing the IL-8 transcriptional activators followed by semiquantitative and/or quantitative PCR (Figure 1C and D and Supplementary Figure 1, respectively).
Figure 1C shows that virus infection induces binding of the p65 subunit of NF-κB to the IL-8 promoter in HeLa, but not in Namalwa cells (compare lanes 1, 2 with lanes 3, 4). This is not due to defective Namalwa chromatin preparation, because virus inducible binding of p65 was detected on the IFN-β promoter using the same immunoprecipitated chromatin-derived DNA as a template (lanes 5 and 6). Furthermore, the constitutively active transcription factor C/EBPβ was detected on the IL-8 enhancer/promoter in HeLa cells (lanes 3 and 4) but not in Namalwa cells (lanes 1 and 2). As a control, we showed that C/EBP did not bind to the IFN-β enhancer/promoter in HeLa or Namalwa cells (lanes 5–8). Interestingly, a Fos/Jun heterodimer was detected on the IL-8 promoter upon virus infection in HeLa (lanes 3 and 4) but not in Namalwa cells (lanes 1 and 2), a result consistent with the role of AP1 in IL-8 gene transcription (Hoffmann et al, 2005). Unexpectedly, ATF-2 and JunD, presumably as a heterodimer, were found to associate with the IL-8 promoter before virus infection in Namalwa but not in HeLa cells and this binding was slightly enhanced upon virus infection in Namalwa cells (Figure 1C, lanes 1–4, Supplementary Figure 1). Thus, both NF-κB and C/EBP cannot bind to the IL-8 promoter in Namalwa cells, whereas ATF-2/JunD associates with the IL-8 promoter in the nonexpressing Namalwa cells.
Components of the basal transcriptional machinery (PolII and TBP) were recruited to the IL-8 promoter in HeLa but not in Namalwa cells (Figure 1C, lanes 1–4). Interestingly, we found that the GCN5 histone acetyltransferase (HAT) was recruited to the IL-8 promoter in Namalwa but not in HeLa cells (lanes 1–4), thus suggesting that this recruitment is mediated by the ATF-2/JunD heterodimer (see also below). By contrast, CBP and the SWI/SNF complex were recruited only in HeLa cells (lanes 3 and 4).
The cell-specific recruitment of GCN5 and CBP histone acetyltransferases correlates with a distinct histone acetylation pattern of the IL-8 promoter in Namalwa or HeLa cells (Figure 1D). In summary, the experiments described so far show that (1) NF-κB and C/EBP, the main IL-8 transcriptional activators, cannot bind to the IL-8 promoter in Namalwa cells, whereas the same factors can bind to the promoter of other genes in the same cells; (2) the ATF-2/JunD heterodimer binds to the IL-8 promoter only in the nonexpressing Namalwa cells and (3) the CBP and GCN5 HAT proteins are differentially recruited to the IL-8 promoter in HeLa and Namalwa cells, respectively, printing a distinct pattern of histone acetylation. Interestingly, despite the fact that histone H3 is acetylated at K9 and K14, presumably by GCN5, in Namalwa cells, this acetylation did not mediate recruitment of the TFIID complex, as it is the case for the IFN-β gene promoter (Agalioti et al, 2002). On the other hand, the lack of H4 acetylation at K8 and K12 correlates with the inability of the SWI/SNF complex to be recruited to the IL-8 promoter in Namalwa cells (Figure 1C, lanes 1 and 2)
The inability of the NF-κB and C/EBP activators to access the IL-8 promoter in Namalwa cells prompted us to determine its nucleosomal organization in this cell type. Namalwa and HeLa cells were mock or virus infected for 6 h followed by formaldehyde crosslinking of the histone DNA contacts and micrococcal nuclease treatment of the isolated nuclei. DNA extracted from the resulting mononucleosomes was annealed with primers A (−117 to −98) and B (+47 to +70) spanning the IL-8 enhancer/promoter, followed by primer extension and ligation-mediated PCR to detect the extended DNA fragments. Figure 2 (lanes 7 and 8) shows that when primer B was annealed with mononucleosomal DNA prepared from HeLa cells, the final product was 163 bp, indicating that the nucleosome's 3′ border is at +186, a result consistent with our previous studies (Lomvardas and Thanos, 2002). However, when the same primer was annealed with mononucleosomal DNA prepared from Namalwa cells, it did not produce an extended fragment (lanes 3 and 4), thus indicating that the nucleosome present in HeLa cells between +39 and +186 relative to the start site of IL-8 transcription is absent in Namalwa cells. Remarkably, when primer A was annealed with Namalwa cell-derived mononucleosomal DNA, a fragment of 134 bp was produced (lanes 1 and 2), indicating the presence of a nucleosome with boundaries at −153 and −7, at least in a significant population of cells. No such product was detected when HeLa cell-derived mononucleosomal DNA was used as a template (lanes 5 and 6). Taken together, these experiments demonstrate that the default chromatin architecture of the IL-8 enhancer/promoter region is distinct in two different cell types. In HeLa cells, where the IL-8 gene can be induced, the enhancer promoter is nucleosome-free, thus allowing the assembly of the transcriptional apparatus, whereas in Namalwa cells, where the gene remains silent, the enhancer/promoter is masked by a nucleosome, thus making it inaccessible to the transcriptional machinery (Figure 2, bottom).
Figure 2.
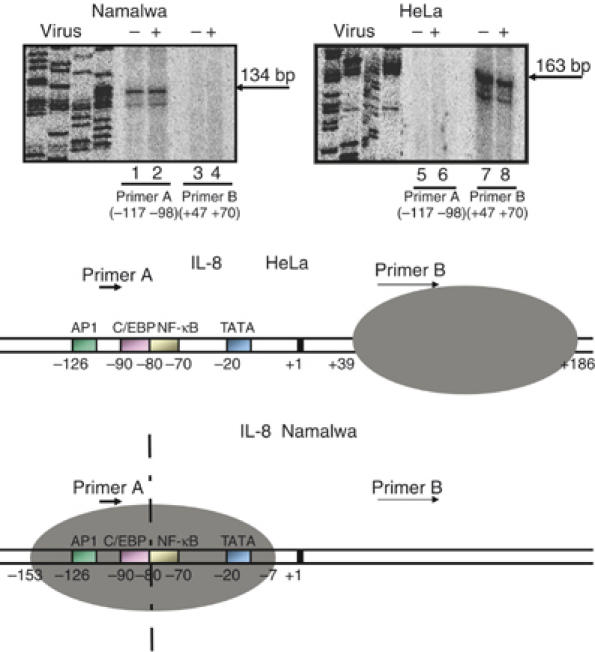
The chromatin architecture of the IL-8 gene enhancer/promoter differs between distinct cell types. A diagrammatic illustration depicting the distinct chromatin architecture of the IL-8 enhancer/promoter in Namalwa and HeLa cells is shown at the bottom of the figure. The dotted vertical line indicates the axis of dyad symmetry of the nucleosome in Namalwa cells. HeLa and Namalwa cells were mock or virus infected for 6 h followed by formaldehyde crosslinking and micrococcal nuclease treatment of the isolated chromatin. The DNA was purified and annealed with primers A and B followed by extension and ligation-mediated PCR using 32P-dCTP in the reaction to detect the extended products. A sequencing gel containing the extended products run side-by-side with the DNA sequencing reactions serving as size markers is shown.
The histone variant macroH2A marks IL-8 for transcriptional repression in B cells
The determination of the chromatin architecture of the IL-8 gene enhancer/promoter in Namalwa B cells revealed that the C/EBP and NF-κB-binding sites immediately flank the dyad axis of the nucleosome (Figure 2). A previous in vitro study had shown that NF-κB sites positioned next to the dyad symmetry of canonical nucleosomes do not inhibit NF-κB DNA binding (Angelov et al, 2003). However, replacement of H2A with the histone variant macroH2A in these nucleosomes caused the NF-κB DNA-binding sites to be inaccessible to the transcription factor. The remarkable similarity of these in vitro observations, using synthetic templates with our in vivo data on the IL-8 promoter in the natural chromatin context, prompted us to examine whether the nucleosome masking the IL-8 promoter in Namalwa cells contains macroH2A. To accomplish this, we carried out ChIP experiments using an antibody specific for macroH2A1.2 and chromatin prepared from HeLa and Namalwa cells. Figure 3A (lanes 5 and 6) shows that the histone variant macroH2A1.2 is indeed a constituent of the nucleosome masking the IL-8 promoter in Namalwa cells. No macroH2A was identified on the IL-8 promoter in HeLa cells (lanes 7 and 8). Furthermore, we found that following virus infection, the amount of IL-8 promoter precipitated with the macroH2A antibody was slightly enhanced, thus indicating that either the macroH2A–promoter DNA interactions are strengthened or the abundance of macroH2A is increased (compare lane 5 with lane 6, see also Supplementary Figure 1). As a control, we showed that the IFN-β promoter, which is induced in both Namalwa and HeLa cells, does not contain macroH2A (Figure 3A, lanes 13–16). MacroH2A is also part of nucleosomes present in the coding region of the IL-8 gene in Namalwa but not in HeLa cells (lanes 9–12). However, when the same immunoprecipitated chromatin DNA was used as a PCR template with primers amplifying the genomic region located further upstream of the IL-8 enhancer (−690 to –405), it did not produce a product (lanes 1 and 2), thus indicating the absence of macroH2A in this region. The polarized distribution of macroH2A on the IL-8 locus is not due to the general absence of nucleosomes in the area upstream of the enhancer/promoter, as an antibody against the histone H3 C-terminus precipitated the corresponding chromatin DNA (Figure 3A, lanes 1–2) with efficiencies comparable to those of the promoter and coding regions (lanes 5, 6 and 9, 10). The distinct nucleosomal organization and histone composition of the IL-8 promoter in HeLa and Namalwa cells was verified by using mononucleosomes in ChIP experiments and sets of PCR primers spanning the regions of the IL-8 promoter occupied by nucleosomes in HeLa and Namalwa cells (Figure 3B).
Figure 3.
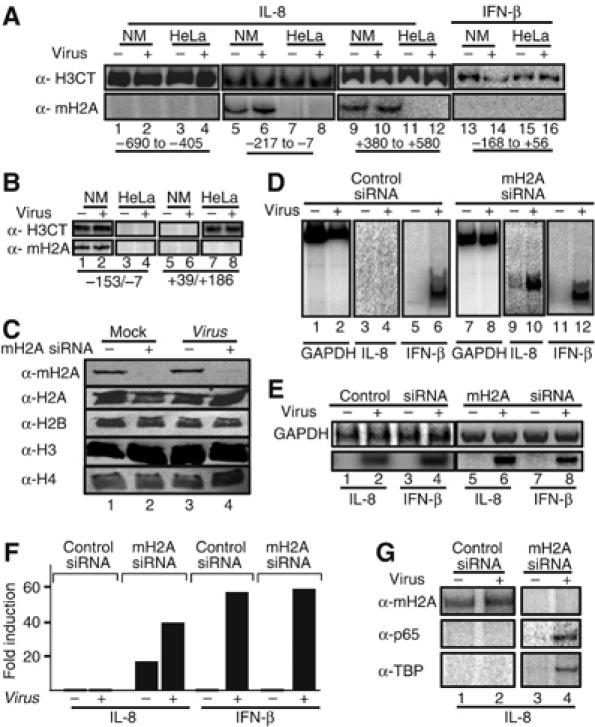
The histone variant macroH2A marks the IL-8 gene for repression in B cells. (A) HeLa and Namalwa cells were mock or virus infected for 6 h followed by formaldehyde crosslinking and immunoprecipitation using antibodies specific for macroH2A1.2 and the carboxyl-terminus of histone H3. The precipitated chromatin-bound DNA was detected using the indicated set of primers amplifying the upstream IL-8 region (−690 to −405, lanes 1–4), the enhancer/promoter (−217 to −7, lanes 5–8) and the coding region (+380 to +580, lanes 9–12) or the IFN-β enhancer (lanes 13–16). The relative position of the amplified regions is shown at the bottom of the gel. (B) Same as in (A) except the chromatin used was composed of mononucleosomes prepared after digestion with micrococcal nuclease. (C) A Western blot using whole-cell extracts prepared from mock- or virus-infected HeLa cells transfected with a control siRNA (lanes 1, 3) or the macroH2A-specific siRNA (lanes 2 and 4). Our transfection efficiency in HeLa cells is approximately 90%. The abundance of macroH2A1.2 and histones H2A, H2B, H3 and H4 was determined by Western blotting using specific antibodies. (D) Namalwa cells were transfected with the control (lanes 1–6) or the macroH2A-specific siRNA (lanes 7–12) and 48 h later they were infected with Sendai virus for 6 h. The isolated RNA was used as a template in RT–PCR reactions using primers detecting GAPDH, IL-8 and the IFN-β-coding regions. (E) Same as in (D) except the RNAs used in the RT–PCR reactions were prepared from HeLa cells transfected with the indicated siRNAs. (F) Namalwa cells were transfected with luciferase-based reporter plasmids bearing the IL-8 or the IFN-β enhancer promoter along with control or macroH2A siRNAs. At 48 h following transfection, the cells were mock or virus infected for 10 h and the luciferase activity was determined. The average of three experiments is shown and the variability from experiment to experiment was less than 20%. (G) Namalwa cells were transfected with the IL-8 reporter as in (F). At 6 h after virus infection, the cells crosslinked with formaldehyde and the isolated chromatin was immunoprecipitated using the indicated antibodies. PCR was used to detect the precipitated IL-8 promoter fragments using plasmid-specific primers.
To examine the role of macroH2A in IL-8 gene repression, we carried out siRNA depletion experiments in HeLa and Namalwa cells followed by RT–PCR analysis of virus-infected cells. Figure 3C shows that siRNA against macroH2A1.1/1.2 reduced the protein levels below detection, but it did not affect H2A, H2B, H3 or H4 following its introduction into HeLa cells, thus underscoring the specificity of the experiment. Consistent with our data, depletion of macroH2A in HeLa cells did not affect the expression of the IL-8 and IFN-β genes (Figure 3E). Remarkably, however, transfection of the macroH2A siRNA into Namalwa B cells derepressed IL-8 basal level transcription and allowed further transcriptional activation following virus infection (Figure 3D, compare lanes 3 and 4 with lanes 9 and 10).
It should be noted that the transfection efficiency of Namalwa cells under our conditions is approximately 5%, but it appears that the elimination of the endogenous macroH2A protein even in this small percentage of cells sufficed to detect the derepressed IL-8 transcripts in the same cells. To exclude the possibility of nonspecific effects of the macroH2A siRNA, we transfected Namalwa cells with IL-8 and IFN-β reporter constructs along with control or macroH2A-specific siRNAs followed by ChIP experiments. Consistent with the results presented above, siRNA against macroH2A derepressed the IL-8 enhancer/promoter activity of the cotransfected reporter, but it did not affect IFN-β expression (Figure 3F). The ChIP experiment of Figure 3G shows that this derepression correlates with the absence of macroH2A from the IL-8 promoter in the same cells, and to the ability of NF-κB and TBP to associate with the promoter following treatment with the macoH2A-specific siRNA and virus infection.
To determine whether the transcriptional activity or inactivity of the IL-8 gene in HeLa and Namalwa cells, respectively, is restricted to these cell lines or it is a general characteristic of many epithelial, fibroblast and B-cell lines, we tested four additional B-cells lines, one epithelial, one fibroblast cell line and fresh human B cells isolated from surgically removed tonsils. As seen in Supplementary Figure 2, IL-8 expression is prohibited in all B cells tested because of the presence of the macroH2A nucleosome on the enhancer/promoter.
Recruitment of the macroH2A nucleosome requires the ATF/enhancer DNA complex
The experiments described above showed that the IL-8 gene is transcriptionally repressed in Namalwa B cells because the critical DNA regulatory regions are masked by a nucleosome containing the histone variant macroH2A. Therefore, the question arises how the macroH2A-containing nucleosome is specifically recruited to the IL-8 promoter in Namalwa but not in HeLa cells. We noticed that binding of the ATF-2/JunD heterodimer and macroH2A with the IL-8 promoter correlate with repression in Namalwa cells, and these interactions are enhanced slightly upon virus infection (Figures 1C, 3A, Supplementary Figure 1). Although no proteins have been identified yet that affect macroH2A recruitment, we hypothesized that the ATF/AP1 site may play a role in positioning the macroH2A nucleosome in Namalwa cells. To test this hypothesis, we generated an IL-8 promoter in which the ATF/AP1 site was mutated (ΔAP1). Figure 4A shows that mutation of the AP1/ATF site led to a significant derepression of the basal levels of expression and to a further activation of the IL-8 promoter following virus infection in Namalwa cells and not in HeLa cells, consistent with the fact that the AP1/ATF site is not critical for IL-8 expression in epithelial cells (Hoffmann et al, 2002).
Figure 4.
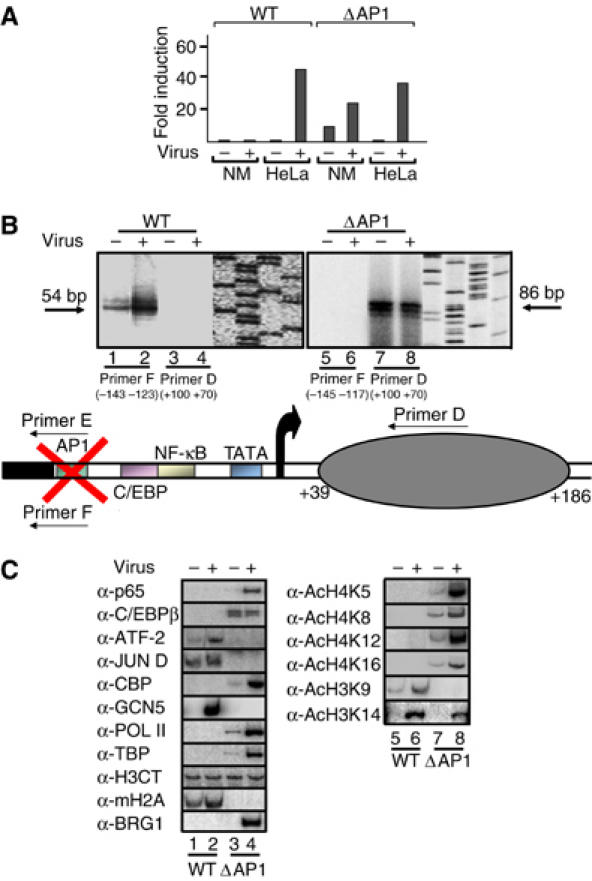
The ATF/AP1 site of the IL-8 enhancer is required for the recruitment and positioning of the macroH2A-containing repressing nucleosome. (A) HeLa and Namalwa cells were transfected with either the WT or the ΔAP1 IL-8 enhancer/promoter-luciferase fusion constructs followed by virus infection for 10 h. Shown is the average of seven experiments and the variation from experiment to experiment was less than 35%. (B) Namalwa cells were transfected with the WT (−127/+59) (lanes 1–4) or an isogenic IL-8 enhancer/promoter construct bearing a mutation in the ATF/AP1 site (ΔAP1) (lanes 5–8) followed by mock or virus infection. Mononucleosomal DNA was purified and annealed with the indicated primers followed by ligation-mediated PCR. To distinguish between the endogenous and the exogenous WT and mutant templates, we used promoter-luciferase fusion constructs. Thus, primer D hybridizes to luciferase sequences and therefore detects the exogenous genes only. Primer E hybridizes to the mutant AP1 site only and the nearby vector sequences, whereas primer F recognizes the WT AP1 site and the nearby vector sequences. The diagrammatic illustration at the bottom of the figure illustrates the position of the nucleosome in the ΔAP1 template in Namalwa cells. (C) The WT and the ΔAP1 IL-8 enhancer/promoter templates were transfected into Namalwa cells followed by virus infection for 6 h. Crosslinked chromatin was immunoprecipitated with the indicated antibodies and the IL-8 enhancer/promoter was detected by PCR using vector-specific primers and 32P-dCTP in the reaction.
To investigate whether deletion of the AP1/ATF site has altered the nucleosomal organization of the IL-8 promoter, we carried out nucleosome mapping experiments using mononucleosomes prepared from Namalwa cells transfected with the WT or ΔAP1 templates. Remarkably, mutation of the AP1 site resulted in the loss of the nucleosome masking the IL-8 enhancer (Figure 4B, lanes 5 and 6). Furthermore, we found that the nucleosomal organization of the ΔAP1 template in Namalwa cells is similar to that of the WT IL-8 enhancer/promoter in HeLa (Figure 4B, lanes 7 and 8), as primer D produced a product of 86 base pairs, suggesting the existence of a nucleosome between +39 and +186. In other words, we mapped a nucleosome between +39 to +186, and this nucleosome lacks macroH2A (see below). Importantly, this region is nucleosome-free in the WT IL-8 gene in Namalwa cells (Figure 4B, lanes 3 and 4). We conclude that the ATF/AP1 site is critical for the cell-type-specific recruitment of a nucleosome on the IL-8 promoter.
Figure 4C shows that both NF-κB and C/EBP access their binding sites on the ΔAP1 template, leading to the recruitment of both CBP and SWI/SNF (lanes 3 and 4). As a result, PolII and TBP are recruited to the promoter, thus leading to transcriptional activation. In addition, GCN5 was not recruited to this template, thus underscoring the role of ATF proteins in recruiting GCN5 (Figure 4C, lanes 3 and 4). Finally, the histone acetylation pattern of the transcriptionally active ΔAP1 template in Namalwa is similar to that of the WT gene in HeLa cells (compare lanes 7 and 8 with Figure 1D). Taken together, these experiments suggest that the constitutive DNA binding of an ATF/JunD heterodimer to the IL-8 enhancer in Namalwa cells correlates with the presence of a macroH2A nucleosome, which prohibits the access of other transcription factors and consequently the recruitment of the basal transcriptional machinery.
One of the predictions derived from the experiments described above is that ATF-2 may function as a repressor of IL-8 gene expression in HeLa cells. To test this idea, we transfected ATF-2 into HeLa cells and measured IL-8 and IFN-β gene expression of the endogenous genes after virus infection. Figure 5A shows that ATF-2 causes two opposite effects on the endogenous IL-8 and IFN-β gene expression: it represses IL-8 and it super-activates IFN-β transcription. This repression on IL-8 transcription correlates with the recruitment of macroH2A on the endogenous IL-8 promoter, which in turn prohibits the binding of NF-κB (Figure 5B, lanes 1–4). This effect is due to the direct binding of ATF-2 to the IL-8 enhancer, as deletion of the ATF site abolished both macroH2A recruitment and transcriptional repression (Figures 4C and 5C). Thus, consistent with the data presented above, virus infection induced binding of NF-κB to the WT IL-8 enhancer, whereas ATF-2 and macroH2A were not found on the enhancer (Figure 5D, lanes 1 and 2). As a control we showed that overexpressed p65 did not recruit macroH2A (lanes 5 and 6). We conclude that ATF-2 is responsible for the recruitment of the macroH2A nucleosome on the IL-8 enhancer. As predicted from the above, depletion of ATF-2 by siRNA in Namalwa cells derepressed IL-8 gene expression (Figure 5E, lanes 3 and 4, Figure 5F) and recruitment of macroH2A to the IL-8 enhancer (Figure 5G), thus indicating that ATF-2 is indeed responsible for the recruitment of macroH2A and subsequently for the inability of the IL-8 gene to be induced in B cells.
Figure 5.
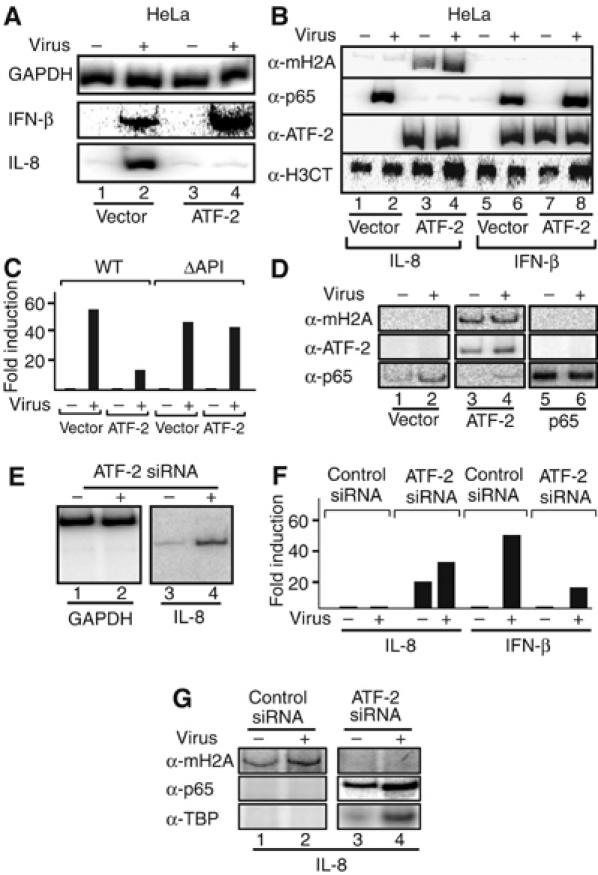
ATF-2 functions as a repressor of IL-8 transcription. (A) HeLa cells were transfected with empty or ATF-2-expressing vector and 40 h following transfection the cells were infected with Sendai virus for 6 h. RNA was prepared and the abundance of GAPDH, IL-8 and IFN-β mRNAs was determined by RT–PCR. (B) HeLa cells were transfected and infected with Sendai virus as in (A). Following virus infection, the cells were crosslinked with formaldehyde and the isolated chromatin was precipitated with the indicated antibodies. PCR was used to detect the precipitated endogenous IL-8 and IFN-β promoters. (C) HeLa cells were transfected with the WT or the ΔAP1 IL-8 enhancer/promoter luciferase fusion constructs and were mock or virus infected for 10 h. Shown is the average of three experiments and the variability from experiment to experiment was less than 25%. (D) HeLa cells were transfected with the WT IL-8 luciferase reporter along with empty (lanes 1 and 2) or ATF-2 (lanes 3 and 4) or p65-expressing vectors (lanes 5 and 6). Following transfection, the cells were virus infected for 6 h, crosslinked with formaldehyde and the isolated chromatin was immunoprecipitated using the indicated antibodies. PCR was used to detect the precipitated IL-8 promoter fragments using plasmid-specific primers. (E) Namalwa cells were transfected with an siRNA against ATF-2 and 48 h later the cells were infected with Sendai virus followed by RNA isolation and RT–PCR analysis using IL-8-specific primers. (F) Namalwa cells were transfected with the IL-8 or IFN-β luciferase reporters along with control or ATF-2 siRNA. At 48 h following transfection, the cells were mock or virus infected for 8 h and the luciferase activity was determined. Shown is the average of two experiments and the variability from experiment to experiment was less than 25%. (G) Namalwa cells were transfected with the IL-8 reporter as in (F). At 6 h after virus infection, the cells were crosslinked with formaldehyde and the isolated chromatin was immunoprecipitated using the indicated antibodies. PCR was used to detect the precipitated IL-8 promoter fragments using plasmid-specific primers.
Our experiments predict that placement of the IL-8 ATF site to a different position should lead to a simultaneous relocation of the macroH2A nucleosome. To test this possibility, we mutated the IL-8 AP1 site and inserted an oligonucleotide bearing a copy of the WT site to a new position 90 bp further upstream (Figure 6A). This template (AP1 OUT) was introduced into Namalwa cells and its nucleosomal map was determined as described above. Figure 6A shows that the macroH2A nucleosome present on the WT IL-8 enhancer between −153 to −7 has been relocated covering the region between −243 to −97, thus exposing the critical regulatory elements of the IL-8 enhancer/promoter to transcription factors and the basal transcriptional apparatus. Interestingly, the relative position of the AP1 site and the nucleosome has remained the same, thus indicating that the AP1 site not only functions by nucleating the assembly of the macroH2A-containing nucleosome but also by determining its precise position along the DNA. As expected, the AP1 OUT template can be activated by virus infection (Figure 6B), and this activation correlates with the recruitment of transcription factors and the basal transcriptional machinery (Figure 6C).
Figure 6.
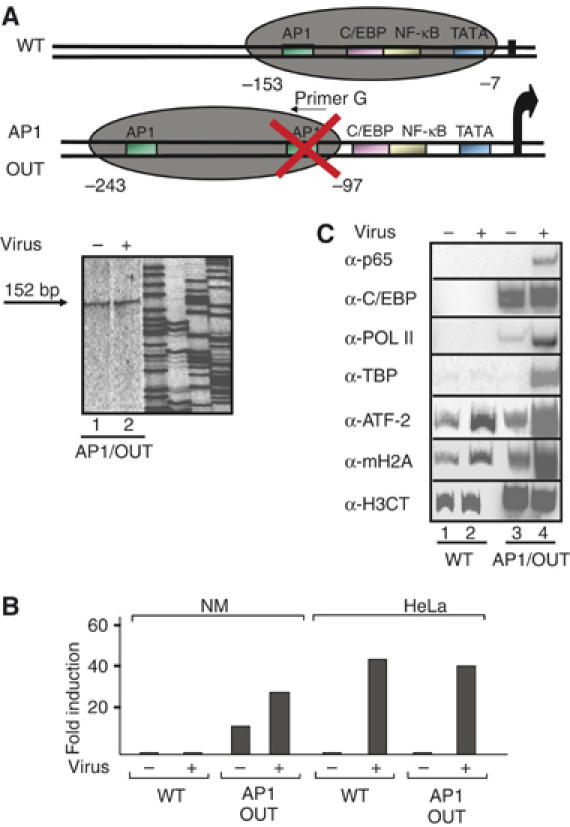
Altering the chromatin architecture of the IL-8 promoter by relocating the ATF site. (A) Namalwa cells were transfected with the AP1 OUT IL-8 reporter followed by isolation of mononucleosomes. The DNA was purified and annealed with the indicated primer G (hybridizes to the mutated AP1 site) followed by ligation-mediated PCR. The diagrammatic illustration at the top of the figure illustrates the position of the nucleosome in the AP1 OUT as opposed to the WT template in Namalwa cells. (B) Namalwa cells were transfected with the WT or the AP1 OUT IL-8 templates and 48 h later the cells were infected with Sendai virus for 10 h followed by determination of the luciferase activity. Shown is the average of four experiments and the variability from experiment to experiment was less than 30%. (C) Namalwa cells were transfected with the IL-8 reporters as in (B). At 6 h after virus infection, the cells were crosslinked with formaldehyde and the isolated chromatin was immunoprecipitated using the indicated antibodies. PCR was used to detect the precipitated IL-8 promoter fragments using plasmid-specific primers.
ATF-2 bound to the IL-8 but not to the IFN-β enhancer directly recruits macroH2A-containing nucleosomes
To investigate whether ATF-2 directly recruits macroH2A, we carried out protein–protein interaction assays using recombinant proteins. Figure 7A demonstrates that macroH2A, and not any of the other histones, was specifically retained on glutathione beads containing either full-length ATF-2 or the bzip domain (lanes 1–3). Interestingly, the macro-domain of macroH2A sufficed for this interaction. Thus, ATF-2 interacts specifically with macroH2A and therefore it could recruit it directly to the IL-8 enhancer. This possibility was tested by carrying out the experiment shown in Figure 7B. Biotinylated promoter fragments spanning the region from −211 to +245 of the IL-8 gene or the −143 to +183 region from the IFN-β gene served as templates in nucleosome reconstitution experiments using donor chromatin prepared from HeLa cells in the absence or in the presence of recombinant ATF-2 or NF-κB. Following nucleosome reconstitution, the beads were concentrated, washed extensively, and the histone composition was determined by Western blotting using antibodies specific for macroH2A and canonical histones. Figure 7B shows that when ATF-2 was omitted from the nucleosome reconstitution reaction, the assembled chromatin did not contain macroH2A either on the IL-8 or the IFN-β promoters (compare lanes 1 with 2 and 5 with 6). However, addition of recombinant ATF-2 during the nucleosome reconstitution reaction resulted in the recruitment of a macroH2A nucleosome on the IL-8 enhancer (lane 3) but not on the IFN-β enhancer (lane 7). This effect was specific for ATF-2, as NF-κB could not recruit macroH2A nucleosomes to either enhancer (lanes 4 and 8). Thus, ATF-2 bound to the IL-8 but not to the IFN-β enhancer site recruits a macroH2A nucleosome, which is responsible for transcriptional repression. However, ATF-2 bound on the IL-8 enhancer cannot recruit free macroH2A, despite the fact that both proteins can interact in the absence of DNA (Figure 7C). The control experiment of Supplementary Figure 3 demonstrates that under the salt dialysis conditions used in the nucleosome reconstitution experiments, ATF-2 DNA binding is observed at relatively high salt concentrations.
Figure 7.
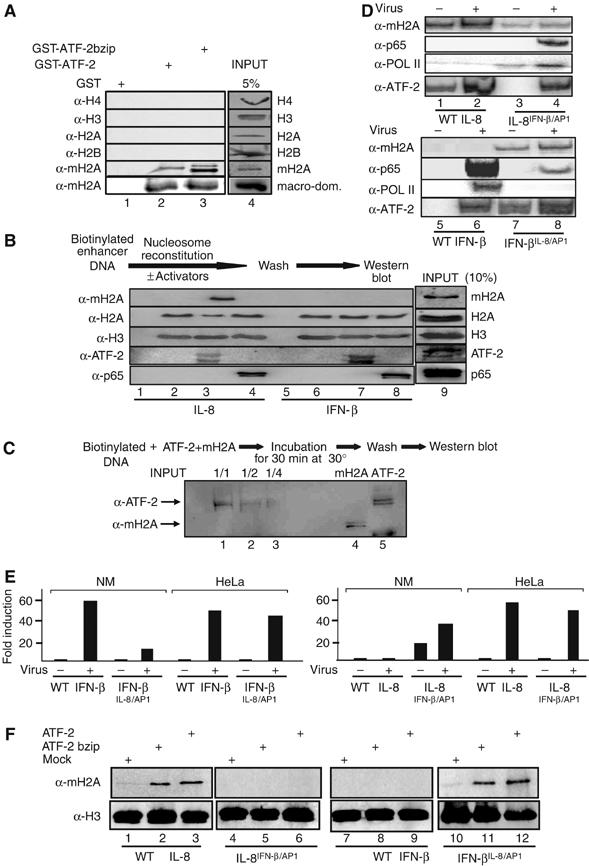
Direct recruitment of macroH2A nucleosomes to the IL-8 enhancer by ATF-2. (A) Glutathione beads containing immobilized GST, GST-ATF2 or GST-ATF2bzip were incubated with recombinant and purified histones H4, H3, H2A, H2B, macroH2A or the macro-domain of the macroH2A. Bound proteins were detected by Western blots using the indicated antibodies. (B) Biotinylated IL-8 (lanes 1–4) or IFN-β (lanes 5–8) promoter fragments were attached to Dyna beads and reconstituted into chromatin using HeLa donor chromatin in the absence (lanes 2 and 6) or in the presence of recombinant ATF-2 (lanes 3 and 7) or p65 (lanes 4 and 8). Lanes 1 and 5 correspond to beads only. Next, the fragments were washed three times before Western blot analysis using the indicated antibodies. (C) A biotinylated IL-8 enhancer/promoter DNA fragment was incubated with the indicated amounts of ATF-2 and free recombinant macroH2A followed by washes and Western blot. The last two lanes on the right depict the amounts of proteins used as input in the experiment (1/1). (D) Namalwa cells were transfected as in (E). At 6 h after virus infection, the cells were crosslinked with formaldehyde and the isolated chromatin was immunoprecipitated using the indicated antibodies. PCR was used to detect the precipitated IL-8 and IFN-β promoter fragments using plasmid-specific primers. (E) Luciferase-based reporter constructs bearing the WT IL-8 and IFN-β and their swapped derivatives were transfected into HeLa or Namalwa cells followed by virus infection. The corresponding luciferase activities were determined from three independent experiments with less than 35% variability from experiment to experiment. (F) Biotinylated IL-8 (lanes 1–3), IL-8IFNβ/AP1 (lanes 4–6), IFN-β (lanes 7–9) or IFNβIL-8/AP1 (lanes 10–12) promoter fragments were attached to Dyna beads and reconstituted into chromatin using HeLa donor chromatin in the absence (lanes 1, 4, 7 and 10) or in the presence of recombinant ATF-2 bzip (lanes 2, 5, 8 and 11) or full-length ATF-2 (lanes 3, 6, 9 and 12). Next, the fragments were washed three times before Western blot analysis using the indicated antibodies.
The ATF sites of IL-8 (TGACTCAG) and IFN-β (TGACATAG) differ in two nucleotides (underlined). Previous studies have shown that bzip proteins acquire distinct three-dimensional conformations when bound to different sites. Therefore, we investigated whether swapping the ATF sites between IL-8 and IFN-β could alter both the nucleosomal organization and the transcriptional activation properties of these genes. We generated reporter genes bearing the IFN-β ATF (PRDIV) site in an otherwise intact IL-8 enhancer promoter (IL-8IFN-βAP1) and an IFN-β enhancer bearing the IL-8 ATF site in the place of PRDIV (IFN-βIL–8AP1). Figure 7E shows that conversion of PRDIV to the IL-8 ATF site caused a dramatic decrease of virus-induced IFN-β expression in Namalwa but not in HeLa cells. Remarkably, replacement of the IL-8 ATF site with the PRDIV element led to derepression of IL-8 transcription in Namalwa cells. Figure 7D (lanes 5 and 6) shows that there is no macroH2A on the WT IFN-β enhancer, a result consistent with our previous data (Figure 3). However, conversion of the IFN-β ATF site to the IL-8 ATF site led to the recruitment of macroH2A to the IFN-β enhancer (lanes 7 and 8). This resulted in a strong reduction in NF-κB DNA binding and PolII association with the promoter, a finding correlating with the strong decrease in transcriptional activation. Conversely, the presence of the IFN-β ATF site in the context of an otherwise intact IL-8 enhancer, strongly decreased the recruitment of macroH2A (compare lanes 1and 2 with lanes 3 and 4) and as a result it allowed both NF-κB DNA binding, PolII recruitment (Figure 7D) and gene activation (Figure 7E).
To demonstrate whether the differential in vivo recruitment of macroH2A-containing nucleosomes depends on the binding of ATF-2 to distinct ATF sites, we repeated the in vitro nucleosome reconstitution assays using side-by-side the WT IL-8 and IFN-β enhancers along with their swapped counterparts and HeLa-derived donor chromatin. Figure 7F demonstrates that either full-length ATF-2 or its bZip domain can recruit a macroH2A nucleosome when bound to the IL-8 but not to the IFN-β enhancer (compare lanes 1–3 with 7–9). However, macroH2A-containing nucleosomes were not recruited by ATF-2 on the IL-8 template bearing the ATF site taken from the IFN-β enhancer (lanes 4–6). By contrast, ATF-2 bound to the IFN-β template bearing the IL-8 ATF site efficiently recruits a macroH2A nucleosome (lanes 10–12). These results demonstrate that (1) the bZip DNA-binding domain of ATF-2 is responsible for the recruitment of macroH2A-containing nucleosomes and (2) this recruitment depends on the specific sequence of the ATF DNA site, thus implying a DNA-induced protein allostery mechanism.
Discussion
We have uncovered a previously unrecognized mechanism of establishing a stable gene expression pattern by using widely expressed components, that is, a transcription factor and a histone variant, mixed and matched in a specific manner designed for precise cell-specific gene expression patterns (Figure 8A and B). The use of sequence-specific transcription factors, such as the ATF-2/JunD heterodimer, provides the means for accurate targeting of specialized-nucleosomes, thus ensuring selective control of gene expression.
Figure 8.
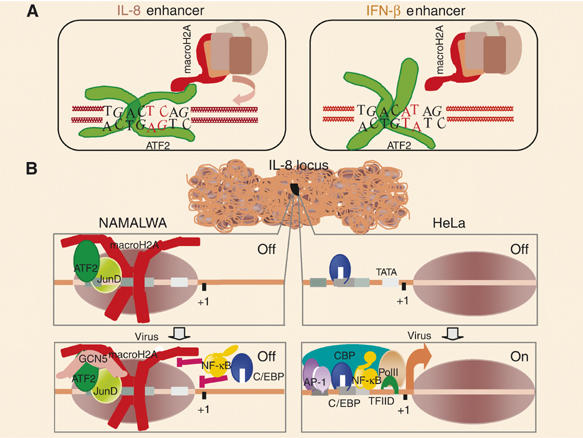
A model for the cell-type-specific expression of the IL-8 gene. (A) A model depicting distinct configurations of ATF-2 bzip domain bound to the IL-8 and IFN-β sites, correlating with the recruitment of macoH2A-containing nucleosomes. (B) The default chromatin architecture of the IL-8 enhancer/promoter differs between distinct cell types. Namalwa B cells are incapable of inducing the gene in response to virus infection because a nucleosome containing the histone variant macroH2A masks the enhancer/promoter region and inhibits the association of the IL-8 activators. This nucleosome is recruited via direct protein–protein interactions between the constitutively bound ATF-2/JunD heterodimer and the macro-domain of macroH2A. In contrast, ATF-2/JunD don not associate with the IL-8 enhancer in HeLa cells and therefore the macroH2A nucleosome is not recruited, thus allowing transcriptional activation.
The specificity in the targeting mechanism is due to the fact that not all ATF-binding sites in the genome promote the recruitment of macroH2A nucleosomes nearby. ATF-2 bound to the IFN-β enhancer site does not recruit macroH2A nucleosomes in vitro or in vivo. As a result, ATF-2 functions as a classical activator of IFN-β transcription and as a repressor of IL-8 transcription by influencing directly the local chromatin architecture through the recruitment of a macroH2A-containing nucleosome via direct protein–protein interactions (Figure 8A). Thus, a transcription factor can establish and orchestrate a stable epigenetic mark by altering directly chromatin structure. In contrast to the previously well-characterized mechanisms of stable (static) epigenetic gene silencing through the assembly of heterochromatic structures, the plasticity of the molecular mechanism we describe here allows for an accurate and rapid selection of target genes for repression and this repression can be propagated to daughter cells and/or reversed by the removal of the transcription factor from the promoter. Our proposal that some transcription factors have been equipped with an additional function besides the classical promotion of gene activation or repression is also supported by the observation that HNF3, a liver-specific transcription factor, can open highly compacted chromatin through direct protein–protein interactions with histones, at least in vitro (Cirillo et al, 2002).
Most likely, the process of transcriptional silencing begins with the binding of ATF-2 to its genomic sites. As ATF-2 recognizes short DNA motifs that occur many times in the genome, cells must possess a mechanism whereby some but not all ATF-2 DNA-binding sites will nucleate macroH2A-containing nucleosomes. How is ATF-2 able to recruit macroH2A nucleosomes at specific sites? The determination of the crystal structure of the ATF/Jun DNA-binding domains bound to the IFN-β site (TGACATAG), which deviates from the IL-8 site (TGACTCAG) in two positions, revealed significant structural differences when compared to the structure of bzip proteins bound to the consensus CRE site (Panne et al, 2004). We propose that the bzip region of ATF-2/JunD bound to the IL-8 site adopts a unique conformation that favors the interaction of the bzip region with the macro-domain of macroH2A nucleosomes. It is quite remarkable that such a small change in the DNA sequence can have profound effects in gene transcription by altering chromatin architecture on enhancers and promoters. The ATF site functions as a module bearing all the information necessary for switching on or off IL-8 transcription. Replacement of the IFN-β ATF site by the IL-8 site led to repression of IFN-β transcription, because the IFN-β promoter was occupied by macroH2A nucleosomes. Furthermore, relocation of the IL-8 ATF site to a new position further upstream also led to IL-8 transcriptional activation because the macroH2A nucleosome was also relocated to the new position, thus exposing enhancer and promoter to the transcriptional machinery. Remarkably, these radical changes in the organization of the chromatin are mediated via a simple protein–protein interaction between ATF-2's bzip domain and the macro-domain of macroH2A. Previous studies have shown that DNA-induced allosteric conformations of the DNA-binding domain of the glucocorticoid receptor expose distinct protein–protein surfaces, leading to the recruitment of coactivators or corepressors of transcription (Rogatsky et al, 2003). Similarly, different NF-κB sites may impose distinct transcription factor configurations leading to the recruitment of unique cofactor complexes, thus affecting transcription in a gene-specific manner (Chen-Park et al, 2002; Leung et al 2004). Therefore, a likely possibility is that the particular IL-8 ATF site imposes a configuration on the bound ATF2/JunD heterodimer that promotes the stable recruitment of macroH2A-containing nucleosomes and not free macroH2A. Our model is also supported by the observation that specific DNA sequences can affect the displacement of histones H2A and H2B at the MMTV promoter after activation (Vicent et al, 2004).
We still do not know why the ATF-2/JunD heterodimer binds constitutively to the IL-8 enhancer in B cells and not in HeLa cells despite the fact that these proteins are present at equivalent amounts (Supplementary Figure 4). One possibility is that B cells contain additional factors, not necessarily transcriptional regulatory proteins, facilitating ATF-2/JunD DNA binding. Furthermore, it is known that the regulation and the relative ratio of AP1 and ATF proteins differs between distinct cell types and this could result in the selection of specific heterodimers for DNA binding in this enhancer in B cells. For example, in the IFN-β case, binding of HMGI(Y) to the ATF site promotes the association of the ATF-2/cJun instead of the ATF-2/JunD heterodimer (Du et al, 1993).
The aberrant expression of IL-8 in inappropriate cell types, like B cells, leads to chronic inflammatory responses and to disease. Therefore, the mechanism we describe here ensures that IL-8 expression will be restricted only to the correct type of cells and not to every cell type as would have been predicted by the ubiquitous expression of the IL-8 activators. Taken together, our experiments suggest that the epigenetic information stored on the IL-8 promoter in the form of a specialized chromatin structure is encoded on the DNA and it is translated differentially by families of transcription factors. Thus, what is really transmitted from one cell to another is the DNA sequence together with the molecular mechanism(s) that ensures sequence-specific DNA binding of ATF proteins to the IL-8 enhancer (Figure 8). Put it simply, it is the unique association of transcription factors with DNA elements that contain the blueprints for setting up the epigenome in this locus.
Our experiments do not exclude the possibility that specialized histone chaperones may also assist in delivering macroH2A to the IL-8 locus in B cells. Previous studies have shown that the HIRA and Swr1 histone chaperones catalyze the replacement of H3.3 and H2A.Z in mammalian and yeast cells, respectively (Krogan et al, 2003; Mizuguchi et al, 2004; Tagami et al, 2004). Furthermore, the formation of macroH2A-containing senescence-associated heterochromatin foci appear to be driven by the chaperon complexes containing ASF1a and HIRA (Zhang et al, 2005). However, the HIRA/ASF1a complex does not seem to directly deposit macroH2A. We showed that the IL-8 locus is selected for repression in B cells by the ability of the ATF-2/JunD heterodimer to associate constitutively with the IL-8 enhancer, leading to the direct recruitment of a macroH2A nucleosome (Figure 8). This signal could be propagated along the body of the gene by the subsequent recruitment of histone chaperones, which exchange H2A for macroH2A.
MacroH2A-containing nucleosomes occur in every 200–500 nucleosomes in the human genome (S Dimitrov, personal communication) and are evenly distributed through out the different structures of chromatin (Abbott et al, 2005). It appears that the transcriptional repressive potential/nature of macroH2A nucleosomes is due to both their tighter folding and to the presence of the macro-domain, which protrudes from the octamer core (Chakravarthy et al, 2005) and which is an ADP-ribose-binding module (Karras et al, 2005). Tighter folding could be responsible for the inability of transcription factors such as NF-κB and C/EBP to access their binding sites when located near the dyad axis. Altogether, we propose that unique nucleosome positions and specific chromatin architectures can work as an epigenetic filter that efficiently selects target genes for expression by exposing or occluding the transcription factor target sites associated with distinct genes.
Materials and methods
Cell transfection and plasmid construction
HeLa cells were transfected with the calcium phosphate method essentially as described before. The cells were harvested 48–60 h following transfection. Namalwa cells were transfected by electroporation using 5 × 107 cells and 5 μg of reporter plasmid at 240 mV and 850 μF. siRNAs were transfected into HeLa or Namalwa cells using lipofectamine following the manufacturer's instructions (Life Technologies). The sequence of the macroH2A siRNA is 5′ AAGCAGGGUGAAGUCAGUAAG, whereas the control siRNA sequence is 5′ AACGUACGCGGAAUACUUCGA 3′. The ATF-2 siRNA was purchased from Santa Cruz.
The WT IL-8 enhancer/promoter luciferase reporter was generated by subcloning the −211 to +59 region or the −127 to +59 of the human IL-8 promoter into the PGL3 basic vector (Promega). The ΔAP1, AP1 OUT, IFN-βATFIL-8, and IL-8IFNβATF mutants were generated by site-directed mutagenesis using standard PCR methodology (Lomvardas and Thanos, 2002). The GST-macroH2A construct was generated by cloning the macroH2A1.2 open reading frame into pGEX5x-1 vector (Pharmacia). All constructs were verified by DNA sequencing. The GST-ATF-2 constructs have been previously described (Du et al, 1993).
Nucleosome mapping, nucleosome reconstitution and ChIP
HeLa and Namalwa cells (1.5 × 107) were mock or virus infected with Sendai virus followed by formaldehyde fixing with 1.1% formaldehyde for 30 min at room temperature. The cells were harvested and mononucleosomes were prepared as described previously (Lomvardas and Thanos, 2002). Mononucleosomal DNA was purified using the Qiagen gel extraction kit followed by primer extension. The primer extension mix including 0.5 μl (1 μg/μl) gene-specific primer, 0.5 μl (2000 U/ml) Vent polymerase, 10 μl (1 mM) dNTPs, 3 μl (25 mM) MgCl2, 5 μl 10 × thermo buffer , 30.5 μl H2O and 1 μg of mononucleosomal DNA. The adaptor used for ligation-mediated PCR contained the sequence 5′gcggtgacccgggagatctgaattc-3′ (top strand) and 5′-P-gaattcagatct and it was ligated to the extension product for 16 h in 16°C followed by PCR using the gene-specific and the top strand adaptor primer. The primers used to map nucleosomes in this study are shown in Supplementary Figure 5.
HeLa donor chromatin and PCR-generated DNA fragments were used in nucleosome reconstitution experiments in the presence or absence of bacterial expressed NF-κB and ATF-2 proteins (10 μg). In total, 1 μg of DNA attached to Dyna magnetic beads was diluted in 1 × reconstitution buffer (50 mM HEPES pH 7.9, 1 mM EDTA, 5 mM DTT, 1 mM PMSF) and 1 M NaCl in the presence of 0.15 μg BSA followed by the addition of 12.5 μg donor chromatin in a final volume of 45 ìl. The mixture was incubated in 30°C and serial dilutions were carried out to a final concentration of 0.1 M NaCl. In each case the mixture was incubated for at least 30 min and finally the reaction was diluted in a buffer containing Tris–HCl pH 7.6, 0.1% NP-40, 20% glycerol, 1 mM EDTA, 5 mM DTT, 1 mM PMSF, and 100 μg/ml BSA. After aspiration, the beads washed three times with BC-50 (50 mM NaCl, 10 mM HEPES pH 7.9, 10% glycerol 0.1 mM EDTA, 0.25 mM DTT, 0.25 mM PMSF) followed by Western blotting.
ChIPs were carried out as described previously (Lomvardas and Thanos, 2002). The primers used for ChIPs and RT–PCR are shown in Supplementary Figure 5. The antibodies used were obtained from Santa Cruz (p65, C/EBPβ, ATF-2, JunD, cFos, cJun, CBP, GCN5, PolII, BRG1 and BRM), Upstate (acH4-tetra, acH4K5, acH4K8, acH4K12, acH4K16, acH3, acH3K9, acH3K14 and macroH2A) and Abcam (H2B, H3 C-terminus and H4).
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Acknowledgments
We thank Stefan Dimitrov and Flore Mietton for recombinant macroH2A protein, antibodies, siRNAs and for helpful discussions throughout this work. We thank Athina Antonaki, Effie Apostolou and Joseph Papamatheakis for help with the transfection experiments, Paschalis Sideras for help with the isolation of primary B cells, Vaggelis Harokopos for help with the real-time PCR analysis and Deppy Papadopoulou for help in preparing the figures. We also thank Tom Maniatis, George Mosialos, Richard Mann, Terry Orr-Weaver, George Panayotou, Nicole Francis and Stavros Lomvardas for critical reading of the manuscript. This work was supported by the March of Dimes, Leukemia and Lymphoma Society of America, Greek Secretariat for Research and Technology (PENED 01ED225), Philip Morris USA Inc. and Philip Morris International.
References
- Abbott DW, Chadwick BP, Thambirajah A, Ausio J (2005) Beyond the Xi: macroH2A chromatin distribution and post-translational modification in an avian system. J Biol Chem 280: 16437–16445 [DOI] [PubMed] [Google Scholar]
- Abbott DW, Laszczak M, Lewis JD, Su H, Moore SC, Hills M, Dimitrov S, Ausio J (2004) Structural characterization of macroH2A containing chromatin. Biochemistry 43: 1352–1359 [DOI] [PubMed] [Google Scholar]
- Agalioti T, Chen G, Thanos D (2002) Deciphering the transcriptional histone acetylation code for a human gene. Cell 111: 381–392 [DOI] [PubMed] [Google Scholar]
- Angelov D, Molla A, Perche PY, Hans F, Cote J, Khochbin S, Bouvet P, Dimitrov S (2003) The histone variant macroH2A interferes with transcription factor binding and SWI/SNF nucleosome remodeling. Mol Cell 11: 1033–1041 [DOI] [PubMed] [Google Scholar]
- Chakravarthy S, Gundimella SK, Caron C, Perche PY, Pehrson JR, Khochbin S, Luger K (2005) Structural characterization of the histone variant macroH2A. Mol Cell Biol 25: 7616–7624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen-Park FE, Huang DB, Noro B, Thanos D, Ghosh G (2002) The kappa B DNA sequence from the HIV long terminal repeat functions as an allosteric regulator of HIV transcription. J Biol Chem 277: 24701–24708 [DOI] [PubMed] [Google Scholar]
- Cirillo LA, Lin FR, Cuesta I, Friedman D, Jarnik M, Zaret KS (2002) Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol Cell 9: 279–289 [DOI] [PubMed] [Google Scholar]
- Costanzi C, Pehrson JR (1998) Histone macroH2A1 is concentrated in the inactive X chromosome of female mammals. Nature 393: 599–601 [DOI] [PubMed] [Google Scholar]
- Du W, Thanos D, Maniatis T (1993) Mechanisms of transcriptional synergism between distinct virus-inducible enhancer elements. Cell 74: 887–898 [DOI] [PubMed] [Google Scholar]
- Ercan S, Carrozza MJ, Workmann JL (2004) Global nucleosome distribution and the regulation of transcription in yeast. Genome Biol 5: 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaus A, Owen-Hughes T (2004) Mechanisms of ATP-dependent chromatin remodeling: farewell to the tuna-can octamer? Curr Opin Genet Dev 14: 165–173 [DOI] [PubMed] [Google Scholar]
- Henikoff S, Furuyama T, Ahmad K (2004) Histone variants, nucleosome assembly and epigenetic inheritance. Trends Genet 20: 320–326 [DOI] [PubMed] [Google Scholar]
- Hoffmann E, Dittrich-Breiholz O, Holtmann H, Kracht M (2002) Multiple control of interleukin-8 expression. J Leukoc Biol 72: 847–854 [PubMed] [Google Scholar]
- Hoffmann E, Thiefes A, Buhrow D, Dittrich-Breiholz O, Schneider H, Resch K, Kracht M (2005) MEK1-dependent delayed expression of Fos-related antigen-1 counteracts c-Fos and p65 NF-κappaB-mediated interleukin-8 transcription in response to cytokines or growth factors. J Biol Chem 280: 9706–9718 [DOI] [PubMed] [Google Scholar]
- Jaenisch R, Bird A (2003) Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Gen Suppl 33: 245–254 [DOI] [PubMed] [Google Scholar]
- Kamakaka RT, Biggins S (2005) Histone variants: deviants? Genes Dev 19: 295–310 [DOI] [PubMed] [Google Scholar]
- Karras GI, Kustatscher G, Buhecha HR, Allen MD, Pugieux C, Sait F, Bycroft M, Ladurner AG (2005) The macro domain is an ADP-ribose binding module. EMBO J 24: 1911–1920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogan NJ, Keogh MC, Datta N, Sawa C, Ryan OW, Ding H, Haw RA, Pootoolal J, Tong A, Canadien V, Richards DP, Wu X, Emili A, Hughes TR, Buratowski S, Greenblatt JF (2003) A Snf2 family ATPase complex required for recruitment of the histone H2A variant Htz1. Mol Cell 12: 1565–1576 [DOI] [PubMed] [Google Scholar]
- Leung TH, Hoffmann A, Baltimore D (2004) One nucleotide in a kB site can determine cofactor specificity for NF-κB dimmers. Cell 118: 453–464 [DOI] [PubMed] [Google Scholar]
- Lomvardas S, Thanos D (2002) Modifying gene expression programs by altering core promoter chromatin architecture. Cell 110: 261–271 [DOI] [PubMed] [Google Scholar]
- Luger K (2003) Structure and dynamic behavior of nucleosomes. Curr Opin Genet Dev 13: 127–135 [DOI] [PubMed] [Google Scholar]
- Mermoud JE, Costanzi C, Pehrson JR, Brockdorff N (1999) Histone macroH2A1.2 relocates to the inactive X chromosome after initiation and propagation of X-inactivation. J Cell Biol 147: 1399–1408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuguchi G, Shen X, Landry J, Wu WH, Sen S, Wu C (2004) ATP-driven exchange of histone H2AZ variant catalyzed by SWR1 chromatin remodeling complex. Science 303: 343–348 [DOI] [PubMed] [Google Scholar]
- Morgan HD, Santos F, Green DW, Reik W (2005) Epigenetic reprogramming in mammals. Hum Mol Gen 14: R47–R58 [DOI] [PubMed] [Google Scholar]
- Panne D, Maniatis T, Harrison SC (2004) Crystal structure of ATF-2/c-Jun and IRF-3 bound to the interferon-beta enhancer. EMBO J 23: 4384–4393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pehrson JR, Fried VA (1992) MacroH2A, a core histone containing a large non histone region. Science 257: 1398–1400 [DOI] [PubMed] [Google Scholar]
- Rogatsky I, Wang JC, Derynck MK, Nonaka DF, Khodabakhsh DB, Haqq CM, Darimont BD, Garabedian MJ, Yamamoto KR (2003) Target-specific utilization of transcriptional regulatory surfaces by the glucocorticoid receptor. Proc Natl Acad Sci USA 100: 13845–13850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarma K, Reinberg D (2005) Histone variants meet their match. Nat Rev Mol Cell Biol 6: 139–149 [DOI] [PubMed] [Google Scholar]
- Tagami H, Ray-Gallet D, Almouzni G, Nakatani Y (2004) Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell 116: 51–61 [DOI] [PubMed] [Google Scholar]
- Turner BM (2002) Cellular memory and the histone code. Cell 111: 285–291 [DOI] [PubMed] [Google Scholar]
- Vicent GP, Nacht AS, Smith CL, Peterson CL, Dimitrov S, Beato M (2004) DNA instructed displacement of histones H2A and H2B at an inducible promoter. Mol Cell 16: 439–452 [DOI] [PubMed] [Google Scholar]
- Zhang R, Poustovoitov MV, Ye X, Santos HA, Chen W, Daganzo SM, Erzberger JP, Serebriiskii IG, Canutescu AA, Dunbrack RL, Pehrson JR, Berger JM, Kaufman PD, Adams PD (2005) Formation of MacroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev Cell 8: 19–30 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5