

Abstract
Membrane protein complexes can support both the generation and utilisation of a transmembrane electrochemical proton potential (‘proton-motive force'), either by transmembrane electron transfer coupled to protolytic reactions on opposite sides of the membrane or by transmembrane proton transfer. Here we provide the first evidence that both of these mechanisms are combined in the case of a specific respiratory membrane protein complex, the dihaem-containing quinol:fumarate reductase (QFR) of Wolinella succinogenes, so as to facilitate transmembrane electron transfer by transmembrane proton transfer. We also demonstrate the non-functionality of this novel transmembrane proton transfer pathway (‘E-pathway') in a variant QFR where a key glutamate residue has been replaced. The ‘E-pathway', discussed on the basis of the 1.78-Å-resolution crystal structure of QFR, can be concluded to be essential also for the viability of pathogenic ɛ-proteobacteria such as Helicobacter pylori and is possibly relevant to proton transfer in other dihaem-containing membrane proteins, performing very different physiological functions.
Keywords: bioenergetics, membrane proteins, quinol:fumarate reductase, transmembrane proton potential, transmembrane proton transfer
Introduction
According to chemiosmotic theory (see Mitchell, 1979, for a review), the energy released upon the oxidation of electron donor substrates, in both aerobic and anaerobic respiration, is transiently stored in the form of a proton potential, Δp, across the energy-transducing membranes, which can then be used by the ATP synthase for ADP phosphorylation with inorganic phosphate. Fundamentally, there are two mechanisms (Richardson and Sawers, 2002) by which integral membrane proteins can act as catalysts in this coupling of electron transfer reactions to Δp generation. Firstly, the redox loop mechanism essentially involves transmembrane electron transfer coupled to protolytic reactions on opposite sides of the membrane. In a simple form, this mechanism is represented by the formate dehydrogenase (Jormakka et al, 2002) and membrane-bound nitrate reductase (Bertero et al, 2003) enzymes of anaerobic respiration and in a more complicated form by the Q-cycle of the cytochrome bc1 complex, complex III of the aerobic respiratory chain (Mitchell, 1976). Secondly, the proton pump mechanism involves transmembrane proton translocation by a Grotthuss-type mechanism (von Grotthuss, 1806; Agmon, 1995), driven by the transfer of electrons between sites of catalysis via the prosthetic groups of the membrane protein complex. Such a mechanism is thought to be operative, for example, in cytochrome c oxidase, complex IV of the respiratory chain (Faxén et al, 2005).
In a specific case of a single respiratory membrane protein complex, the experimental results support (Haas et al, 2005; Lancaster et al, 2005; Mileni et al, 2005) both of these mechanisms being harnessed together, so as to ensure the counterbalancing of their effects for energetic reasons. The membrane protein in question is the quinol:fumarate reductase (QFR) from the anaerobic ɛ-proteobacterium Wolinella succinogenes. QFR contains two haem b groups oriented along the membrane normal and bound by the transmembrane subunit C (see Figure 1A), which are termed the ‘proximal haem', bP, and the ‘distal haem', bD, according to their relative proximity to the hydrophilic subunits A and B. It is the terminal enzyme of fumarate respiration, a form of anaerobic respiration that allows anaerobic bacteria to use fumarate instead of dioxygen as the terminal electron acceptor (Kröger, 1978; Lancaster, 2004). QFR couples the two-electron reduction of fumarate to succinate to the two-electron oxidation of quinol to quinone. This reaction is part of an electron transfer chain that enables the bacterium to grow with various electron donor substrates such as formate or hydrogen (Supplementary Figure S1).
Figure 1.
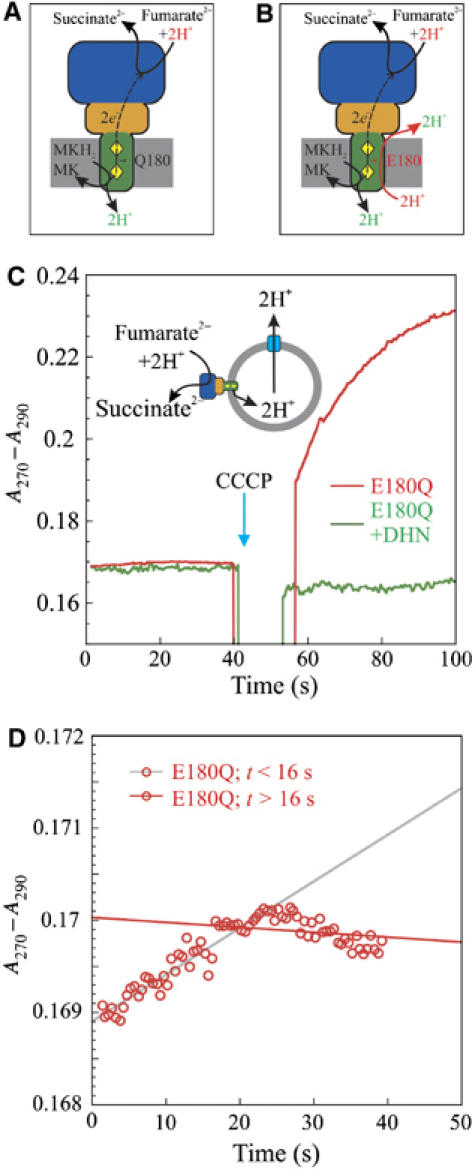
Addition of the uncoupler CCCP stimulates the oxidation of DMNH2 by fumarate as catalysed by proteoliposomal E180Q-QFR. (A) Non-functionality of the ‘E-pathway' gives rise to electrogenicity of E180Q-QFR. Subunit A is shown in blue, subunit B in orange, and subunit C in green. The haem groups are shown as yellow diamonds, with the upper haem corresponding to bP and the lower one to bD. Protons bound are shown in red, protons released in green. (B) Overall electroneutrality in wild-type QFR as explained by the ‘E-pathway'. For clarity, protons are indicated to be released to and taken up from the bulk solvent phase on both sides of the membrane. However, it can presently not be ruled out that, on either side of the membrane, the protons are transported along the protein surface from the respective proton exit sites to those of proton entry, without being released to the bulk, as argued elsewhere (Mulkidjanian et al, 2006). (C) DMNH2 oxidation as (not) catalysed by E180Q-QFR reconstituted in proteoliposomes (500 μg). The traces were recorded under the same conditions as for Figure 2A, except that DMNH2 (20 μM) was used as the electron donor and the fumarate concentration was 40 μM. Catalytic activity was significantly detectable only after the addition of 25 μM of the protonophore CCCP. (D) Enlarged section of the left half of (C). Linear fit of the data points of ΔA at t<16 s (grey) and t>16 s (red).
Although experiments performed with inverted vesicles and proteoliposomes containing QFR demonstrated that the reaction catalysed by the dihaem-containing QFR from W. succinogenes is not associated directly with Δp generation (Geisler et al, 1994; Biel et al, 2002; Kröger et al, 2002), the three-dimensional structure of QFR, initially solved at 2.2 Å resolution (Lancaster et al, 1999), revealed locations of the active sites of fumarate reduction (Lancaster et al, 2001) and of menaquinol oxidation (Lancaster et al, 2000) that are oriented towards opposite sides of the membrane (Figure 1A), thus indicating that menaquinol oxidation by fumarate, as catalysed by W. succinogenes QFR, should be associated directly with the establishment of a Δp. To reconcile these apparently conflicting experimental observations, the so-called ‘E-pathway hypothesis' (Lancaster, 2002) was proposed (Figure 1B). According to this hypothesis, the transfer of two electrons via the two QFR haem groups is strictly coupled to a compensatory, parallel transfer of two protons across the membrane via an unprecedented proton transfer pathway, which is transiently open during the reduction of the two haem groups and closed in the oxidised state of the enzyme. The latter is a requirement for the strict coupling of transmembrane proton transfer to electron transfer and for preventing the QFR from operating as an uncoupler of fumarate respiration (Supplementary Figure S1). The two most prominent constituents of the proposed pathway were suggested to be the haem bD ring C propionate and the amino-acid residue Glu C180, which is conserved in dihaem-containing QFR from ɛ-proteobacteria (Lancaster, 2002; Supplementary Figure S2) and after which the ‘E-pathway' was named.
Since the proposal of this hypothesis, a number of theoretical (Haas and Lancaster, 2004) and experimental results (Haas et al, 2005; Lancaster et al, 2005; Mileni et al, 2005) have been obtained that supported it, but did not prove it. Here we provide, for the first time, evidence for the presence of such a compensatory ‘E-pathway' in the wild-type enzyme and the non-functionality of the ‘E-pathway' in a variant enzyme lacking Glu C180.
Results and discussion
In its detergent-solubilised state, the enzyme variant E180Q, obtained by replacing Glu C180 with Gln, exhibited only 1/10 of the specific activity of the wild-type enzyme for 2,3-dimethyl,1,4-naphthoquinol (DMNH2) oxidation by fumarate, whereas the Michaelis constant KM for DMNH2 was unchanged (Lancaster et al, 2005). However, compared to the wild-type enzyme, a significant increase of approximately 50 mV in the oxidation–reduction midpoint potential of haem bP was found for the E180Q variant (EM7=+39 mV; Lancaster et al, 2005). Considering that the [3Fe-4S] cluster (Supplementary Figure S1) has been reported to exhibit a redox midpoint potential of −61 mV (Mileni et al, 2006), such an increase in the midpoint potential makes electron transfer from bP to the [3Fe-4S] cluster two times more energetically unfavourable than the corresponding process in the wild-type enzyme. In addition, the redox midpoint potential of haem bP in the E180Q variant is higher than that of the fumarate/succinate couple (EM7=25 mV; Ohnishi et al, 2000). Both of these features serve as explanations for the reduced activity of the detergent-solubilised E180Q variant. However, when reconstituted into proteoliposomes and therefore bound by sealed membrane vesicles, the E180Q variant exhibited even less and ostensibly negligible enzymatic activity of DMNH2 oxidation by fumarate (left half of Figure 1C). Closer inspection (Figure 1D) indicated that this lack of activity was only obvious after approximately 20 s. Prior to that, the membrane-bound variant enzyme was weakly catalytically active, which, according to the scheme in Figure 1A, should lead to the generation of a Δp, against which the reaction could no longer be catalysed (see Supplementary discussion for a more quantitative analysis). We attributed this rapid loss in activity to be caused by the small difference in oxidation–reduction midpoint potential between the 2,3-dimethyl,1,4-naphthoquinone (DMN)/DMNH2 couple routinely used (EM7.3=−35 mV; see Table I) and the fumarate/succinate couple, rendering the overall reaction only mildly exergonic (ΔG≈−12 kJ/mol) under standard conditions (at pH 7), and apparently not exergonic enough to support sustained Δp generation (Figure 1A). These conclusions are supported by our finding that addition of the protonophore carbonyl cyanide m-chlorophenylhydrazone (CCCP) to ostensibly inactive proteoliposomal E180Q-QFR enabled the catalysis of DMNH2 oxidation (right half of Figure 1C).
Table 1.
H+/e− ratio measurements with proteoliposomes and associated specific activities of the respective QFR as reconstituted into the proteoliposomes
| Quinol substrate | In situ redox midpoint potential EM7.3 (mV) | QFR enzyme | H+/e− ratio | Specific activity of quinol oxidation by fumarate (U mg−1 protein) | KM (mM) |
|---|---|---|---|---|---|
| DMNH2 | −35 | WT | 0.0 | 5.4 | 0.1 |
| E180Q | ND | Variablea | 0.1 | ||
| AMNH2 | ND | WT | 0.7 | 1.1 | |
| E180Q | 0.1 | 1.1 | |||
| MMANH2 | −124 | WT | 0.0 | 5.7 | 0.1 |
| E180Q | 1.0 | 1.3 | 0.1 | ||
| One unit (U) corresponds to 1 μmol of substrate turned over per minute. The Michaelis constants KM for the quinols were determined with the respective QFR solubilised in dodecyl-β-D-maltoside (0.01% w/v) and decyl-β-D-maltoside (0.1%). The in situ redox midpoint potentials were determined as described in the Supplementary data and are quoted with reference to the standard hydrogen electrode at pH 7.3. | |||||
| ND: not determined. | |||||
| aSpecific activities for the oxidation of DMNH2 by fumarate as catalysed by proteoliposomal E180Q-QFR apparent from Figures 1C and D (ranging from ⩽0.01 to 1.5 U/mg) are estimated to be considerably higher than for the other measurements due to different experimental conditions. | |||||
Two components contribute to Δp. These are the concentration difference of protons across the membrane, ΔpH, and the difference in electrical potential between the two membrane-separated aqueous phases, ΔΨ. In principle, according to Figure 1A, we should have measured the generation of a ΔpH based on the acidification of the interior of proteoliposomes containing E180Q-QFR and the generation of a ΔΨ by monitoring the uptake of tetraphenylborate (TPB−) with a TPB−-selective electrode. However, due to the small energy difference of the redox couples involved, the resulting low rates (prior to the addition of uncoupler) and the short duration of turnover, any detection of ΔpH or ΔΨ for this reaction was not reliably possible with our experimental setup.
These developments prompted us to design and synthesise a substrate analogue with suitable substituents to lower the redox midpoint potential of the respective naphthoquinone/naphthoquinol couple, thus increasing the driving force of the catalysed oxidation by fumarate. Based on textbook literature (Fieser and Fieser, 1965), we expected substituents such as amino or methylamino groups (Supplementary Figure S4), when replacing the 2-methyl group of DMN to lower the redox midpoint potential. As expected, both 2-amino-3-methyl-1,4-naphthoquinone (AMN) and 2-methyl-3-methylamino-1,4-naphthoquinone (MMAN) exhibited lower redox midpoint potentials (see Supplementary data). However, the Michaelis constant KM (see Table I) for AMNH2 as a QFR substrate was an order of magnitude higher than the others, resulting in a correspondingly lower specific enzymatic activity of quinol oxidation (see Table I), so that further characterisation focussed on DMNH2 and MMANH2. The in situ redox midpoint potential for the protein-bound MMAN/MMANH2 couple was found to be approximately 90 mV lower than that of the DMN/DMNH2 couple (Table I), rendering the overall reaction of MMANH2 an estimated two-and-half times more exergonic (ΔG≈−30 kJ/mol) under standard conditions at pH 7 and possibly exergonic enough to support measurable Δp generation.
We determined ΔpH by monitoring the acidification of the interior of QFR-containing proteoliposomes in the presence of valinomycin and 100 mM KCl. We found that, despite being catalytically active (Figure 2A), wild-type QFR did not contribute to the generation of a ΔpH (Figure 2B), irrespective of whether DMNH2 or MMANH2 was provided as substrate (see Table I). When wild-type QFR was replaced by the E180Q variant, the membrane-bound enzyme did not significantly support the oxidation of DMNH2 (left half of Figure 1C). It did, however, catalyse the oxidation of MMANH2 by fumarate (Figures 2A and B), establishing a ΔpH of 1.6 pH units (see Supplementary data). The H+/e− ratios associated with the quinol oxidation by fumarate were determined from the ratios of the rate constant of acidification of the proteoliposomal interior and that of quinol oxidation in the same experiment. We found that the oxidation of 1 mol MMANH2 by E180Q-QFR resulted in an increase of 2.0 mol H+ (Supplementary Table SII), corresponding to an H+/e− ratio of 1.0 (Table I). Specific inhibition at the quinol oxidation site upon addition of the competitive inhibitor 2-decyl-3-hydroxy-1,4-naphthoquinone (DHN) with an IC50 of 8 μM (see Supplementary data) abolished the effects observed for E180Q-QFR (Figures 1C, 2A and B). The apparent slowing and eventual reversal of the MMANH2 oxidation process after approximately 30 s (Figure 2A) can be attributed to the protonation of the product MMAN to MMANH+. The latter has a much lower extinction coefficient Δɛ280−320 versus MMANH2 than MMAN (Supplementary Figure S5A). This is supported by the observation that the rate of acidification of the proteoliposomal interior (Figure 2B) is still significant, even after 50 s, but is eventually only one fifth of the initial rate (see also Supplementary Figure S8). We conclude that, after significant acidification of the proteoliposomal interior, one of the two protons liberated upon oxidation of MMANH2 to MMAN is no longer released to the interior of the proteoliposomes but instead protonates the product MMAN to MMANH+.
Figure 2.
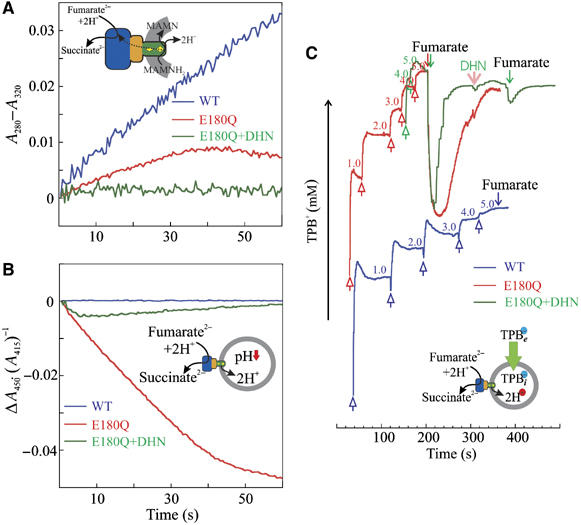
Catalytic activity (A), and monitoring of any generation of ΔpH (B) and ΔΨ (C) by wild type and E180Q-QFR. (A) Normalised, apparent progress curves of MMANH2 oxidation by fumarate as catalysed by wild-type QFR (blue trace) and by E180Q-QFR (red) and not catalysed by E180Q-QFR in the presence of the inhibitor DHN (20 μM, green). Proteoliposomes (200 μg) containing the respective QFR enzyme and MMANH2 (10 mM/mg phospholipid) were suspended in N2-flushed buffer. The reaction was started by the addition of fumarate (20 μM). The absorbance difference A280−A320 was monitored as a function of time. The traces were normalised by setting the absorption difference of A280−A320 to zero and the time of fumarate addition to zero. The progress curves were recorded in the same experiment as in B using a diode array spectrophotometer. (B) Normalised progress curves of the pyranine absorbance ratio offset as an indicator of the acidification of the proteoliposomal interior upon MMANH2 oxidation by fumarate. The colour-coding is as defined for (A). The absorbance ratio offset Δ(A450/A415) depended only on the H+ concentration in the liposomes and was monitored as a function of time. The reaction was started by the addition of fumarate with the time set to 0. The traces were normalised by setting the initial absorbance ratio of A450/A415 to zero. (C) Recording of the external TPB− concentration (logarithmic scale) in a suspension of proteoliposomes during fumarate reduction by MMANH2. To a solution containing proteoliposomes (200 μg in 50 mM HEPES, pH 7.5) with reconstituted wild-type QFR (blue trace) or E180Q-QFR (red trace), MMANH2 (10 mM/mg phospholipid), TPB− was added in 1 mM aliquots for calibration (open arrows pointing upwards). Reaction was started by the addition of 20 μM fumarate (arrows pointing downwards). The E180Q-QFR in the presence of 20 μM DHN (green trace, addition of DHN indicated by the pink arrow) did not generate any appreciable ΔΨ. The TPB− uptake was calculated from the amplitude between the first fumarate addition and the level where the TPB− concentration reached a minimum. The concentration inside the proteoliposomes was corrected according to Equation (1). Because the electrodes had different Nernst factors, the respective TPB− concentrations are indicated in the plot in the respective colour.
We determined ΔΨ by monitoring the uptake of TPB− with a TPB−-selective electrode (Figure 2C). Catalysis of MMANH2 oxidation by fumarate by E180Q-QFR established a ΔΨ of 86 mV (positive inside, Table II). This was inhibited by the addition of DHN (see Figure 2C and Supplementary discussion). No ΔΨ was detected during analogous catalysis by wild-type QFR. In the case of both the wild type and the E180Q enzyme, no generation of a ΔΨ by means of TPB− uptake could be detected after replacement of MMANH2 with DMNH2.
Table 2.
TPB− accumulation in proteoliposomes in the steady state of electron transport
| Quinol substrate | QFR enzyme | Ts (μM g phospholipids−1) | Ti (μM) | Te (μM) | ΔΨ (mV) |
|---|---|---|---|---|---|
| DMNH2 | WT | No TPB− uptake detected | 5000 | 0 | |
| DMNH2 | E180Q | No TPB− uptake detected | 5000 | 0 | |
| MMANH2 | WT | No TPB− uptake detected | 5000 | 0 | |
| MMANM2 | E180Q | 4.4 | 248 | 9.1 | 86 |
| Proteoliposomes containing wild-type QFR or E180Q-QFR were subjected to the experiment using different e− donors, first column. The reaction was started by the addition of fumarate, the e− acceptor. TPB− was accumulated in the proteoliposomes in the steady state of the electron transport. Ts represents the maximum TPB− amount taken up by the proteoliposomes in the steady state of electron transport, Te represents the external TPB− concentration in the medium (see Figure 2C), Ti represents the internal TPB− concentration in the proteoliposome as calculated according to Equation (1). ΔΨ was calculated from Te and Ti according to the Nernst equation. | |||||
Considering the measurements of both ΔpH and ΔΨ, Δp generation can be demonstrated only for MMANH2 oxidation by fumarate as catalysed by E180Q-QFR. Comparison to catalysis by the wild-type enzyme clearly proves the presence of the compensatory ‘E-pathway' in the wild-type enzyme and its non-functionality in E180Q-QFR. Comparison of these results to the inability of membrane-bound E180Q-QFR to support sustained DMNH2 oxidation in the absence of CCCP demonstrates that this latter observation is due to the insufficient driving force of DMNH2 oxidation by fumarate in the absence of a compensatory E-pathway. In contrast, catalysis of the same reaction by the membrane-bound wild-type enzyme demonstrates the facilitation of transmembrane electron transfer by transmembrane proton transfer.
The X-ray crystal structure, refined at 1.78-Å resolution (see Supplementary Table S1), of the oxidised W. succinogenes QFR at pH 6 most importantly reveals the side chain of Glu C180 to be found in two approximately equally populated alternate conformations, one a so-called ‘intermediate' orientation, oriented between the two haem groups, and a second conformer in a so-called ‘distal' conformation, oriented more towards the distal haem group (Figure 3A and B). These conformers are analogous to those predicted by multiconformation continuum electrostatic calculations (Haas and Lancaster, 2004), where their relative occupancy and degree of protonation are predicted to depend on the redox state of the haem groups, and therefore be functionally relevant within the context of the E-pathway hypothesis. These theoretical results are supported by experimental results from redox-induced Fourier-transform infrared difference spectroscopy (Haas et al, 2005), which indicate that the side chain of Glu C180 undergoes a combination of a change in protonation and its environment upon reduction of the enzyme.
Figure 3.
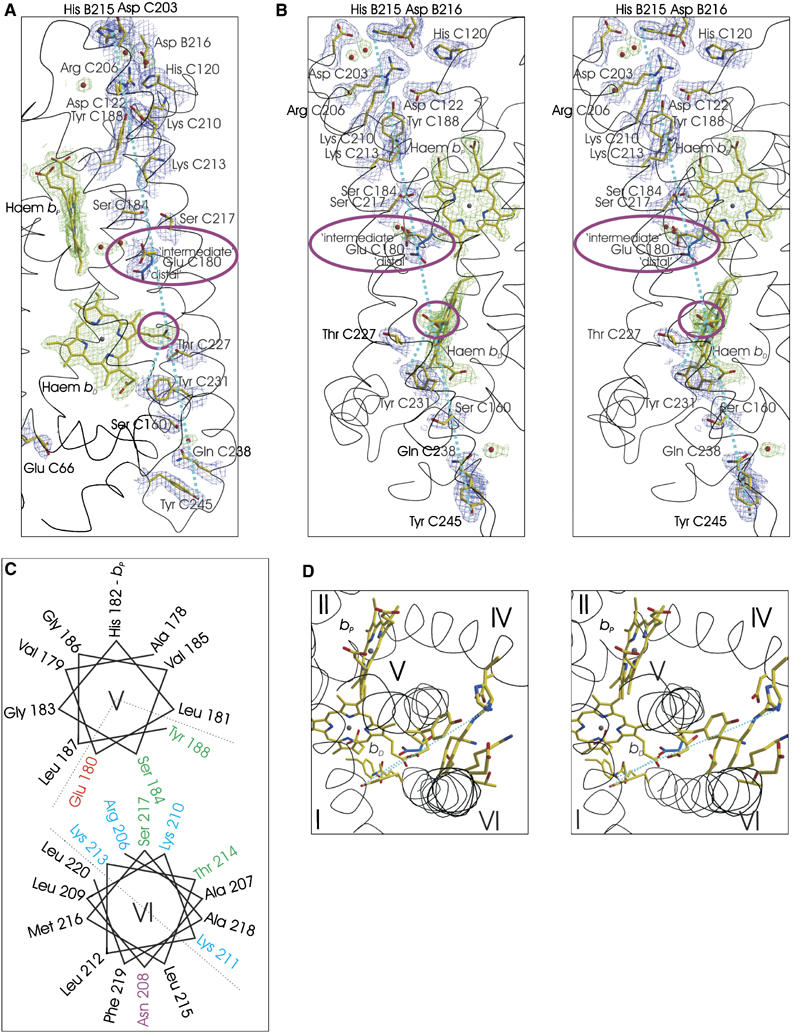
The ‘E-pathway' in dihaem-containing QFR. Possible elements of the ‘E-pathway' as observed in the crystal structure of wild-type QFR refined at 1.78 Å resolution (PDB entry 2BS2). To facilitate orientation between various panels, dashed light blue lines connect the hydroxyl group of residue Tyr C245 to the Nɛ atom of His-B215 and the hydroxyl group of Tyr C231 to the Cγ atom of the bD ring C propionate. Along these dashed lines, a large number of polar and protonatable residues can be found. (A, B (stereo)) Perpendicular views with the periplasm at the bottom, the cytoplasm at the top and the membrane spanning region indicated by the two haem groups. In general, carbon, nitrogen and oxygen atoms are shown in yellow, blue and red, respectively. In the case of Glu C180, the ‘distal' conformer contains light blue carbon atoms. The groups whose role in such an ‘E-pathway' has received experimental support, Glu C180 (Lancaster et al, 2005; Haas et al, 2005) and the bD ring C propionate (Mileni et al, 2005) are highlighted by purple ellipsoids. The 2∣Fo∣−∣Fc∣ electron density map, contoured at 1.0 standard deviations (σ) above the mean density of the map, is shown in blue for the protein and in green for non-protein groups such as haem groups and water molecules. (C, D) The transmembrane region as viewed from the cytoplasmic side. (C) A schematic representation of the packing of transmembrane helices V and VI, whereas (D) (stereo) depicts the corresponding view in the structure. The residues shown on transmembrane helix V are Glu C180 (alternate conformers), Ser C184 and Tyr C188. Residues shown on transmembrane helix VI are Tyr C231, Ser C217, Thr C214, Lys C213, Lys C210, Arg C206 and Asp C203. The residue shown on transmembrane helix IV is Asp C122.
Although all available QFR crystal structures are those of the oxidised enzyme and the E-pathway is required to be non-functional in the oxidised state, inspection of the crystal structure provides clues as to which further groups could contribute to the ‘E-pathway' (Figure 3A and B). Large parts of transmembrane helix VI and a significant part of transmembrane helix V are amphipathic (Figure 3C), and their hydrophilic sides are oriented towards one another, thus providing, in addition to the previously proposed and supported roles of the transmembrane helix V residue Glu C180 and the haem bD ring C propionate, further possible constituents (Figure 3D). The role of these additional residues as possible constituents of the ‘E-pathway' is currently being investigated by site-directed mutagenesis. However, the results presented here, in agreement with previous theoretical and experimental results, strongly support the view that Glu C180 is the central ‘switch' in the coupling of transmembrane electron and proton transfer.
What we have now proved to be the ‘E-pathway' has been demonstrated to be essential for the growth of W. succinogenes with fumarate as the terminal electron acceptor (Lancaster et al, 2005). In turn, QFR has been shown to be essential for the viability of the pathogenic ɛ-proteobacterium Helicobacter pylori in the murine stomach (Ge et al, 2000). It is therefore reasonable to conclude that the ‘E-pathway' is also essential for the viability of H. pylori in its host.
Succinate oxidation by menaquinone, an endergonic reaction under standard conditions, is catalysed by dihaem-containing succinate:menaquinone reductases in several Gram-positive bacteria, for example, B. subtilis. These enzymes are similar to W. succinogenes QFR also with respect to the location of the four histidine residues ligating the haem groups (Hägerhäll and Hederstedt, 1996), the conservation of an acidic residue at the position of Glu C66, demonstrated to be selectively essential for menaquinol oxidation (Lancaster et al, 2000), the location of the site of menaquinone reduction close to the haem bD (Matsson et al, 2000), but different in that they lack an acidic residue at the position of Glu C180 (Supplementary Figure S2). Experimental results on whole cells and crude membranes indicated that succinate oxidation by menaquinone results in charge imbalance across the membrane, that is, is electrogenic, in B. subtilis and is driven by Δp (Schirawski and Unden, 1998), and that catalysing the reaction in the opposite direction generates a Δp (Schnorpfeil et al, 2001). In this context, the results presented here on E180Q-QFR also present the first experimental evidence on electrogenic catalysis by an isolated and reconstituted succinate:menaquinone reductase.
The electric compensation of transmembrane electron transfer against membrane potential may be a more general phenomenon. Under conditions where the free energy gap of electron transfer is small, it appears that even in the cytochrome bc1 complex, transmembrane electron transfer can be electroneutral (Mulkidjanian et al, 1991). However, this electroneutrality need not involve proton transfer and other principles of electric compensation (Lancaster, 2003) may be applicable in this case (Mulkidjanian, 2005). On the other hand, transmembrane proton transfer pathways similar to the ‘E-pathway' may be relevant also in other, hitherto less well-studied membrane proteins, containing two haem groups, which are oriented along the membrane normal so as to support transmembrane electron transfer. These membrane proteins can have very different physiological roles, such as NADPH oxidases, which, through the generation of superoxide and its derivatives, are thought to be involved in a variety of important cellular functions in phagocytic and non-phagocytic cells and whose involvement in transmembrane proton translocation is currently highly disputed (DeCoursey et al, 2002; Henderson and Meech, 2002; Maturana et al, 2002).
Materials and methods
QFR production, crystallisation, data collection and processing, and crystallographic refinement of wild-type QFR
Wild-type and E180Q QFR were produced and purified as described earlier (Lancaster et al, 1999; Lancaster et al, 2005). Wild-type QFR was crystallised also as described earlier (Lancaster et al, 1999). X-ray diffraction data were collected at the European Synchrotron Radiation facility (ESRF, Grenoble, France). The crystal structure of QFR was determined to 1.78 Å resolution and refined to R and Rfree values of 0.229 and 0.237, respectively (Supplementary Table S1). The atomic coordinates of the oxidised QFR wild-type enzyme have been deposited in the Protein Data Bank with the accession number 2BS2.
Synthesis of naphthoquinones
DMN was synthesised as described earlier (Lancaster et al, 2005). The synthesis of AMN, MMAN and 2-decyl-3-hydroxy-1,4-naphthoquinone is described in Supplementary methods. The oxidation–reduction midpoint potentials of DMN, AMN and MMAN in solution were determined (see Supplementary methods) to be 20, 50 and 80 mV lower, respectively, than that of menaquinone-4 (vitamin K2).
Enzymatic assays
The oxidation of quinols by fumarate was recorded as the absorbance difference. For DMNH2, the difference in absorption at 270 nm minus that at 290 nm (Δɛ270−290=15.2 mM−1 cm−1) (Lancaster et al, 2000), for MMANH2 the difference in absorption at 280 nm minus that at 320 nm (Δɛ280−320=27.6 mM−1 cm−1), and for AMNH2 the difference in absorption at 270 nm minus that at 310 nm (Δɛ270−310=24.6 mM−1 cm−1) were used (Supplementary Figure S5).
Proteoliposomes
Sonicated liposomes were prepared with phosphatidylcholine (Avanti Polar Lipids) and di-palmitoyl phosphatidate (Fluka, Buchs, Switzerland) as described (Biel et al, 2002), except that the liposome film was resuspended in 5 mM HEPES buffer (adjusted to pH 7.5 with NaOH) containing 100 mM KCl. For the acidification measurements, 10 mM pyranine (Molecular Probes, Leiden, NL) was added to the buffer. The suspension of the liposomes contained 10 g phospholipids/l. Proteoliposomes with reconstituted wild-type or E180Q-QFR from W. succinogenes were prepared as described earlier (Biel et al, 2002). Sonicated liposomes were treated with dodecyl-β-D-maltoside (0.8 g/g phospholipid) in a suspension containing 5 mM HEPES buffer (adjusted to pH 7.5 with NaOH) and the naphthoquinone analogues for 3 h under constant stirring. The following addition of wild-type QFR or E180Q-QFR (0.16 g/g phospholipids) was carried out stepwise, stirring was continued for 1 h. The detergent was removed with Bio-Beads SM-2 (Bio-Rad) (0.24 g ml−1) under continued stirring for 1 h. The suspension was sonicated for 20 s at 0°C before use. All steps were carried out using anaerobised buffers. The solution and the Bio-Beads were separated by gentle centrifugation (30 s at 5000 g). The proteoliposomes were concentrated by harsh centrifugation (1 min at 16 100 g), and the supernatant was removed. The concentration of the proteoliposomes was then adjusted with fresh buffer to 10 g phospholipids/l.
Acidification measurements, H+/e− ratio
The proteoliposomes were added to 50 mM HEPES buffer (adjusted to pH 7.5 with NaOH) containing 100 mM KCl and 5 μM valinomycin per gram of phospholipid. MMAN was reduced with NaBH4. The reaction was started by addition of 40 μM fumarate. The MMANH2 oxidation by fumarate was recorded as the absorbance difference at 280 nm minus 320 nm (Supplementary methods). The ΔpH inside the proteoliposomes was recorded as the amplitude of the absorbance ratio at 450 and 415 nm calibrated with 119.3 mM HCl (Supplementary methods). The values expressing the ratio of the acidification of the proteoliposomal interior per reduced quinone (H+/e−) were calculated from the time constants monitored under steady-state conditions. A diode-array UV/VIS spectrophotometer (Agilent 4853) was used for recording different wavelengths simultaneously; therefore, no further scaling was needed.
Measurement of ΔΨ
The TPB− electrode was constructed as described (Karlovsky and Dadak, 1982). Proteoliposomes were suspended in 15 mM HEPES buffer (adjusted to pH 7.5 with NaOH) containing 100 mM LiCl. The buffers were anaerobised and flushed with N2 prior to use. The ΔΨ was calculated by means of the TPB− uptake. The concentration of TPB− within the proteoliposomes (Ti) and in the medium (Te) was calculated using the Nernst equation. Ti was calculated from the maximum amount of TPB− (Ts in mol/g phospholipid) taken up from the medium in the steady state of electron transport according to Equation (1) (Zaritsky et al, 1981; Geisler et al, 1994; Biel et al, 2002).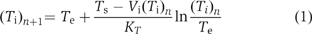 where Vi (3.5 ml g phospholipids−1) (Biel, 2002) represents the average internal volume of the proteoliposomes and KT is the binding constant for the reporter ion (3050 ml/g phospholipids) (Biel, 2002). The internal reporter ion concentration (Ti)n+1 was calculated by the iteration of an assumed value for (Ti)1 (Equation (1)) until (Ti)n+1 was equal to (Ti)n.
where Vi (3.5 ml g phospholipids−1) (Biel, 2002) represents the average internal volume of the proteoliposomes and KT is the binding constant for the reporter ion (3050 ml/g phospholipids) (Biel, 2002). The internal reporter ion concentration (Ti)n+1 was calculated by the iteration of an assumed value for (Ti)1 (Equation (1)) until (Ti)n+1 was equal to (Ti)n.
Supplementary Material
Supplementary Material
Acknowledgments
We thank O Schürmann and A Roth for technical assistance, T Prisner for access to the cyclic voltammetry equipment, B Trumpower for providing an initial sample of 2-decyl-3-hydroxy-1,4-naphthoquinone, H Belrhali and S Monaco for being respective local contacts at ESRF beamlines ID14-EH3 and -EH1, the Deutsche Forschungsgemeinschaft (SFB 472 ‘Molecular Bioenergetics') and the Max Planck Society for funding.
References
- Agmon N (1995) The Grotthuss mechanism. Chem Phys Lett 244: 456–462 [Google Scholar]
- Bertero MG, Rothery RA, Palak M, Hou C, Lim D, Blasco F, Weiner JH, Strynadka NC (2003) Insights into the respiratory electron transfer pathway from the structure of nitrate reductase A. Nature Struct Biol 10: 681–687 [DOI] [PubMed] [Google Scholar]
- Biel S (2002) Rekonstitution der gekoppelten Fumarat-Atmung von Wolinella succinogenes. Doctoral Thesis, Johann Wolfgang Goethe-Universität, Frankfurt am Main, Germany
- Biel S, Simon J, Groß R, Ruiz T, Ruitenberg M, Kröger A (2002) Reconstitution of coupled fumarate respiration in liposomes by incorporating the electron transport enzymes isolated from Wolinella succinogenes. Eur J Biochem 269: 1974–1983 [DOI] [PubMed] [Google Scholar]
- DeCoursey TE, Morgan D, Cherny VV (2002) The gp91(phox) component of NADPH oxidase is not a voltage-gated proton channel. J Gen Physiol 120: 773–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faxén K, Gilderson G, delroth P, Brzezinski P (2005) A mechanistic principle for proton pumping by cytochrome c oxidase. Nature 437: 286–289 [DOI] [PubMed] [Google Scholar]
- Fieser LF, Fieser M (1965) Organische Chemie. Weinheim: Verlag Chemie. pp 1038–1043 [Google Scholar]
- Ge Z, Feng Y, Dangler CA, Xu S, Taylor NS, Fox JG (2000) Fumarate reductase is essential for Helicobacter pylori colonization of the mouse stomach. Microb Pathog 29: 279–287 [DOI] [PubMed] [Google Scholar]
- Geisler V, Ullmann R, Kröger A (1994) The direction of the proton exchange associated with the redox reactions of menaquinone during electron transport in Wolinella succinogenes. Biochim Biophys Acta 1184: 219–226 [Google Scholar]
- Haas AH, Lancaster CRD (2004) Calculated coupling of transmembrane electron and proton transfer in dihemic quinol:fumarate reductase. Biophys J 87: 4298–4315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas AH, Sauer US, Gross R, Simon J, Mäntele W, Lancaster CRD (2005) FTIR difference spectra of Wolinella succinogenes quinol:fumarate reductase support a key role of Glu C180 within the ‘E-pathway hypothesis' of coupled transmembrane electron and proton transfer. Biochemistry 44: 13949–13961 [DOI] [PubMed] [Google Scholar]
- Hägerhäll C, Hederstedt L (1996) A structural model for the membrane-integral domain of succinate: quinone oxidoreductases. FEBS Lett 389: 25–31 [DOI] [PubMed] [Google Scholar]
- Henderson LM, Meech RW (2002) Proton conduction through gp91(phox). J Gen Physiol 120: 759–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jormakka M, Törnroth S, Byrne B, Iwata S (2002) Molecular basis of proton motive force generation: structure of formate dehydrogenase-N. Science 295: 1863–1868 [DOI] [PubMed] [Google Scholar]
- Karlovsky P, Dadak V (1982) Tetraphenylborate-sensitive electrode for measuring membrane potential. Folia Microbiol 27: 460–464 [DOI] [PubMed] [Google Scholar]
- Kröger A (1978) Fumarate as terminal acceptor of phosphorylative electron transport. Biochim Biophys Acta 505: 129–145 [DOI] [PubMed] [Google Scholar]
- Kröger A, Biel S, Simon J, Groß R, Unden G, Lancaster CRD (2002) Fumarate respiration of Wolinella succinogenes: enzymology, energetics and coupling mechanism. Biochim Biophys Acta 1553: 23–38 [DOI] [PubMed] [Google Scholar]
- Lancaster CRD (2002) Wolinella succinogenes quinol:fumarate reductase—2.2 Å resolution crystal structure and the E-pathway hypothesis of coupled transmembrane proton and electron transfer. Biochim Biophys Acta 1565: 215–231 [DOI] [PubMed] [Google Scholar]
- Lancaster CRD (2003) The role of electrostatics in proton-conducting membrane protein complexes. FEBS Lett 545: 52–60 [DOI] [PubMed] [Google Scholar]
- Lancaster CRD (2004) Structure and function of succinate:quinone oxidoreductases and the role of quinol:fumarate reductases in fumarate respiration. In Respiration in Archaea and Bacteria. Volume 1: Diversity of Prokaryotic Electron Transport Carriers, Zannoni D (ed). The Netherlands, Dordrecht: Kluwer Scientific pp 57–85 [Google Scholar]
- Lancaster CRD, Kröger A, Auer M, Michel H (1999) Structure of fumarate reductase from Wolinella succinogenes at 2.2 Å resolution. Nature 402: 377–385 [DOI] [PubMed] [Google Scholar]
- Lancaster CRD, Groß R, Haas A, Ritter M, Mäntele W, Simon J, Kröger A (2000) Essential role of Glu-C66 for menaquinol oxidation indicates transmembrane electrochemical potential generation by Wolinella succinogenes. Proc Natl Acad Sci USA 97: 13051–13056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster CRD, Groß R, Simon J (2001) A third crystal form of Wolinella succinogenes quinol:fumarate reductase reveals domain closure at the site of fumarate reduction. Eur J Biochem 268: 1820–1827 [PubMed] [Google Scholar]
- Lancaster CRD, Sauer US, Groß R, Haas AH, Graf J, Schwalbe H, Mäntele W, Simon J, Madej MG (2005) Experimental support for the ‘E-pathway hypothesis' of coupled transmembrane e− and H+ transfer in dihemic quinol:fumarate reductase. Proc Natl Acad Sci USA 102: 18860–18865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsson M, Tolstoy D, Aasa R, Hederstedt L (2000) The distal heme center in Bacillus subtilis succinate:menaquinone reductase is crucial for electron transfer to menaquinone. Biochemistry 39: 8617–8624 [DOI] [PubMed] [Google Scholar]
- Maturana A, Krause KH, Demaurex N (2002) NOX family NADPH oxidases: do they have built-in proton channels? J Gen Physiol 120: 781–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mileni M, Haas AH, Mäntele W, Simon J, Lancaster CRD (2005) Probing heme propionate involvement in transmembrane proton transfer coupled to electron transfer in dihemic quinol:fumarate reductase by 13C-labeling and FTIR difference spectroscopy. Biochemistry 44: 16718–16728 [DOI] [PubMed] [Google Scholar]
- Mileni M, MacMillan F, Tziatzios C, Zwicker K, Haas AH, Mäntele W, Simon J, Lancaster CRD (2006) Heterologous production in Wolinella succinogenes and characterisation of the quinol:fumarate reductase enzymes from Helicobacter pylori and Campylobacter jejuni. Biochem J 395: 191–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell P (1976) Possible molecular mechanisms of the protonmotive function of cytochrome systems. J Theor Biol 62: 327–367 [DOI] [PubMed] [Google Scholar]
- Mitchell P (1979) Keilin's respiratory chain concept and its chemiosmotic consequences. Science 206: 1148–1159 [DOI] [PubMed] [Google Scholar]
- Mulkidjanian AY (2005) Ubiquinol oxidation in the cytochrome bc complex: reaction mechanism and prevention of short-circuiting. Biochim Biophys Acta 1709: 5–34 [DOI] [PubMed] [Google Scholar]
- Mulkidjanian AY, Heberle J, Cherepanov DA (2006) Protons at interfaces: implications for biological energy conversion, Biochim. Biophys. Acta, in press, accessible online as DOI: 10.1016/j.bbabio.2006.02.015 [DOI] [PubMed] [Google Scholar]
- Mulkidjanian AY, Mamedov MD, Drachev LA (1991) Slow electrogenic events in the cytochrome bc1-complex of Rhodobacter sphaeroides: the electron transfer between cytochrome b hemes can be non-electrogenic. FEBS Lett 284: 227–231 [DOI] [PubMed] [Google Scholar]
- Ohnishi T, Moser CC, Page CC, Dutton PL, Yano T (2000) Simple redox-linked proton-transfer design: new insights from structures of quinol-fumarate reductase. Structure 8: R23–R32 [DOI] [PubMed] [Google Scholar]
- Richardson D, Sawers G (2002) Structural biology. PMF through the redox loop. Science 295: 1842–1843 [DOI] [PubMed] [Google Scholar]
- Schirawski J, Unden G (1998) Menaquinone-dependent succinate dehydrogenase of bacteria catalyzes reversed electron transport driven by the proton potential. Eur J Biochem 257: 210–215 [DOI] [PubMed] [Google Scholar]
- Schnorpfeil M, Janausch IG, Biel S, Kröger A, Unden G (2001) Generation of a proton potential by succinate dehydrogenase of Bacillus subtilis functioning as a fumarate reductase. Eur J Biochem 268: 3069–3074 [DOI] [PubMed] [Google Scholar]
- von Grotthuss CJT (1806) Mémoire sur la decomposition de l'eau et des corps qu'elle tient en dissolution à l'aide de l'electricité galvanique. Ann Chim Phys (Paris) 58: 54–74 [Google Scholar]
- Zaritsky A, Kihara M, Macnab RM (1981) Measurement of membrane potential in Bacillus subtilis: a comparison of lipophilic cations, rubidium ion, and cyanine dye as probes. J Membr Biol 63: 215–231 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material