

Abstract
K28 is a viral A/B toxin that traverses eukaryotic cells by endocytosis and retrograde transport through the secretory pathway. Here we show that toxin retrotranslocation from the endoplasmic reticulum (ER) requires Kar2p/BiP, Pdi1p, Scj1p, Jem1p, and proper maintenance of Ca2+ homeostasis. Neither cytosolic chaperones nor Cdc48p/Ufd1p/Npl4p complex components or proteasome activity are required for ER exit, indicating that K28 retrotranslocation is mechanistically different from classical ER-associated protein degradation (ERAD). We demonstrate that K28 exits the ER in a heterodimeric but unfolded conformation and dissociates into its subunits as it emerges into the cytosol where β is ubiquitinated and degraded. ER export and in vivo toxicity were not affected in a lysine-free K28 variant nor under conditions when ubiquitination and proteasome activity was blocked. In contrast, toxin uptake from the plasma membrane required Ubc4p (E2) and Rsp5p (E3) and intoxicated ubc4 and rsp5 mutants accumulate K28 at the cell surface incapable of toxin internalization. We propose a model in which ubiquitination is involved in the endocytic pathway of the toxin, while ER-to-cytosol retrotranslocation is independent of ubiquitination, ERAD and proteasome activity.
Keywords: A/B toxin, ERAD, retrotranslocation, ubiquitination, yeast
Introduction
K28 is a protein toxin secreted by Saccharomyces cerevisiae strains infected with a toxin-coding double-stranded RNA killer virus. The α/β heterodimeric protein kills susceptible yeasts by blocking DNA synthesis resulting in both G1/S cell cycle arrest and caspase-mediated apoptotic cell death (Reiter et al, 2005). Like microbial A/B toxins, K28 enters the cytosol of a target cell by retrograde transport through the secretory pathway. Members of this family usually consist of a single catalytic A or α-subunit and one or more B or β-subunit(s) responsible for toxin binding to the cell surface and translocation of the cytotoxic A/α-subunit to the cytosol (Falnes and Sandvig, 2000). While some A/B toxins enter mammalian cells from and through acidic endosomes (like Anthrax toxin, Diphteriae toxin, Chlostridia neurotoxins), others are taken up by receptor-mediated endocytosis and transported retrograde to the cytosol by travelling through the Golgi apparatus and the endoplasmic reticulum (ER) (Vago et al, 2005). As member of this toxin family, K28 is a α/β heterodimer whose subunits are covalently linked by a single disulfide bond (Riffer et al, 2002). In vivo, intoxification of a sensitive cell requires initial toxin binding to a cell wall receptor and subsequent translocation to the cytoplasmic membrane where the toxin interacts with a so far unknown membrane receptor. Uptake and retrograde transport of the toxin depend on the existence of a short amino-acid motif (HDEL) at the C-terminus of β which serves as intracellular targeting signal that is bound by the HDEL receptor Erd2p of the host cell. It has been proposed that within the secretory pathway of intoxicated yeast, Erd2p recognizes the β-C-terminus and catalyzes α/β toxin redistribution from an early Golgi compartment back to the ER (Eisfeld et al, 2000; Riffer et al, 2002). Several other A/B toxins (like Pseudomonas exotoxin A, cholera toxin, Escherichia coli HLT) also contain C-terminal H/KDEL or H/KDEL-like targeting motifs which ensure toxin delivery to the cytosol (Lencer et al, 1995). However, how precisely the toxins exit the ER and translocate to the cytosol is still unclear, although it has been proposed that the toxins disguise themselves as misfolded proteins that are recognized as substrate for ER-associated protein degradation (ERAD) (Hazes and Read, 1997; Lord et al, 2003). In the ERAD pathway, misfolded and/or misassembled proteins undergo quality control in the ER and, if misfolded persistently, are targeted to retrotranslocation and proteasomal degradation (for reviews, see Ellgaard and Helenius, 2003; Jarosch et al, 2003; Meusser et al, 2005).
In this study, we show that ER export of the K28 virus toxin depends on the assistance of Kar2p/BiP, Pdi1p, Scj1p, and Jem1p and present evidence that cellular Ca2+ homeostasis in the secretory pathway is critical for K28 toxicity. We show that K28 exits the ER in its heterodimeric but unfolded conformation and dissociates into its subunits after entering the cytosol. The β-subunit is subsequently ubiquitinated and degraded while α transmits the lethal signal into the nucleus. In yeast ubc4 and rsp5 mutants, the toxin can no longer enter the cell rendering these mutants toxin resistant. A mechanistic model is proposed describing the most crucial events in toxin uptake and retrograde transport, indicating that K28 represents a powerful tool in identifying novel components essentially involved in eukaryotic cell function.
Results
Kar2p and Pdi1p assist toxin exit from the ER
We have previously shown that the Hsp70 chaperone Kar2p/BiP is required for proper targeting of internalized K28 toxin to the ER luminal side of the Sec61 complex (Eisfeld et al, 2000). As shown in Figure 1A, Western analysis of cell fractions from a toxin-treated kar2-203ts mutant grown at the restrictive temperature confirmed that K28 accumulates in intracellular membranes (most likely ER membranes) and can no longer exit the ER, explaining why kar2 mutants are toxin resistant.
Figure 1.
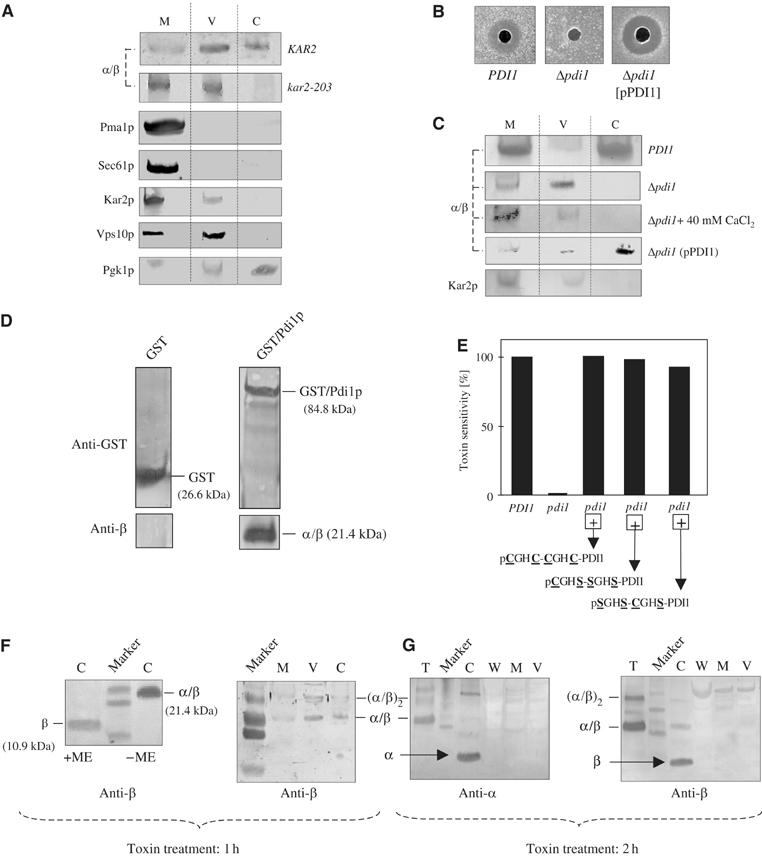
Kar2p and Pdi1p ensure toxin retrotranslocation from the ER. (A) Immunoblot of cell fractions of a kar2-203 mutant and its wild type (KAR2) treated with toxin for 1 h and grown at 35°C. Cell extracts were separated by nonreducing SDS–PAGE and probed with K28-specific anti-β (M, membrane fraction (cytoplasmic, ER and Golgi membranes); V, endosomal vesicle fraction; C, cytosol). Marker proteins were probed with anti-Pma1p, anti-Sec61p, anti-Kar2p, anti-Vps10p, and anti-Pgk1p. (B) K28 resistance in a Δpdi1 mutant and its complementation to hypersensitivity after coexpression of PDI1 from pPDI1. (C) Immunoblot of cell fractions from toxin-treated PDI1 wild-type and Δpdi1 mutant cells before and after coexpression of PDI1 from the centromer vector pPDI1. (D) Physical interaction between K28 and a GST-Pdi1p fusion protein evidenced by coelution from a glutathione sepharose matrix. Bound proteins were subjected to SDS–PAGE and probed with anti-GST or anti-β. (E) Toxin sensitivity is fully restored in a K28-resistant Δpdi1 mutant after coexpression of either wild-type Pdi1p (pCGHC-CGHC-PDI1) or its active site mutants pCGHS-SGHS-PDI1 and pSGHS-CGHS-PDI1. (F) Cell fractions of wild-type yeast treated with toxin for 1 h separated by SDS–PAGE in the presence or absence of β-mercaptoethanol (+ME, −ME) and probed with anti-β. (G) Western analysis of cell fractions of wild-type yeast treated with toxin for 2 h and separated by SDS–PAGE under nonreducing conditions. Blots were probed with anti-α or anti-β (T, toxin control; C, cytosol; W, cell wall fraction; M, intracellular membrane fraction; V, endosomal vesicle fraction).
As Kar2p and Pdi1p are known to be central components in delivering misfolded proteins to the Sec61 export channel in the ER membrane, we asked if Pdi1p as part of this quality control is also involved in K28 toxicity. In yeast, simultaneous deletion of PDI1 and its homologous genes MPD1, MPD2, EPS1 and EUG1 is lethal, but cell viability and cell growth can be restored by single-copy expression of MPD1 under PDI1 promoter control (Norgaard et al, 2001). As shown in Figure 1B, such a pdi1 mutant is completely toxin resistant while Pdi1+ cells are sensitive. Interestingly, retransformation with PDI1 on a centromer vector (pPDI1) fully restored sensitivity and resulted in a slight increase in toxin sensitivity compared to wild type (Figure 1B). Western analysis further revealed that ER-to-cytosol export of K28 is completely blocked in the pdi1 mutant while it was fully restored after providing PDI1 from a single-copy plasmid (Figure 1C). As Pdi1p is a known calcium-binding protein in the ER, and retrotranslocation competence of K28 is severely affected by calcium levels in the secretory pathway (see below), we aimed to exclude any indirect effect by additionally analyzing toxin export to the pdi1 mutant cytosol in the presence of 40 mM calcium. As illustrated in Figure 1C, the addition of extra calcium did not restore toxin retrotranslocation in the pdi1 mutant, confirming that it is the absence of Pdi1p that causes toxin accumulation in the ER. The important role of Pdi1p in toxin retrotranslocation was further supported in a GST pull-down experiment by demonstrating physical interaction between Pdi1p and K28 (Figure 1D). As both proteins coeluted from a glutathione sepharose matrix, it can be concluded that K28 and Pdi1p also interact during toxin retrotranslocation in vivo.
Pdi1p ensures ER retrotranslocation competence of the toxin
For cholera toxin (CT) it was proposed that Pdi1p acts as a redox-dependent chaperone that depends on the oxidase activity of Ero1p (Tsai and Rapoport, 2002). However, as it was recently argued that Pdi1p binding to its substrate might not at all be regulated by the redox state of Pdi1p (Lumb and Bulleid, 2002), we investigated whether or not Pdi1p must be re-oxidized by Ero1p to release K28 in the ER. Interestingly, toxin sensitivity in an ero1 mutant was identical both at the permissive and at the restrictive temperature (data not shown), indicating that the effect of Pdi1p on K28 export is different from its proposed unfolding activity during ER exit of CT.
The precise effect of Pdi1p on K28 toxicity was further analyzed by testing a set of mutants coexpressing Pdi1p variants with active site mutations. As member of the thioredoxin family, Pdi1p contains two catalytically active Cys-Gly-His-Cys (CGHC) sites, which are both involved in disulfide bond formation. Within both domains, only the first cysteine is essential for isomerization reactions between Pdi1p and its substrate (Holst et al, 1997; Solovyov et al, 2004). We therefore tested two Pdi1p mutants (CGHS-SGHS-[PDI] and SGHS-CGHS-[PDI]) defective as protein oxidant but still capable to catalyze disulfide bond isomerization. To do so, the Δpdi1 null mutant (carrying wild-type PDI1 on a URA3 plasmid) was cotransformed with a TRP1-containing plasmid expressing either of the two Pdi1p variants. After cotransformation, cells were forced to lose the wild-type PDI1-encoding plasmid by plating onto 5-fluoro orotic acid agar. As shown in Figure 1E, both active site mutants restored toxin sensitivity in the Δpdi1 mutant, indicating that it might be a partial unfolding by Pdi1p (rather than a redox reaction), which is required for K28 toxin export to the cytosol.
In wild-type cells treated with toxin for 1 h, K28 was present in the cytosol in its α/β conformation (Figure 1F), indicating that the single disulfide bond between Cys56 in α (10.5 kDa) and Cys340 in β (10.9 kDa) (Riffer et al, 2002) is sufficiently stable in the reducing environment of the cytosol to prevent or delay rapid toxin dissociation. However, the heterodimeric toxin in the intracellular membrane fractions and in the cytosol almost disappeared when cells were treated with toxin for 2 h; under such conditions, the intracellular ‘stores' of the α/β toxin were depleted and both subunits, α and β, became detectable and predominated in the cytosol; consequently, only traces of cytosolic α/β toxin remained nondissociated (Figure 1G). From these findings, three conclusions can be drawn: (i) K28 is capable to exit the ER in its α/β conformation, (ii) complete toxin unfolding (i.e. reduction of its single disulfide bond) is not a prerequisite for ER retrotranslocation, and (iii) toxin dissociation into its subunits occurs after the heterodimer entered the cytosol.
Essential role of DnaJ-domain chaperones in ER export of K28
DnaJ domain-containing Hsp40 co-chaperones facilitate export of ERAD substrates into the cytosol by interacting with Kar2p in the ER lumen and preventing aggregation of misfolded proteins before retrotranslocation (Nishikawa et al, 2001). To check if the Hsp40s Scj1p and Jem1p have any effect on K28 toxicity, single and double deletion mutants were tested for K28 sensitivity. While Scj1+ Jem1+ wild-type cells as well as the single deletion mutants were sensitive, the Δscj1/Δjem1 double mutant showed complete toxin resistance (Figure 2A). Furthermore, K28 was absent from the mutant cytosol and accumulated in intracellular membranes, indicating that ER exit of the toxin is blocked in the genetic background of a Δscj1/Δjem1 double knockout (Figure 2B).
Figure 2.

ERAD-independent toxin retrotranslocation requires proper maintenance of Ca2+ homeostasis and DnaJ co-chaperone activity in the ER. (A) Methylene blue agar diffusion assay illustrating K28 toxin resistance in a Δscj1/Δjem1 double knockout mutant. (B) Western analysis of cell fractions from wild-type (SCJ1 JEM1) and its isogenic Δscj1 Δjem1 double mutant after 1 h toxin treatment. (C) K28 resistance in a Δpmr1 mutant lacking the Ca2+/Mn2+ ion pump Pmr1p and functional complementation to toxin sensitivity by increasing extracellular Ca2+ levels. Toxin sensitivity in a Δspf1/cod1 mutant lacking the second P-type ATPase Spf1p/Cod1p is decreased to 36% of wild type. (D) Immunoblot of cell fractions of a toxin-treated Δpmr1 mutant grown in the absence or presence of 40 mM CaCl2 (Kar2p was used as ER luminal marker). (E) Toxin sensitivity in single and double Δubc1-7 and/or Δhrd1 mutants is impaired in the genetic background of a Δubc4 deletion. Note that already in a heterozygous diploid (ubc4/UBC4 ubc5/UBC5), toxin sensitivity is 60% reduced compared to wild type.
Cytosolic chaperones are not involved in toxin retrotranslocation
The cytosolic AAA-ATPase Cdc48p/p97 is involved in dislocating ubiquitinated and nonubiquitinated ERAD substrates for proteasomal degradation (Rabinovich et al, 2002; Ye et al, 2003). To analyze if Cdc48p/p97 together with its cofactors Ufd1p and Npl4p is involved in extracting K28 from the ER, yeast mutants defective in the corresponding genes were tested for toxin sensitivity. As illustrated in Table I, cdc48-1/cdc48-3 as well as npl4-1, npl4-2, and ufd1-1 mutants all displayed wild-type sensitivity indicating that toxin retrotranslocation is not affected in mutants defective in Cdc48p/Ufd1p/Npl4p complex components. Consistently, yeast Δdsk2 and Δrad23 mutants (blocked in delivering ubiquitinated proteins to the proteasome (Medicherla et al, 2004)) as well as mutants defective in the chymotrypsin-like activity of the proteasome (pre1-1, pre2-2) were likewise not impaired in toxin sensitivity (Table I), indicating that ER export of K28 is mechanistically different from ‘classical' ERAD. The same sensitive phenotype was also seen in mutants lacking cytosolic Hsp70s of the Ssa and Ssb chaperone family (Table I), demonstrating that even cytosolic chaperones are not involved in K28 retrotranslocation.
Table 1.
Toxin sensitivity is not impaired in mutants defective in cytosolic chaperones, Cdc48p/Ufd1p/Npl4p complex components or proteasome delivery factors
| S. cerevisiae strain (plasmid) | Relevant genotypea | Toxin sensitivity (%)b |
|---|---|---|
| W303 | CDC48 | 100 |
| WPY106 | cdc48-3 (at 30°C) | 100 |
| MLY1763 | cdc48-1 (at 20°C) | 100 |
| S288C | NPL4 | 100 |
| PSY2340 | npl4-1 (at 25°C) | 100 |
| PSY2341 | npl4-2 (at 25°C) | 100 |
| BWG1-7a | UFD1 | 100 |
| PM373 | ufd1-1 | 100 |
| BY4742 | DSK2 | 100 |
| YMR276w | Δdsk2 | 100 |
| BY4742 | RAD23 | 100 |
| YEL037c | Δrad23 | 100 |
| JN55 | SSA SSB wild type | 100 |
| JN212 | Δssb1,Δssb2 (at 35°C) | 100 |
| JB67 | ssa1-45 ts, ssa2∷LEU2 | 100 |
| ssa3∷TRP1, ssa4∷LYS2 | ||
| WCG4α | PRE1 PRE2 | 100 |
| WCG4-11a | pre1-1 (at 35°C) | 100 |
| WCG4-22a | pre2-2 (at 35°C) | 100 |
| WCG4α [pWO21] | PRE1 PRE2 overexpressing mutant ubiquitin (Ub-RR48/63) | 100 |
| The mutant phenotype in a cdc48-1, npl4-1, npl4-2, or cdc48-3 mutant already manifests at the indicated temperatures of 20, 25, and 30°C. | ||
| In each strain, toxin sensitivity was measured on MBA plates (pH 4.7) against 104 units K28 (corresponding to 1 pmol (32 ng) purified protein toxin) resulting in growth inhibition zones of 12–18 mm in diameter. |
Changes in Ca2+ homeostasis in the secretory pathway prevent K28 toxicity
Given that in a Δpmr1 mutant misfolded CPY* is significantly stabilized (Durr et al, 1998), we hypothesized that Pmr1p might also affect K28 toxicity and we therefore analyzed a Δpmr1 null mutant and its isogenic wild type. In contrast to toxin susceptible Pmr1+ cells, Δpmr1 mutant cells were completely resistant, accumulated internalized toxin in intracellular membranes and were incapable to release the toxin from the ER (Figure 2C and D). Interestingly, toxin sensitivity and ER export could be fully restored by stepwise enhancing exogenous calcium in the medium, giving rise to full sensitivity and regained retrotranslocation competence at 40 mM Ca2+ (Figure 2C and D). We conclude from these experiments that Ca2+ levels in the ER play a crucial role in toxin translocation to the cytosol and that proper maintenance of Ca2+ homeostasis is an essential prerequisite for the in vivo activity of the toxin. In contrast to K28 resistance in a Δpmr1 strain, mutants defective in Spf1p/Cod1p, the second P-type ATPase involved in ERAD and maintenance of Ca2+ homeostasis in the ER (Cronin et al, 2002) were killed by the toxin, but sensitivity in the Δspf1/cod1 mutant was significantly decreased to 36% of wild-type level (Figure 2C).
Ubiquitination and K28 toxicity
Most ERAD substrates undergo polyubiquitination during retrotranslocation from the ER before proteasomal degradation. To examine if Ubc proteins participate in toxin retrotranslocation, a set of ubc deletion mutants was tested against K28. We hypothesized that mutants such as Δubc1, Δubc6, and Δubc7 (which are defective in ER-to-cytosol export of ERAD substrates) should become toxin resistant if K28 was recognized as ERAD substrate in the ER. However, unexpectedly, neither of the tested ubc single or double knockouts was toxin resistant, instead all mutants displayed wild-type sensitivity (Figure 2E). The same was true for mutants defective in Hrd1p, a ubiquitin protein ligase (E3) required for the ubiquitination of ERAD substrates in conjunction with Ubc7p and/or Ubc1p (Bays et al, 2001). In contrast, Δubc4 mutants showed a dramatic decrease in toxin sensitivity and all double mutant combinations in conjunction with Δubc4 were completely K28 resistant (Figure 2E).
To clarify if toxin ubiquitination is involved in or possibly required for K28 toxicity, the cytosol of wild-type cells treated with toxin for 2 h (and thus containing toxin that already dissociated into its subunit components; see Figure 1F) was immunoprecipitated with anti-α or anti-β and probed with anti-Ub. Interestingly, only the β-subunit was ubiquitinated, whereas no such signal was detectable for α (Figure 3A). Additional controls (Figure 3B) excluded any nonspecific staining of the α/β toxin or its monomeric subunits by the anti-ubiquitin antibody. In contrast to wild-type toxin, a mutant variant (α*/β) in which the potential EH (Eps15 Homology) domain 16SWG18 in α had been destroyed and converted into 16TFA18 (in order to exclude that this motif has any in vivo relevance within the clathrin-mediated endocytic pathway of K28) resulted in a biologically inactive toxin that was capable to enter a cell and to retrotranslocate into the cytosol. However, in contrast to wild-type α/β toxin, the mutant α-subunit (α*) was now substrate for ubiquitination explaining why α*/β is nontoxic (Figure 3A and C).
Figure 3.

Ubiquitination and in vivo toxicity of a lysine-free K28 variant. (A) Immunoprecipitation (IP) and anti-Ub blot of cytosolic fractions of untreated cells (control) or cells treated for 2 h with wild-type toxin (α/β) or its mutant variant α*/β. As indicated, IPs were performed with polyclonal anti-α or anti-β, and the resulting IP was probed with monoclonal anti-Ub. Mono- and polyubiquitinated toxin signals (α*-Ub, [α*]-Ubx, β-Ub, [β]-Ubx) are indicated and positions of immunoglobulin light and heavy chains are marked with IgG-LC and IgG-HC, respectively. (B) Immunoblot excluding nonspecific anti-Ub staining of the α/β toxin or its monomeric subunits. Purified K28 toxin was separated by SDS–PAGE under reducing or nonreducing conditions in the presence or absence of β-mercaptoethanol (β-ME) and probed with anti-Ub or anti-β. (C) Anti-β immunoblot of cytosolic fractions of a K28-sensitive yeast either untreated (control) or treated for 1 h with wild-type α/β toxin or a biologically inactive mutant variant (α*/β). In contrast to wild-type toxin, cytosolic α*/β toxin migrates as protein ladder most likely representing ubiquitination products (indicated as [α*/β]-Ubx). (D) Schematic outline of the α/β wild-type toxin and positions of the endogenous lysine residues that had been converted into arginine in the mutant variant αK-0/βK-0. (E) In vivo killing activity (top) of wild-type α/β toxin and its lysine-free mutant αK-0/βK-0. Relative toxin secretion (bottom) in yeast expressing the indicated toxin variant determined by Western analysis of a 50-fold concentrated cell-free culture supernatant probed with anti-β (purified K28 wild-type toxin (20 ng) was used as positive control). (F) Western analysis of cell fractions of yeast treated for 1 h with α/β wild-type toxin or its mutant variant αK-0/βK-0. (G) Anti-Ub blot of the cytosol of wild-type yeast either untreated (toxin-free control) or treated for 1 h with αK-0/βK-0 and probed with anti-β or anti-Ub. Position of αK-0/βK-0 and nonspecific polyubiquitination signals (Ubx) in the cytosol are indicated (S, marker protein standard; C, cytosolic fraction).
A lysine-free toxin variant still exits the ER and kills in vivo
The presented results suggested to us that ubiquitination of β is a secondary effect occurring only after the K28 heterodimer entered the cytosol and dissociated into its subunits; thus, toxin ubiquitination might not at all be required for ER export. This assumption was confirmed by a toxin variant in which all four internal lysines had been converted to arginine (Figure 3D). Such a toxin (αK-0/βK-0) lacks any endogenous lysine residue and internal ubiquitination should be prevented. If ubiquitination generated the driving force for ER export, αK-0/βK-0 should accumulate in the ER and toxicity should be impaired. However, toxicity was not affected and both wild-type toxin and its αK-0/βK-0 variant showed almost identical in vivo killing activity (Figure 3E). Cell fractionation on yeast treated with wild-type toxin or its αK-0/βK-0 mutant variant confirmed that in either case the α/β toxin is present in the cytosol (Figure 3F), demonstrating that retrotranslocation is not prevented in a toxin devoid of internal ubiquitination sites. Furthermore, as toxin-specific ubiquitination was not detectable in the cytosol of αK-0/βK-0-treated cells, N-terminal ubiquitination at the α- and/or β-subunit can also be excluded (Figure 3G). Interestingly and in contrast to in vivo toxicity, relative toxin secretion was significantly decreased in the mutant toxin lacking internal lysyl residues (Figure 3E); however, it was still capable to exit the ER without being ubiquitinated; thus, toxin ubiquitination is apparently not required for extracting K28 from the ER. This is further supported by the finding that simultaneous overexpression of mutant ubiquitin (Ub-RR48/63)—that can no longer form polyubiquitin chains and thus prevents or minimizes proteasomal toxin degradation—as well as yeast proteasome mutants pre1-1 and pre2-2 likewise did not affect toxicity (Table I), confirming that ubiquitination and proteasomal degradation is a secondary effect, not required for ER retrotranslocation of K28.
Ubc4p (E2) and Rsp5p (E3) are essential components for K28 toxicity
Based on the toxin-resistant phenotype of a yeast Δubc4 mutant, we hypothesized that Ubc4p might be involved in a process upstream from ER dislocation, possibly in receptor-mediated toxin internalization at the level of the plasma membrane. To address this aspect, we focused our attention to the HECT domain Ub ligase Rsp5p as it has already been shown to receive ubiquitin from the ubiquitin-conjugating E2s Ubc4p and Ubc5p (Haynes et al, 2002; Reggiori and Pelham, 2002).
We tested a set of rsp5 mutants defective in the formation of ubiquitin–thioester intermediates (rsp5-1) and/or α-factor internalization (rsp5-2) (Wang et al, 1999). Both mutants are temperature sensitive for growth at 37°C and were, therefore, tested against K28 at 20 and 35°C (Figure 4A). While the Rsp5+ wild-type was sensitive at all temperatures tested, the rsp5-1 mutant showed a significant decrease in K28 sensitivity already at 20°C and became fully resistant at 35°C (Figure 4A). Cell fractionation analysis indicated that the toxin is only detectable in the plasma membrane fraction of the rsp5-1 mutant and neither in the endosomal vesicle fraction nor in the cytosol (Figure 4B), indicating that Rsp5p-deficient cells are blocked in toxin internalization from the plasma membrane. Mutations within the WW domains of Rsp5p (such as WW-1 and WW-3), which have been shown to negatively affect fluid-phase endocytosis of lucifer yellow and its transport to the vacuole (Dunn and Hicke, 2001b), also caused a significant decrease in toxin sensitivity; however, the effect was not as pronounced as in the genetic background of a rsp5-1 and/or rsp5-2 mutation, which both caused resistance at the restrictive temperature (Figure 4A). Most strikingly, an entirely analogous phenotype was seen in toxin-treated cells of a Δubc4 mutant which likewise accumulated K28 at the plasma membrane and were completely deficient in toxin internalization (Figure 4B). To confirm that both mutants are impaired in toxin uptake from the plasma membrane, spheroplasts of Δubc4 and rsp5-1 mutants were treated with purified toxin and at different time intervals aliquots were removed from each sample and the amount of unbound toxin in the cell-free culture supernatant was determined by Western analysis. In contrast to UBC4 RSP5 wild-type cells, in which toxin uptake continuously increased during the time course of the experiment, toxin uptake was completely blocked in Δubc4 and rsp5-1 mutant cells (Figure 4C). The same phenotype could be confirmed in an agar diffusion assay by measuring residual toxin activity in the cell-free culture supernatant of toxin-treated Ubc4+ wild-type and Δubc4 mutant cells (Figure 4D).
Figure 4.

Ubc4p and Rsp5p mediate toxin internalization from the plasma membrane. (A) Mutations in Rsp5p (E3) cause a significant decrease in toxin sensitivity at the permissive/semirestrictive temperature (20°C) and show complete resistance at the restrictive temperature (35°C), whereas RSP5 wild-type cells are killed by the toxin. (B) Immunoblot of cell fractions from rsp5-1 and Δubc4 mutant cells and their isogenic wild type after toxin treatment for 1 h. Cell extracts were separated by SDS–PAGE and probed with anti-β (cell fractions as described in Figure 1A). (C) Toxin uptake by yeast cell spheroplasts is severely impaired in Δubc4 and rsp5-1 mutants. Toxin uptake was determined indirectly via SDS–PAGE and Western analysis (probed with anti-β) by detecting the amount of unbound toxin remaining in the cell-free culture supernatant after incubating the indicated yeast at 30°C with wild-type toxin for up to 5 h (input resembles the amount of K28 at the beginning of the experiment). (D) Residual unbound killer activity in the cell-free culture supernatant of a Δubc4 mutant and its UBC4 wild-type after incubating 5 × 107 cells/ml at 20°C in the presence of K28 toxin. At the indicated time points, aliquots (20 μl) were removed, cells were pelleted and killer activity remaining in the supernatant was determined on MBA plates. (E) Experimental set up for the in vivo expression of a suicidal K28 α-variant in the ER. Wild-type yeast (or the mutants shown in (F)) were transformed with a suicidal 2μ vector expressing the K28 α-subunit including its N-terminal secretion signal (SS) from the GAL1 promoter (PGAL1) and terminator (TGAL1) sequences. After shifting the cells from raffinose to galactose, the toxic K28 variant (SS-α) is imported into the ER, the N-terminal signal sequence is removed by signal peptidase (SP) cleavage, and α is retrotranslocated to the cytosol from where it enters the nucleus and causes cell death. (F) Genetic screen indicating that neither wild-type nor rsp5 or Δubc4 mutant cells tolerate galactose-induced expression of K28-α in the ER. In contrast, signal peptidase mutants Δspc1 and Δspc2 (internal positive control) survive K28-α expression in the ER. For unknown reasons, rsp5 mutants showed an intermediate phenotype characterized by a delay in cell growth for 4 days when cultivated on galactose agar at the restrictive temperature (35°C).
To clarify whether or not Ubc4p and Rsp5p are also involved in toxin retrotranslocation from the ER, a lethal K28 α variant was introduced into the ER (Figure 4E) and the corresponding transformants were screened for survival and cell growth after galactose-induced α-toxin expression. As illustrated in Figure 4F, wild-type cells showed the expected suicidal phenotype and were rapidly killed by the expression of α in the ER. The same suicidal phenotype was seen in the Δubc4 mutant, whereas the signal peptidase mutants Δspc1 and Δspc2 (which had been included as internal positive controls) survived because in these mutants α can no longer exit the ER and translocate into the cytosol (Figure 4F). Interestingly, rsp5-1 and rsp5-2 mutants showed a somewhat intermediate phenotype characterized by a significant delay in cell growth after α expression in the ER. In both rsp5 mutants, cell growth restored only after an incubation for 4 days at 30°C under α-inducing conditions (Figure 4F). Although such a genetic screen is only an indirect mean to identify cellular proteins potentially involved in K28-α toxin retrotranslocation from the ER, together these data indicate that toxin resistance in both mutants (rsp5 and ubc4) is likely caused by a defect in toxin uptake from the plasma membrane rather than by a block in toxin retrotranslocation from the ER.
Discussion
A/B toxins such as Bordetella pertussis toxin (PT), Pseudomonas exotoxin A (PE), and Shigella toxin translocate their catalytically active toxic domain into the host cell cytosol. Some of these toxins developed a unique strategy for cell entrance and penetration by using receptor-mediated endocytosis and retrograde transport through the secretion pathway. Within the ER lumen, these toxins translocate to the cytosol by masquerading as ERAD substrates and thus exploiting the ER quality control system of the host. The fact that all toxins entering the cytosol from the ER are extremely low in lysine may point to an evolutionary conserved strategy to escape ubiquitination and degradation (Lord et al, 2003). The yeast K28 virus toxin as member of this family is a secreted α/β heterodimer that likewise is low in lysine, containing only three lysyl residues in α and just a single lysine in β (Eisfeld et al, 2000). However, in contrast to the majority of bacterial and plant A/B toxins (which usually act within the cytosol), the lethal α-subunit of K28 enters the nucleus to cause cell death, and until now it was not known how precisely the toxin traverses its host (Reiter et al, 2005).
In this study, we show that K28 co-opts the ER chaperones Kar2p, Pdi1p, Scj1p, Jem1p, and Pmr1p of the target cell to translocate across the ER membrane. Physical interaction between the toxin and Pdi1p was demonstrated in a GST pull-down experiment by coelution of K28 and GST-Pdi1p. Furthermore, using a linear α–β protein fusion of K28 as bait, additional two hybrid analyses also confirmed that the toxin interacts with Kar2p and with its Hsp40 co-chaperones Scj1p and Jem1p (data not shown). In contrast to other A/B toxin family members, ER retrotranslocation of K28 neither required the AAA ATPase complex Cdc48p/p97-Ufd1p-Npl4p (Ye et al, 2001), nor cytosolic proteins (Rad23p and Dsk2p) involved in delivering ubiquitinated ERAD substrates to the proteasome (Medicherla et al, 2004), nor proteasome activity. However, consistent with the function of Jem1p and Scj1p in stabilizing misfolded pro-α-factor (ΔGpαF) and carboxypeptidase Y (CPY*) in the ER, we show here that the absence of both Hsp40s leads to toxin resistance caused by a block in toxin retrotranslocation to the cytosol. In addition to the essential role of Kar2p and its Hsp40 co-chaperones in ensuring ER exit of the toxin, proper maintenance of Ca2+ homeostasis in the ER is equally important for K28 toxicity. Yeast Δpmr1 (and to a lesser extent Δspf1/cod1) mutants are toxin resistant and incapable to release K28 into the cytosol. However, toxin retrotranslocation and K28 sensitivity could be fully restored by stepwise increasing the extracellular Ca2+ concentration up to 40 mM. The Ca2+/Mn2+ ion pump Pmr1p resident in a medial Golgi compartment plays a major role in maintaining Ca2+ levels in the secretory pathway (Rudolph et al, 1989), and yeast Δpmr1 null mutants show a 50% decrease in free Ca2+ in the ER (Strayle et al, 1999). The presence of Ca2+ in the ER has numerous functions and is required for the retention of resident ER proteins, for proper protein folding as well as for the degradation of ERAD substrates (Durr et al, 1998).
In most cases, ER retrotranslocation of misfolded proteins and A/B toxins is linked to polyubiquitination at the cytosolic surface of the ER membrane. By analyzing the cytosol of intoxicated cells, we demonstrate that K28 exits the ER in its α/β conformation, thereafter it dissociates into its subunits and, within the cytosol, β is ubiquitinated and proteasomally degraded while α enters the nucleus and causes cell death. In K28-treated cells, the toxin is initially detectable as heterodimer in the cytosol, indicating that it is sufficiently stable in the reducing environment of the cytosol to prevent rapid dissociation. This finding is consistent with a recent study on human superoxide dismutase (SOD) that similarly appears to have the ability to maintain intrasubunit disulfide bonds in the cytosol (Lindberg et al, 2004). In case of K28, this also implies that the heterodimer must be capable to traverse the ER membrane, consistent with a recent report demonstrating that protein unfolding is not a prerequisite for ER dislocation (Tirosh et al, 2003). Among A/B toxins, however, K28 is the first example of a protein that exits the ER in a heterodimeric conformation, although retrotranslocation itself (as shown here) critically depends on the unfolding activity of Pdi1p. In vivo, expression of extra copies of Pdi1p caused a significant increase in toxin sensitivity, resulting in a K28 hypersensitive phenotype in the retransformed pdi1 mutant. This increase in toxin sensitivity might have been caused by the absence of PDI-like proteins in the multiple pdi1 mutant, as it was recently shown (by using a semipermeabilized cell system) that PDI-like proteins play opposing roles during retrotranslocation of CT from the ER: CT retrotranslocation was facilitated by Pdi1p, whereas ER retention of the toxin was mediated by PDI-like proteins (Forster et al, 2006). For CT—and in contrast to K28 toxin—Pdi1p-dependent reductive dissociation in the ER was demonstrated for the A chain of CT (CTA) which likewise contains intramolecular disulfide bonds. In CTA, the A1 fragment is translocated to the cytosol after Pdi1p has unfolded the toxin once its A chain has been cleaved. In this case, it has been suggested that Pdi1p acts as a redox-driven chaperone that (in its reduced state) binds to the A chain by recognizing hydrophobic domains transiently exposed by thermal fluctuation of the polypeptide chain (Tsai et al, 2001). However, results of a more recent study argued that Pdi1p substrate binding might not at all be regulated by its redox state (Lumb and Bulleid, 2002).
Toxin unfolding is also required for ER retrotranslocation of ricin A (RTA), and introducing a disulfide bond into RTA greatly inhibits its in vivo toxicity (Wesche et al, 1999). In analogy to CTA and RTA, we demonstrate here that Pdi1p as part of the ER quality control is also involved in K28 toxicity by interacting with the viral toxin and assisting its targeting to the Sec61 export channel (Eisfeld et al, 2000). Based on the toxin-resistant phenotype of a Δpdi1 mutant and its conversion to sensitivity after coexpression of either wild-type Pdi1p or Pdi1p-active-site mutants, we conclude that it probably is the unfolding activity of Pdi1p rather than its oxidase activity that is required to ensure translocation competence of the α/β heterodimeric toxin. Similar to the unaltered in vivo toxicity of K28 after cysteine mutagenesis (Riffer et al, 2002), replacement of all four lysines in the K28 heterodimer likewise did not affect its retrotranslocation competence nor its biological activity, although in vivo toxin ubiquitination was completely prevented. Thus, based on what we have shown here we propose that toxin ubiquitination is a secondary effect, apparently not required for ER-to-cytosol retrotranslocation. In fact, ER exit of K28 might be mechanistically similar to retrotranslocation of calreticulin which has recently been shown to be likewise independent of ubiquitination and proteasome activity (Afshar et al, 2005). However, how precisely both proteins—K28 in yeast and calreticulin in mammalian cells—emerge into the cytosol remains unknown. Possibly, both proteins pull themselves out of the ER because they can rapidly refold (either spontaneously or catalyzed by post-translational modifications) as they enter the cytosol. A similar mechanism has been discussed for the retrotranslocation of CT A1 chain (Kothe et al, 2005) and pre-pro-α-factor (Lee et al, 2004); however, both proteins require ATPase activities in the 19S proteasome to drive ER export. This is in major contrast to K28 which apparently can emerge into the cytosol even in a pre1/pre2 background deficient in the chymotrypsin-like activity of the proteasome complex. With respect to ER retrotranslocation, it seems obvious that the underlying mechanism(s) can differ for each particular ERAD substrate. As proposed by Vashist and Ng (2004), it is likely that misfolded proteins are sorted by intrinsic checkpoints of ER quality control. In this model, misfolded membrane proteins are recognized by a first checkpoint in the ER and immediately degraded in the cytosol, whereas misfolded soluble proteins bypass this first checkpoint but face a second checkpoint and must undergo a cycling between the Golgi and the ER before degradation in the cytosol can occur. Owing to the great diversity of ERAD substrates, the general mechanism for and the components involved in ERAD might differ in specific cases (Römisch, 2005). In this respect, the K28 virus toxin seems to be a powerful tool in identifying novel components involved in this process, and we are currently using yeast microsomes and a tagged toxin variant (α/βcmyc-HDEL) to identify additional cellular components involved in K28 retrotranslocation. So far, we succeeded to affinity-purify ER proteins such as Kar2p, Pdi1p, and Sec61p that—as we have shown here—are involved in toxin retrotranslocation to the cytosol; however, we also obtained additional proteins that are now being identified by MALDI-TOF mass spectrometry (Sendzik and Schmitt, unpublished).
In contrast to ER retrotranslocation, toxin uptake from the plasma membrane strictly depends on Ubc4p and Rsp5p. As candidate involved in K28 toxicity, Rsp5p is, in particular, interesting as it has been shown to be involved in multiple steps of Ub-mediated endocytosis at the plasma membrane (Dunn and Hicke, 2001a; Morvan et al, 2004). In case of K28, Rsp5p in conjunction with Ubc4p is required for toxin internalization and, correspondingly, mutants defective in either protein accumulate K28 at the plasma membrane incapable of toxin uptake. Based on the results presented in this study, we propose a model in which Ubc4p and Rsp5p are involved in the initial internalization of the toxin/receptor complex at the plasma membrane, possibly triggered by Ubc4p/Rsp5p-driven monoubiquitination of the K28 membrane receptor (Figure 5A). In contrast to the endocytic pathway, Ubc4p and Rsp5p are apparently not involved in toxin retrotranslocation and dissociation into its subunit components (Figure 5B). Subsequent ubiquitination of β is likely to be a secondary effect not involved in the ER export of the toxin. Future studies will have to show if our current model (Figure 5A) for receptor ubiquitination and K28 toxin internalization holds true and, if correct, our prime goal will be to identify the K28 receptor at the plasma membrane and to analyze if it becomes ubiquitinated and subsequently internalized after toxin binding. Monoubiquitination as distinct signal in receptor endocytosis has been described for a number of mammalian proteins (including epidermal growth factor and platelet-derived growth factor (Haglund et al, 2003)) and also for the endocytotic uptake of anthrax toxin receptor (Abrami et al, 2006). In yeast, Rsp5p-mediated monoubiquitination was shown to be required for the downregulation and internalization of various plasma membrane transporters and pheromone receptors (Galan et al, 1996; Beck et al, 1999; Roth and Davis, 2000; Dunn and Hicke, 2001a, 2001b). That both proteins, Rsp5p and Ubc4p, are also involved in the endoyctotic uptake of the K28 virus toxin is in support of several reports describing that Rsp5p-mediated receptor ubiquitination preferentially requires E2s of the Ubc1p/Ubc4p/Ubc5p family (Hicke and Riezman, 1996). Thus, a genome-wide screen for K28 resistance or hypersensitivity might bring up novel components involved in these important aspects of eukaryotic cell biology; we intend to follow up this strategy in the near future.
Figure 5.

Current model of toxin retrotranslocation from the ER and postulated role of Ubc4p and Rsp5p in toxin uptake from the plasma membrane. (A) After initial toxin binding to its plasma membrane receptor R2, the resulting complex between R2 and K28 α/β is (mono)ubiquitinated by the sequential action of Uba1p (E1), Ubc4p (E2), and Rsp5p (E3), triggering R2/K28 complex internalization and subsequent sorting and retrograde transport of the toxin. (B) ER-to-cytosol retrotranslocation of the toxin through the Sec61 complex essentially requires the assistance of Kar2p, Pdi1p, Scj1p, and Jem1p. Partial unfolding of α/β, catalyzed by Pdi1p, ensures translocation competence of the toxin. Cellular components involved in ‘classical' ERAD dislocation (such as Der3p/Hrd1p, Ubc6p, Ubc7p, and Cue1p) are not involved in K28 retrotranslocation. Within the cytosol, K28 dissociates into its subunits, β is ubiquitinated and proteasomally degraded while α enters the nucleus and causes cell death (see text for details). A color version of this figure is available at The EMBO Journal Online.
Materials and methods
Strains and culture conditions
S. cerevisiae strains used throughout this work are listed in Supplementary Table II (available as online Supplementary data). Yeast cultures were grown at 30°C either in complex YEPD medium or in synthetic medium (YNB) supplemented with the appropriate amino acid/base requirements of each strain.
Genetic techniques
Techniques used for yeast molecular biology and genetics including yeast two-hybrid analyses (using a α–β protein fusion of K28 as bait) and GST pull-down experiments were performed according to standard procedures (Sambrook et al, 1999). The lysine-free K28 toxin variant αK-0/βK-0 was constructed by SOE-PCR (Horton et al, 1989) using the K28 preprotoxin encoding plasmid pPGK-M28-I as template (Schmitt and Tipper, 1995). Oligonucleotide primers used throughout this study are described in Supplementary Table III (available as online Supplementary data). All PCR-amplified constructs were routinely sequenced and subsequently cloned into the galactose-inducible expression vector pYES2.1/V5-His-TOPO according to the instructions of the manufacturer (Invitrogen, Groningen). Overexpression of mutant ubiquitin (Ub-RR48/63) was achieved by transformation of wild-type yeast with plasmid pWO21, containing mutant ubiquitin under transcriptional control of the Cu2+ inducible CUP1 promoter.
Toxicity assay
K28 toxin sensitivity or resistance in the indicated wild-type and mutant strains was determined in an agar diffusion assay on methylene blue agar (MBA; pH 4.7) as previously described (Schmitt and Tipper, 1990). Briefly, an aliquot of a K28-secreting killer strain (105 cells in a total volume of 5 μl) was spotted onto an MBA plate that had been seeded with an overlay of the indicated S. cerevisiae mutant and/or wild-type. If not otherwise stated, plates were incubated for 4 days at 20°C. Alternatively, sensitivity/resistance was determined against the purified K28 virus toxin by pipetting 0.1 ml toxin samples into wells (10 mm in diameter) cut into the MB agar. In each case, the diameter of the resulting cell-free zone of growth inhibition surrounding the well (or the killer cells) is proportional to the logarithm of killer toxin activity. Relative toxin sensitivity of a particular yeast mutant corresponds to the sensitivity of the isogenic wild-type, which was set 100% (portraying growth inhibition zones between 18 and 24 mm determined in an agar diffusion assay on MBA plates).
Toxin-binding/uptake studies and cell fractionation
Toxin binding to yeast cell spheroplasts and its subsequent uptake was examined indirectly by estimating the amount of toxin remaining in solution after incubating 5 × 107 cells/ml in the presence of 104 U purified K28 toxin/ml for up to 3 h at 20°C. Toxin content and/or activity remaining in the supernatant was analyzed by sodium dodecyl sulfate (SDS)–PAGE and Western analysis probed with polyclonal anti-β, or by killing zone assays on methylene blue agar (pH 4.7) as previously described (Breinig et al, 2002). For subcellular toxin localization, yeast cells were grown in YEPD medium to early exponential growth phase (1 × 107 cells/ml), harvested by centrifugation and used for cell fractionation essentially as previously described (Eisfeld et al, 2000).
Immunoprecipitation and GST pull-down experiments
To investigate in vivo ubiquitination of K28 α and/or β, the cytosolic fraction of toxin-treated cells was precipitated with ethanol (70%) and the pellet was resuspended in 1 ml ice-cold lysis buffer (PBS containing 5 mM EDTA and 0.02% sodium azide). The lysate was cleared by centrifugation (15 min, 16 000 g, 4°C) and the supernatant was subjected to immunoprecipitation by using antibody-conjugated protein A sepharose beads. Antibody-coated beads were prepared by mixing 30 μl of a 50% protein A sepharose bead slurry with 0.5 ml ice-cold PBS and 5 μl polyclonal antibody directed against the α- or β-subunit of K28. Thereafter, beads were washed twice with lysis buffer, 10 μl BSA (10%) and the cell lysate were added and immunoprecipitation was carried out for 2 h at 4°C in a rotating device. After centrifugation (5 s, 16 000 g, 4°C), the supernatant (containing unbound proteins) was aspirated and the beads were washed four times with ice-cold washing buffer (0.1% Triton-X 100, 50 mM Tris-HCl, 300 mM NaCl, 5 mM EDTA, 0.02% sodium azide, pH 7.4). In the final step, beads were washed with ice-cold PBS and the immunoprecipitate was used for Western analysis probed with monoclonal (Biomol) or polyclonal (Sigma) anti-ubiquitin. Purification of a GST-Pdi1p fusion protein expressed in E. coli from plasmid pGEX-PDI1 was carried out by standard procedures according to the instructions of the manufacturer (Amersham Pharmacia). Purified GST-Pdi1p was applied to a column of GSH-sepharose and incubated with 5 pmol purified K28 toxin (Reiter et al, 2005). Bound proteins were eluted in 50 mM Tris/HCl containing 10 mM reduced glutathione (pH 8.0), subjected to SDS–PAGE and probed with polyclonal goat anti-GST (Amersham Pharmacia) or polyclonal rabbit anti-β (Eisfeld et al, 2000).
Western blot analysis
SDS–PAGE was performed under nonreducing conditions unless otherwise stated. Protein samples were electrophoretically separated in Tris-tricine SDS–polyacrylamide gels (10%) and blotted onto PVDF membranes in transblot buffer (25 mM Tris, 192 mM glycine, 20% methanol, 0.1% SDS). Blots were incubated with polyclonal antibodies directed against the α- or β-subunit of K28 (Riffer et al, 2002). Alkaline phosphatase-conjugated goat anti-rabbit IgG was used as secondary antibody and colorimetric signal detection of the immunoprecipitate was performed using a NBT/BCIP stock solution (Roche, Mannheim).
Supplementary Material
Supplementary Table I
Supplementary Table II
Supplementary Table III
Acknowledgments
We are grateful to Jakob Winther for comments on the manuscript and we thank Linda Hicke, Tom Rapoport, Wolfgang Seufert, Thomas Sommer, Jakob Winther, and Dieter Wolf for providing plasmid pWO21 and various yeast mutants. Randy Schekman and Peter Kölling kindly provided aliquots of anti-Sec61p, anti-Kar2p, and anti-Pma1p antiserum. Technical assistance of Andrea Karrenbauer and Sabine Prediger is also greatly appreciated. This work was supported by the Deutsche Forschungsgemeinschaft (grants Schm 541/11-2 and GK 845-2).
References
- Abrami L, Leppla SH, van der Goot FG (2006) Receptor palmitoylation and ubiquitination regulate anthrax toxin endocytosis. J Cell Biol 172: 309–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afshar N, Black BE, Paschal BM (2005) Retrotranslocation of the chaperone calreticulin from the endoplasmic reticulum lumen to the cytosol. Mol Cell Biol 25: 8844–8853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bays NW, Gardner RG, Seelig LP, Joazeiro CA, Hampton RY (2001) Hrd1p/Der3p is a membrane-anchored ubiquitin ligase required for ER-associated degradation. Nat Cell Biol 3: 24–29 [DOI] [PubMed] [Google Scholar]
- Beck T, Schmidt A, Hall MN (1999) Starvation induces vacuolar targeting and degradation of the tryptophan permease in yeast. J Cell Biol 146: 1227–1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breinig F, Tipper DJ, Schmitt MJ (2002) Kre1p, the plasma membrane receptor for the yeast K1 viral toxin. Cell 108: 395–405 [DOI] [PubMed] [Google Scholar]
- Cronin SR, Rao R, Hampton RY (2002) Cod1p/Spf1p is a P-type ATPase involved in ER function and Ca2+ homeostasis. J Cell Biol 157: 1017–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn R, Hicke L (2001a) Multiple roles for Rsp5p-dependent ubiquitination at the internalization step of endocytosis. J Biol Chem 276: 25974–25981 [DOI] [PubMed] [Google Scholar]
- Dunn R, Hicke L (2001b) Domains of the Rsp5 ubiquitin-protein ligase required for receptor-mediated and fluid-phase endocytosis. Mol Biol Cell 12: 421–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durr G, Strayle J, Plemper R, Elbs S, Klee SK, Catty P, Wolf DH, Rudolph HK (1998) The medial-Golgi ion pump Pmr1 supplies the yeast secretory pathway with Ca2+ and Mn2+ required for glycosylation, sorting, and endoplasmic reticulum-associated protein degradation. Mol Biol Cell 9: 1149–1162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisfeld K, Riffer F, Mentges J, Schmitt MJ (2000) Endocytotic uptake and retrograde transport of a virally encoded killer toxin in yeast. Mol Microbiol 37: 926–940 [DOI] [PubMed] [Google Scholar]
- Ellgaard L, Helenius A (2003) Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol 4: 181–191 [DOI] [PubMed] [Google Scholar]
- Falnes PO, Sandvig K (2000) Penetration of protein toxins into cells. Curr Opin Cell Biol 12: 407–413 [DOI] [PubMed] [Google Scholar]
- Forster ML, Sivick K, Park YN, Arvan P, Lencer WI, Tsai B (2006) Protein disulfide isomerase-like proteins play opposing roles during retrotranslocation. J Cell Biol 173: 853–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galan JM, Moreau V, Andre B, Volland C, Haguenauer-Tsapis R (1996) Ubiquitination mediated by the Npi1p/Rsp5p ubiquitin-protein ligase is required for endocytosis of the yeast uracil permease. J Biol Chem 271: 10946–10952 [DOI] [PubMed] [Google Scholar]
- Haglund K, Di Fiore PP, Dikic I (2003) Distinct monoubiquitin signals in receptor endocytosis. Trends Biochem Sci 28: 598–603 [DOI] [PubMed] [Google Scholar]
- Haynes CM, Caldwell S, Cooper AA (2002) An HRD/DER-independent ER quality control mechanism involves Rsp5p-dependent ubiquitination and ER–Golgi transport. J Cell Biol 158: 91–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazes B, Read RJ (1997) Accumulating evidence suggests that several AB-toxins subvert the endoplasmic reticulum-associated protein degradation pathway to enter target cells. Biochemistry 36: 11051–11054 [DOI] [PubMed] [Google Scholar]
- Heinemeyer W, Gruhler A, Mohrle V, Mahe Y, Wolf DH (1993) PRE2, highly homologous to the human major histocompatibility complex-linked RING10 gene, codes for a yeast proteasome subunit necessary for chymotryptic activity and degradation of ubiquitinated proteins. J Biol Chem 268: 5115–5120 [PubMed] [Google Scholar]
- Hicke L, Riezman H (1996) Ubiquitination of a yeast plasma membrane receptor signals its ligand-stimulated endocytosis. Cell 84: 277–287 [DOI] [PubMed] [Google Scholar]
- Holst B, Tachibana C, Winther JR (1997) Active site mutations in yeast protein disulfide isomerase cause dithiothreitol sensitivity and a reduced rate of protein folding in the endoplasmic reticulum. J Cell Biol 138: 1229–1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR (1989) Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 77: 61–68 [DOI] [PubMed] [Google Scholar]
- Jarosch E, Lenk U, Sommer T (2003) Endoplasmic reticulum-associated protein degradation. Int Rev Cytology 223: 39–81 [DOI] [PubMed] [Google Scholar]
- Kothe M, Ye Y, Wagner JS, DeLuca HE, Kern E, Rapoport TA, Lencer WI (2005) Role of p97 AAA-ATPase in the retrotranslocation of the cholera toxin A1 chain, a non-ubiquitinated substrate. J Biol Chem 280: 28127–28132 [DOI] [PubMed] [Google Scholar]
- Lee RJ, Liu CW, Harty C, McCracken AA, Latterich M, Römisch K, DeMartino GN, Thomas PJ, Brodsky JL (2004) Uncoupling retro-translocation and degradation in the ER-associated degradation of a soluble protein. EMBO J 23: 2206–2215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lencer WI, Constable C, Moe S, Jobling MG, Webb HM, Ruston S, Madara JL, Hirst TR, Holmes RK (1995) Targeting of cholera toxin and Escherichia coli heat labile toxin in polarized epithelia: role of COOH-terminal KDEL. J Cell Biol 131: 951–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindberg MJ, Normark J, Holmgren A, Oliveberg M (2004) Folding of human superoxide dismutase: disulfide reduction prevents dimerization and produces marginally stable monomers. Proc Natl Acad Sci USA 101: 15893–15898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord JM, Deeks E, Marsden CJ, Moore K, Pateman C, Smith DC, Spooner RA, Watson P, Roberts LM (2003) Retrograde transport of toxins across the endoplasmic reticulum membrane. Biochem Soc Trans 31: 1260–1262 [DOI] [PubMed] [Google Scholar]
- Lumb RA, Bulleid NJ (2002) Is protein disufide isomerase a redox-dependent molecular chaperone? EMBO J 21: 6763–6770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medicherla B, Kostova Z, Schaefer A, Wolf DH (2004) A genomic screen identifies Dsk2p and Rad23p as essential components of ER-associated degradation. EMBO Rep 5: 692–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meusser B, Hirsch C, Jarosch E, Sommer T (2005) ERAD: the long road to destruction. Nat Cell Biol 7: 766–772 [DOI] [PubMed] [Google Scholar]
- Morvan J, Froissard M, Haguenauer-Tsapis R, Urban-Grimal D (2004) The ubiquitin ligase Rsp5p is required for modification and sorting of membrane proteins into multivesicular bodies. Traffic 5: 383–392 [DOI] [PubMed] [Google Scholar]
- Nishikawa SI, Fewell SW, Kato Y, Brodsky JL, EndoT (2001) Molecular chaperones in the yeast endoplasmic reticulum maintain the solubility of proteins for retrotranslocation and degradation. J Cell Biol 153: 1061–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norgaard P, Westphal V, Tachibana C, Alsoe L, Holst B, Winther JR (2001) Functional differences in yeast protein disulfide isomerases. J Cell Biol 152: 553–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovich E, Kerem A, Frohlich KU, Diamant N, Bar-Nun S (2002) AAA-ATPase p97/Cdc48p, a cytosolic chaperone required for endoplasmic reticulum-associated protein degradation. Mol Cell Biol 22: 626–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reggiori F, Pelham HR (2002) A transmembrane ubiquitin ligase required to sort membrane proteins into multivesicular bodies. Nat Cell Biol 4: 117–123 [DOI] [PubMed] [Google Scholar]
- Reiter J, Herker E, Madeo F, Schmitt MJ (2005) Viral killer toxins induce caspase-mediated apoptosis in yeast. J Cell Biol 168: 353–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riffer F, Eisfeld K, Breinig F, Schmitt MJ (2002) Mutational analysis of K28 preprotoxin processing in the yeast Saccharomyces cerevisiae. Microbiology 148: 1317–1328 [DOI] [PubMed] [Google Scholar]
- Römisch K (2005) Endoplasmic reticulum-associated degradation. Annu Rev Cell Dev Biol 21: 435–456 [DOI] [PubMed] [Google Scholar]
- Roth AF, Davis NG (2000) Ubiquitination of the PEST-like endocytosis signal of the yeast a-factor receptor. J Biol Chem 275: 8143–8153 [DOI] [PubMed] [Google Scholar]
- Rudolph HK, Antebi A, Fink GR, Buckley CM, Dorman TE, LeVitre J, Davidow LS, Mao JI, Moir DT (1989) The yeast secretory pathway is perturbed by mutations in PMR1, a member of a Ca2+ ATPase family. Cell 58: 133–145 [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T (1999) Molecular Cloning: A Laboratory Manual, 2nd edn New York: Cold Spring Harbor Laboratory Press [Google Scholar]
- Schmitt MJ, Tipper DJ (1990) K28, a unique double-stranded RNA killer virus of Saccharomyces cerevisiae. Mol Cell Biol 10: 4807–4815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt MJ, Tipper DJ (1995) Sequence of the M28 dsRNA: preprotoxin is processed to an α/β heterodimeric protein toxin. Virology 213: 341–351 [DOI] [PubMed] [Google Scholar]
- Solovyov A, Xiao R, Gilbert HF (2004) Sulfhydryl oxidation, not disulfide isomerization, is the principal function of protein disulfide isomerase in yeast Saccharomyces cerevisiae. J Biol Chem 279: 34095–34100 [DOI] [PubMed] [Google Scholar]
- Strayle J, Pozzan T, Rudolph HK (1999) Steady-state free Ca(2+) in the yeast endoplasmic reticulum reaches only 10 microM and is mainly controlled by the secretory pathway pump pmr1. EMBO J 18: 4733–4743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh B, Furman MH, Tortorella D, Ploegh HL (2003) Protein unfolding is not a prerequisite for endoplasmic reticulum-to-cytosol dislocation. J Biol Chem 278: 6664–6672 [DOI] [PubMed] [Google Scholar]
- Tsai B, Rapoport TA (2002) Unfolded cholera toxin is transferred to the ER membrane and released from protein disulfide isomerase upon oxidation by Ero1. J Cell Biol 159: 207–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai B, Rodighiero C, Lencer WI, Rapoport TA (2001) Protein disulfide isomerase acts as a redox-dependent chaperone to unfold cholera toxin. Cell 104: 937–948 [DOI] [PubMed] [Google Scholar]
- Vago R, Marsden CJ, Lord JM, Ippoliti R, Flavell DJ, Flavell SU, Ceriotti A, Fabbrini MS (2005) Saporin and ricin A chain follow different intracellular routes to enter the cytosol of intoxicated cells. FEBS J 272: 4983–4995 [DOI] [PubMed] [Google Scholar]
- Vashist S, Ng DT (2004) Misfolded proteins are sorted by a sequential checkpoint mechanism of ER quality control. J Cell Biol 165: 41–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel JP, Misra LM, Rose MD (1990) Loss of BiP/GRP78 function blocks translocation of secretory proteins in yeast. J Cell Biol 110: 1885–1895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Yang J, Huibregtse JM (1999) Functional domains of the Rsp5 ubiquitin-protein ligase. Mol Cell Biol 19: 342–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesche J, Rapak A, Olsnes S (1999) Dependence of ricin toxicity on translocation of the toxin A-chain from the endoplasmic reticulum to the cytosol. J Biol Chem 274: 34443–34449 [DOI] [PubMed] [Google Scholar]
- Ye Y, Meyer HH, Rapoport TA (2001) The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 414: 652–656 [DOI] [PubMed] [Google Scholar]
- Ye Y, Meyer HH, Rapoport TA (2003) Function of the p97–Ufd1–Npl4 complex in retrotranslocation from the ER to the cytosol: dual recognition of nonubiquitinated polypeptide segments and polyubiquitin chains. J Cell Biol 162: 71–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table I
Supplementary Table II
Supplementary Table III