

Abstract
The activation of tumor suppressor p53 induces apoptosis or cell cycle arrest depending on the state and type of cell, but it is not fully understood how these different responses are regulated. Here, we show that Puma and Noxa, the well-known p53-inducible proapoptotic members of the Bcl-2 family, differentially participate in dual pathways of the induction of apoptosis. In normal cells, Puma but not Noxa induces mitochondrial outer membrane permeabilization (MOMP), and this function is mediated in part by a pathway that involves calcium release from the endoplasmic reticulum (ER) and the subsequent caspase activation. However, upon E1A oncoprotein expression, cells also become susceptible to MOMP induction by Noxa, owing to their sensitization to the ER-independent pathway. These findings offer a new insight into differential cellular responses induced by p53, and may have therapeutic implications in cancer.
Keywords: apoptosis, BH3-only proteins, endoplasmic reticulum, mitochondria, p53
Introduction
An appropriate response to stress stimuli is crucial for the prevention of cellular transformation as well as the maintenance of normal tissue function. The tumor suppressor p53 plays central roles in the organization of stress responses (Ko and Prives, 1996). The p53 protein accumulates promptly in stressed cells, and is activated as a transcription factor. Activated p53 in turn induces stress responses mainly through the transcriptional regulation of effector molecules, each of which is involved in the execution of a specific response (Vousden and Lu, 2002; Oren, 2003). p53 activation can result in the elicitation of two opposing responses, namely, eliminating a cell by activating the apoptotic pathway or supporting the preservation of a cell by arresting the cell cycle (Vousden and Lu, 2002; Oren, 2003). In effect, the balance between p53-induced apoptosis and cell cycle arrest should be important for the simultaneous achievement of eliminating cancerous cells and saving normal cells.
The outcome of p53 activation can be affected by the cell state as well as the cell type (Vousden and Lu, 2002; Oren, 2003). It has been shown that the apoptotic response is favorably selected in cells expressing oncoproteins such as adenovirus E1A. Mouse embryonic fibroblasts (MEFs) normally undergo p53-mediated cell cycle arrest in response to stresses caused by DNA damage or serum deprivation, whereas MEFs expressing E1A undergo p53-mediated apoptosis under the same conditions (Lowe et al, 1993a). Moreover, several types of cell, including thymocytes (Clarke et al, 1993; Lowe et al, 1993b) and epithelial stem cells in the crypts of the small intestine (Merritt et al, 1994), are ready to apoptose in a p53-dependent manner upon DNA damage, even in the absence of oncoproteins. To date, it remains largely unclear how contextual factors such as the state and the type of cell affect the progression of the p53-mediated apoptotic response.
The molecular pathways of p53-mediated apoptosis are still not fully understood. Several proapoptotic molecules have been shown to be transcriptionally induced by p53; moreover, the transcription-independent role of p53 in the promotion of apoptosis has also been described (Zamzami and Kroemer, 2005). The contribution of each factor to the entire p53-mediated apoptotic response is still under extensive investigation. Among the proapoptotic transcriptional targets of p53, Puma (Nakano and Vousden, 2001; Yu et al, 2001) and Noxa (Oda et al, 2000) belong to the BH3-only group of the Bcl-2 family, and are considered to indirectly induce mitochondrial outer membrane permeabilization (MOMP), known to be induced by the activation of Bax and Bak (Letai et al, 2002): Puma and Noxa interfere with the interaction of prosurvival Bcl-2 family members with the proapoptotic Bax and Bak, through their direct binding to the prosurvival members. Indeed, this BH3-only subgroup is termed by Korsmeyer and colleagues as ‘sensitizers' vis-à-vis ‘activators' that directly engage Bax and Bak (Letai et al, 2002) for their activation and subsequent MOMP induction (Wei et al, 2001). The contributions of Puma and Noxa to p53-mediated apoptosis have been studied using gene-targeting strategies in mice (Shibue et al, 2003; Villunger et al, 2003). Both these factors play crucial roles in the DNA-damage-induced apoptosis of MEFs expressing E1A, whereas only Puma is involved in the X-ray-irradiation-induced apoptosis of thymocytes (Shibue et al, 2003; Villunger et al, 2003). It has been reported that Puma interacts with various prosurvival Bcl-2 family members, whereas Noxa selectively interacts with Mcl-1 and A1 of this family (Chen et al, 2005).
The important roles of Puma and Noxa in p53-mediated apoptosis, together with the observation that the progression of p53-mediated apoptosis is affected by the cellular context, prompted us to further study apoptotic pathways induced by Puma and/or Noxa. We report here the presence of a hitherto unrecognized pathway of p53-mediated apoptosis involving calcium release from the endoplasmic reticulum (ER), which is activated by Puma but not Noxa. Our results may also offer a molecular basis for differential cellular responses to stress stimuli, which are commonly mediated by p53.
Results
Distinct proapoptotic functions of Puma and Noxa
We first generated NIH3T3 fibroblasts that constitutively express adenovirus E1A, cellular oncoprotein E2F1 and papilloma virus E7 (hereafter referred to as E1A-3T3, E2F1-3T3 and E7-3T3 cells, respectively), all of which interfere with the mechanism of retinoblastoma protein-mediated cell cycle regulation, and may therefore alter the fate of cellular responses (Lavia et al, 2003). The effects of Puma and Noxa on these generated and control NIH3T3 cells were studied by the retroviral expression of these genes. Interestingly, Noxa did not induce the death of control NIH3T3, E2F1-3T3 or E7-3T3 cells, but did induce that of E1A-3T3 cells 24 h after its expression, whereas Puma induced the death of all cells at the same time point (Figure 1A). It is worth noting that the level of ectopically expressed Noxa is even lower in E1A-3T3 cells than in control NIH3T3 cells (Supplementary Figure 1A). Therefore, the specific function of Noxa in E1A-3T3 cells likely reflects a change in the cellular state induced by E1A, but not the level of Noxa expression; E1A expression results in the sensitization of cells to Noxa-induced apoptosis. E1A-dependent sensitization to Noxa-induced apoptosis, as well as the differential effects of Puma and Noxa on cells that do not express E1A, was similarly observed in MEFs (Supplementary Figure 2A).
Figure 1.
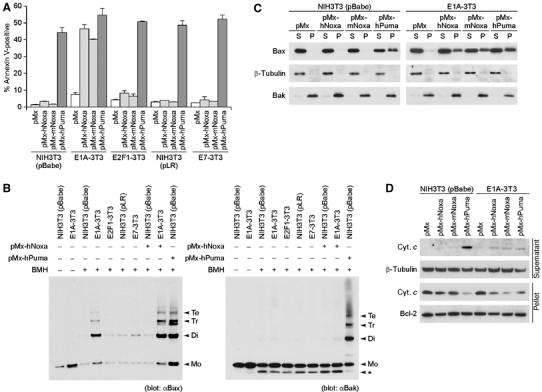
Noxa- and Puma-induced apoptosis in the absence and presence of oncoproteins. (A) Noxa- and Puma-induced apoptosis of NIH3T3 cells expressing various oncoproteins. E1A-3T3, E2F1-3T3 and E7-3T3 cells as well as NIH3T3 cells expressing control vectors (pBabe and pLR) were infected with the retrovirus expressing human (pMx-hNoxa) or mouse Noxa (pMx-mNoxa), human Puma (pMx-hPuma) or the control retrovirus (pMx). Testing with a GFP-expressing pMx construct indicated that the efficiencies of pMx-derived retrovirus infection of control and oncoprotein-expressing NIH3T3 cells were consistently about 80 and 70%, respectively, at 24 h after infection. Values shown are means±s.d. of triplicate samples. (B) Oligomerization of Bax (left panel) and Bak (right panel). Mitochondria-enriched heavy-membrane fractions were prepared from NIH3T3 cells infected with the indicated retrovirus. Obtained protein samples were subsequently treated with the chemical crosslinker 1,6-bismaleimidohexane (BMH) or dimethylsulfoxide (DMSO), and analyzed by immunoblotting. Markers Mo, Di, Tr and Te represent the sizes of monomer, dimer, trimer and tetramer, respectively. (*) An intramolecularly crosslinked Bak monomer. (C, D) MOMP induced by Noxa and Puma in control NIH3T3 cells and E1A-3T3 cells. Membrane insertion of Bax (C) and cytosolic release of cytochrome c (cyt. c) (D) were analyzed. In panel C, cell homogenates were treated with alkali, and subsequently separated by centrifugation. Bax molecules residing in the cytosol or loosely attaching to the membrane are separated into the supernatant (S) fraction, whereas membrane-inserted Bax molecules are separated into the pellet (P) fraction. In panel D, the supernatant and pellet fractions obtained by the digitonin treatment and subsequent centrifugation were analyzed, in which supernatant corresponds to the cytosolic fraction and pellet corresponds to the membrane and nuclear fraction.
To gain further insights into the proapoptotic functions of Puma and Noxa in the above cells, each expressing a distinct oncoprotein, we analyzed events associated with MOMP, which has been suggested to be the rate-limiting step in the mitochondrial pathway of apoptosis (Green and Kroemer, 2004). We first examined the oligomerization of Bax and Bak, one of the hallmarks of the progression of MOMP (Wei et al, 2001). Perhaps surprisingly, we found that Bax is substantially oligomerized in E1A-3T3 cells but not in E2F1-3T3, E7-3T3 or control NIH3T3 cells (Figure 1B). Although Noxa could not induce Bax oligomerization in NIH3T3 cells, it did enhance the oligomerization of Bax in E1A-3T3 cells (Figure 1B). Noxa failed to induce Bak oligomerization in either of these cells, suggesting that Noxa selectively acts on Bax (Figure 1B). Further analysis of the activation of Bax, using an antibody that specifically detects the activation-related conformational change of the Bax protein (Hsu and Youle, 1997), revealed that E1A expression induces this change, which is further enhanced by the expression of Noxa (Supplementary Figure 3). In contrast, Puma effectively induced the oligomerization of Bax and Bak even in the absence of E1A expression (Figures 1B). These observations are consistent with those of other studies showing that Puma functions independently of the cell type by interfering with more Bcl-2 prosurvival members than does Noxa (Chen et al, 2005), and also raise the question of why Puma is more potent than Noxa in inducing apoptosis.
We next studied the membrane insertion of Bax, as well as the cytosolic release of cytochrome c, the events directly associated with MOMP (Liu et al, 1996; Goping et al, 1998). As expected from the above results, Puma but not Noxa induced the membrane insertion of Bax and the cytosolic release of cytochrome c in NIH3T3 cells within 20 h after its expression (Figure 1C and D). Interestingly, both the membrane insertion of Bax and the cytosolic release of cytochrome c were almost undetectable in E1A-3T3 cells, unless Noxa or Puma was expressed (Figure 1C and D). Thus, we interpret that Bax oligomerization detected in E1A-3T3 cells is insufficient to induce Bax membrane insertion for apoptosis and that Noxa and Puma can induce apoptosis in these cells by further promoting the activation of Bax (i.e., enhancing oligomerization to allow its membrane insertion) to trigger cytochrome c release. Although the oligomerization of neither Bax nor Bak was observed in control NIH3T3 cells, Bax and Bak were effectively activated for MOMP induction upon Puma expression (Figure 1B–D). These results in toto suggest that, in addition to the common apoptotic pathway induced by Puma and Noxa, Puma may activate an additional pathway that would converge to MOMP during p53-dependent apoptosis.
Differential requirement of caspases for MOMP induction
Caspases, a group of proteases essential for apoptosis, have been generally supposed to be activated in the downstream of MOMP (Degterev et al, 2003). However, it has also been indicated that several members of the caspase family can be activated before MOMP, and play crucial roles in the progression of MOMP, at least in some forms of apoptosis (Lassus et al, 2002; Marsden et al, 2002). On the basis of these reports, we next examined the role of caspases in Noxa-induced and Puma-induced apoptotic responses. Treatment with zVADfmk, a ubiquitous inhibitor of caspases, markedly decreased the rate of cell death induced by Puma in control NIH3T3 cells, as well as that induced by Noxa or Puma in E1A-3T3 cells (Figure 2A). Interestingly, however, the membrane insertion of Bax and the cytosolic release of cytochrome c were inhibited only in the Puma-induced apoptosis of control NIH3T3 cells (Figure 2B and C). These results in toto suggest that the induction of MOMP is indeed mediated by two pathways. That is, in control NIH3T3 cells, the induction of MOMP is mediated, at least in part, by a zVADfmk-sensitive pathway, which is activated by Puma but not Noxa, whereas in E1A-3T3 cells a zVADfmk-insensitive MOMP-inducing pathway, which is activated by both Noxa and Puma, operates affectively. Time-course analysis of cell viability using another caspase inhibitor, Q-VD-OPh, revealed that the Puma-induced death of control NIH3T3 cells is almost completely inhibited up to 72 h after infection with a Puma-expressing retrovirus, whereas the Noxa- or Puma-induced death of E1A-3T3 cells gradually progresses even in the presence of this inhibitor (Supplementary Figure 1B). These results can be interpreted as follows. In the former case, control NIH3T3 cells remain almost completely alive up to 72 h because MOMP progression is impaired or delayed by Q-VD-OPh treatment. In contrast, MOMP occurs even in the presence of Q-VD-OPh in the latter cases; hence, E1A-3T3 cells eventually start dying, which is consistent with the report showing that MOMP reduces cell viability even in the absence of massive caspase activation (Ekert et al, 2004). These results collectively suggest the fundamental difference between zVADfmk-sensitive and -insensitive pathways of MOMP induction.
Figure 2.
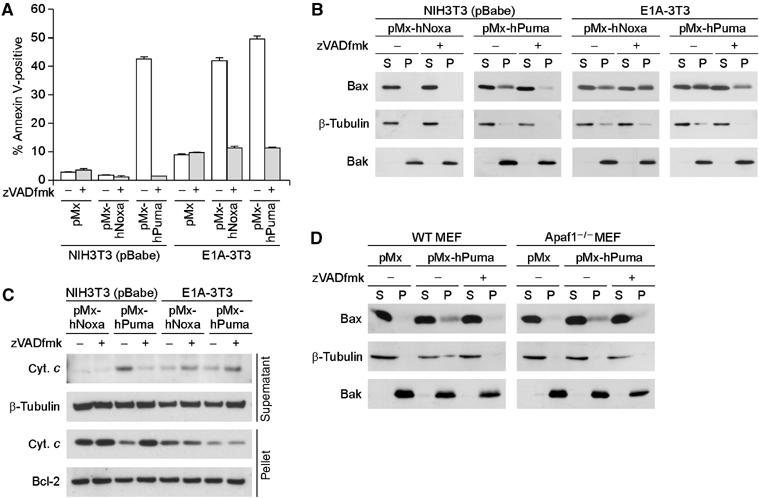
zVADfmk-sensitive and -insensitive pathways of MOMP induction. (A) Effect of zVADfmk on Puma- and Noxa-induced cell death in control NIH3T3 cells and E1A-3T3 cells. Under these experimental settings, zVADfmk treatment did not have clear cytotoxic effects. Values shown are means±s.d. from triplicate samples. (B, C) Inhibitory effect of zVADfmk on MOMP. The effects of zVADfmk treatment on membrane insertion of Bax (B) and cytochrome c release (C) were analyzed. In panel C, the amount of cytochrome c detected in the supernatant fractions of Noxa- or Puma- expressing E1A-3T3 cells was reproducibly increased by the zVADfmk treatment. Because the amount of cytochrome c detected in the pellet fraction decreased to a similar extent regardless of zVADfmk treatment, difference in the supernatant fraction is presumably due to the inhibitory effect of zVADfmk on the progression of apoptosis, that is, zVADfmk-treated cells do not readily lose their plasma membrane integrity and thereby they should retain released cytochrome c molecules in the cytosol for longer periods than untreated cells. (D) Puma-induced Bax membrane insertion in wild-type (WT) and Apaf1-deficient (Apaf1−/−) MEFs. WT and Apaf1−/− MEFs were prepared from the same litter. Notably, Puma-induced membrane insertion of Bax occurred in an Apaf-1-independent but caspase-dependent manner.
To delineate further the Puma-activated, zVADfmk-sensitive MOMP induction pathway, we expressed Puma in MEFs deficient in the Apaf1 gene (Apaf1−/− MEFs), in which the activation of post-MOMP apoptotic events is severely impaired (Yoshida et al, 1998). As shown in Figure 2D, the Puma-induced membrane insertion of Bax was inhibited in both wild-type and Apaf1−/− MEFs following zVADfmk treatment, indicating that a caspase(s) that contributes to the insertion of Bax is activated by Puma upstream of Apaf-1, namely, in the absence of post-MOMP apoptotic events. In contrast, Puma-induced apoptosis was almost completely suppressed in Apaf1−/− MEFs at least 24 h after the expression of Puma (Supplementary Figure 2B), collectively indicating that Puma activates a caspase(s) involved upstream of MOMP induction, whereas Apaf-1 and other caspases (such as caspase-3) are required for post-MOMP apoptotic events.
Involvement of caspase-12 in Puma-induced apoptosis
To identify the caspase(s) responsible for the induction of MOMP, we used peptide-based inhibitors that show specificity for several members of the caspase family (Garcia-Calvo et al, 1998). Among these, only zWEHDfmk inhibited Puma-induced Bax membrane insertion in NIH3T3 cells (Figure 3A). Indeed, whereas zWEHDfmk interfered with Bax membrane insertion and cell death induced by Puma in NIH3T3 cells, it failed to do so in the case of Noxa- and Puma-induced apoptosis of E1A-3T3 cells (Figure 3B and Supplementary Figure 4A). Furthermore, protease activity that cleaves the W-E-H-D peptide was induced by Puma in NIH3T3 cells, but not by Noxa in E1A-3T3 cells (Supplementary Figure 4B), indicating the specific involvement of W-E-H-D peptide-cleaving caspase(s) in the former case.
Figure 3.
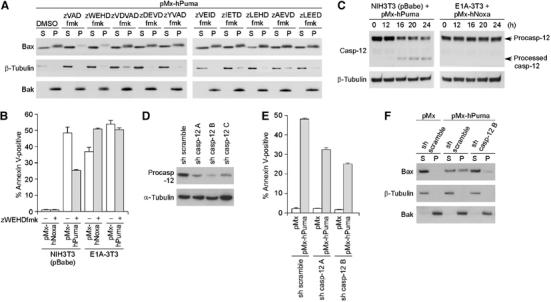
Essential role of caspase-12 in Puma-induced MOMP and apoptosis. (A) Effects of various peptide-based inhibitors of caspases on Puma-induced Bax membrane insertion. NIH3T3 cells expressing Puma were treated with DMSO, a pan-caspase inhibitor zVADfmk, or various peptide-based inhibitors that specifically inhibit several members of the caspase family (from zWEHDfmk to zLEEDfmk). (B) Effect of zWEHDfmk on Noxa- and Puma-induced cell death in control NIH3T3 cells and E1A-3T3 cells. (C) Caspase-12 (casp-12) processing during Puma-induced apoptosis of NIH3T3 cells and Noxa-induced apoptosis of E1A-3T3 cells. Caspase-12 in the total cell lysate of indicated cells was detected by immunoblotting using the anti-caspase-12 rat monoclonal antibody. The processed form of caspase-12, a possible intermediate for the conversion of the precursor (procasp-12) into the active form, was detected specifically during Puma-induced apoptosis of NIH3T3 cells. Cells were harvested at the indicated times after infection with the pMx-derived retrovirus. (D–F) Effect of shRNA-mediated knock-down of caspase-12 on Puma-induced apoptosis of NIH3T3 cells. NIH3T3 cells were infected with lentivirus that expresses either scramble shRNA or shRNA targeting caspase-12. Three different caspase-12-targeting shRNA sequences (sh casp-12 A–C) were tested, and each resulted in 64, 83 and 63% reduction in the level of procaspase-12 expression, respectively (D). The effects of caspase-12-targeting shRNA on Puma-induced cell death (E) and Bax membrane insertion (F) were also analyzed. In panels B and E, values shown are means±s.d. from triplicate samples.
The W-E-H-D peptide is a substrate of caspase-1 (human and mouse), -4 and -5 (human) (Thornberry et al, 1997). These three caspases form a subgroup in the caspase family together with caspase-11 and -12 (mouse), all of which share structural and functional similarities (Degterev et al, 2003). As caspase-1 is not expressed in NIH3T3 cells (Li et al, 1995), remaining candidates in this murine cell line are caspase-11 and -12. For caspase-11, the L-E-H-D peptide is a more preferable substrate than the W-E-H-D peptide (Kang et al, 2000), but zLEHDfmk showed no inhibitory effect on the Puma-induced membrane insertion of Bax (Figure 3A). We therefore focused on caspase-12, and found that this caspase is indeed processed for activation in NIH3T3 cells expressing Puma (Figure 3C). As caspase-12 predominantly resides on the cytoplasmic surface of the ER membrane (Nakagawa et al, 2000), this suggests that caspase-12 is activated by Puma on ER. We further tried to suppress the expression of caspase-12 in NIH3T3 cells using several different short hairpin RNA (shRNA) sequences that specifically interfere with the caspase-12 expression (Figure 3D), and found that Puma-induced cell death was suppressed by up to 50% 24 h after the expression of Puma, which is accompanied by an impaired Bax membrane insertion (Figure 3E and F). Moreover, Puma-induced cell death was partly impaired by caspase-12 deficiency in MEFs (Supplementary Figure 2C). These data collectively indicate the essential role of caspase-12 in Puma-induced MOMP and apoptosis in these fibroblasts, at least at their onset.
Puma-mediated calcium release from ER
We then determined how Puma, which is generally considered to reside on mitochondria, activates caspase-12 presumably on the ER membrane. The most established function of caspase-12 is its contribution to the apoptotic pathway induced by ‘ER stresses' (Orrenius et al, 2003), which include all the insults that interfere with the physiological functions of ER, such as the deregulated mobilization of calcium through the ER membrane and the accumulation of misfolded proteins in the ER lumen (Orrenius et al, 2003). Accordingly, we considered the possibility that Puma also perturbs ER function and thereby activates caspase-12. Calcium release from ER and the resultant increase in cytosolic free-calcium concentration ([Ca2+]c) is commonly triggered by most forms of ER stress stimuli, and is supposed to play central roles in the ER-initiated apoptotic response (Breckenridge et al, 2003). Assuming that Puma activates the apoptotic response through the perturbation of ER function, Puma-induced apoptosis should be affected by the interference of this calcium release.
The release of calcium from ER is primarily achieved through two different types of channel, the inositol 1,4,5-triphosphate receptor (InsP3R) and ryanodine receptor (RyR) families (Breckenridge et al, 2003). We therefore attempted treatment with chemical inhibitors specific for each type of calcium channel, and found that xestospongin C (xestC) and 2-aminoethoxy-diphenylborate (2-APB), both of which are blockers of InsP3Rs, interfered with the Puma-induced apoptosis of NIH3T3 cells (Figure 4A). In contrast, dantrolene, a specific inhibitor of RyRs, did not exert significant effects (Figure 4A). In addition, Puma-induced caspase-12 processing and Bax membrane insertion were both specifically inhibited by the treatment with xestC or 2-APB (Figure 4B and Supplementary Figure 5A). Moreover, shRNA-mediated suppression of type I InsP3R expression caused partial inhibition of Puma-induced death of NIH3T3 cells (Figure 4C and D). We also investigated the link between Puma and InsP3Rs by the immunoprecipitation analysis. As shown in Figure 4E, Puma was not clearly co-precipitated with InsP3Rs; however, in accordance with the previous reports (Chen et al, 2004; White et al, 2005), co-precipitation was observed between InsP3Rs and the prosurvival Bcl-2 family members Bcl-2 or Bcl-XL (Figure 4E). It may be worth noting that levels of this coprecipitation were significantly lower when Puma is expressed (Figure 4E), suggesting the possibility that Puma affects the function of InsP3Rs indirectly through its interaction with prosurvival Bcl-2 family members. These results collectively support the notion that Puma induces calcium release from ER through InsP3Rs, which promotes subsequent activation of caspase-12 and further progression of the apoptotic response.
Figure 4.
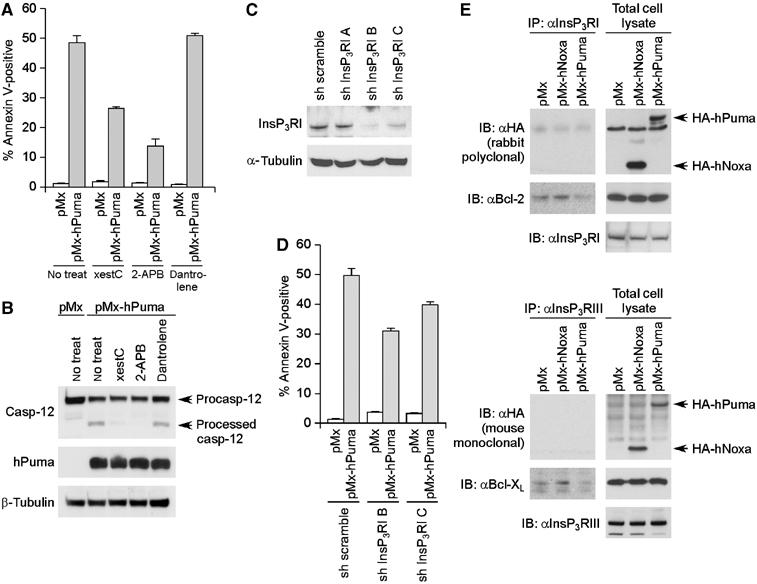
InsP3R-mediated calcium release from ER during Puma-induced apoptosis. (A, B) Effects of the blockade of ER calcium channels on Puma-induced apoptosis of NIH3T3 cells. Puma-induced cell death (A) and caspase-12 processing (B) in NIH3T3 cells were analyzed in the presence of the blockers of InsP3R (xestC and 2-APB) or dantrolene, the blocker of ryanodine receptor (RyR). (C, D) Impairment of Puma-induced apoptosis by the shRNA-mediated knock-down of type I InsP3R expression. NIH3T3 cells were infected with lentivirus that expresses either scramble shRNA or shRNA targeting type I InsP3R. Three different type I InsP3R-targeting shRNA sequences (sh InsP3RI A–C) were tested, and each resulted in 6, 84 and 60% reduction in the level of type I InsP3R expression, respectively (C). The effects of these shRNA on Puma-induced cell death (D) were also analyzed. (E) Interaction between Bcl-2 family proteins and InsP3Rs. NIH3T3 cells infected with Puma-expressing, Noxa-expressing or control retrovirus were analyzed by immunoprecipitation. The lysate from each sample was immunoprecipitated with an antibody to type I InsP3R (InsP3RI) (upper panel) or an antibody to type III InsP3R (InsP3RIII) (lower panel), and subsequently analyzed by immunoblotting. In panels A and D, values shown are means±s.d. from triplicate samples.
To examine directly the effect of Puma on the mobilization of calcium, we used yellow cameleon 3.60 (YC3.60), a cytosolic calcium indicator based on the principle of fluorescent resonance energy transfer (Nagai et al, 2004). As shown by the increased emission ratio of YC3.60, [Ca2+]c was elevated by Puma in NIH3T3 cells, whereas Noxa did not significantly change [Ca2+]c in E1A-3T3 cells (Supplementary Figure 6A). Puma-induced increase in [Ca2+]c was significantly suppressed by the treatment with xestC, supporting the notion that this [Ca2+]c change was caused by the release of ER calcium through InsP3Rs (Supplementary Figure 6B). Moreover, [Ca2+]c change was still observed in the presence of zVADfmk, indicating that calcium mobilization occurs in the upstream of caspases activation (Supplementary Figure 6C). Time-lapse microscopy revealed that NIH3T3 cells expressing Puma typically showed a sustained, intermediate [Ca2+]c increase with a YC3.60 emission ratio of 3–6 (Figure 5A and B). Subsequently, the cells completed the remaining process of apoptosis resulting in an emission ratio of more than 8, which may represent a marked [Ca2+]c increase owing to the loss of plasma membrane integrity (Figure 5A and B). In contrast, E1A-3T3 cells expressing Noxa did not show a sustained, intermediate [Ca2+]c increase (Figure 5A and B). Furthermore, the causal contribution of calcium release from ER in the Puma-induced apoptotic response was confirmed by the observation that the cytosolic calcium chelator BAPTA-AM exerted inhibitory effect specifically on the Puma-induced apoptosis of NIH3T3 cells (Figure 5C).
Figure 5.
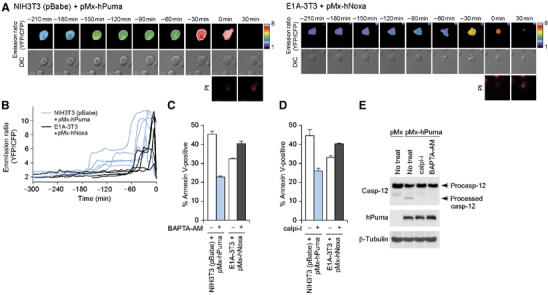
Causal contribution of ER calcium release in Puma-induced apoptosis. (A, B) Time-lapse observation of [Ca2+]c change during Puma-induced apoptosis of control NIH3T3 cells (left panel) and Noxa-induced apoptosis of E1A-3T3 cells (right panel). Cells nucleofected with the YC3.60 expression vector, together with pMx-hPuma or -hNoxa, were analyzed by time-lapse microscopy (see Materials and methods). The time point at which the relevant cell turned PI-positive is set as time 0, and changes in YC3.60 emission ratio (YFP/CFP) were retrospectively displayed. Panel A shows pseudocolored images representing emission ratio calculated on the pixel-by-pixel basis, whereas panel B shows the ratio of fluorescence intensity of CFP and YFP within a region drawn around an individual cell. In panel B, five representative cases are shown from each of control NIH3T3 cells expressing Puma and E1A-3T3 cells expressing Noxa. (C, D) Effects of BAPTA-AM (C) and calpain inhibitor I (calpi-I) (D) on the rate of Puma-induced death of NIH3T3 cells and that of Noxa-induced death of E1A-3T3 cells. Values shown are means±s.d. from triplicate samples. (E) Effects of calpain inhibitor I and BAPTA-AM on Puma-induced caspase-12 processing.
Regarding how this calcium mobilization leads to caspase-12 activation, a previous study implicated m-calpain as a direct activator of caspase-12 (Nakagawa and Yuan, 2000). Calpains belong to the family of cysteine proteases that are activated in a calcium-dependent manner; they translocate from the cytosol to membranes such as the cytoplasmic surfaces of the plasma membrane and ER membrane in response to calcium mobilization (Suzuki et al, 2004). To examine the role of calpains in linking between Puma-induced calcium mobilization and caspase-12 activation, we utilized calpain inhibitor I, a specific inhibitor of calpain also known as ALLN. As expected, calpain inhibitor I treatment specifically interfered with the Puma-induced apoptosis of NIH3T3 cells (Figure 5D), which is accompanied by an impaired Bax membrane insertion and cytochrome c release (Supplementary Figure 5B and C). Both calpain inhibitor I and BAPTA-AM attenuated the Puma-induced processing of caspase-12 (Figure 5E), consistent with the idea that Puma activates caspase-12 by mobilizing calcium and subsequently activating calpain.
Caspase-dependent MOMP in p53-mediated apoptosis of thymocytes and cerebellar granule neurons
The results shown above indicated the presence of dual pathways operating in the p53-mediated apoptotic response: one is the zVADfmk-sensitive, Puma–ER–Ca2+–calpain–caspase-12 pathway and the other, the zVADfmk-insensitive Puma/Noxa pathway (see Figure 6D). Between the two p53-inducible BH3-only molecules, only Puma but not Noxa can activate the former pathway; Puma-induced calcium mobilization from ER and the subsequent activation of caspase-12 specifically contribute to this pathway. To confirm the role of the zVADfmk-sensitive, caspase-12-dependent pathway in the p53-mediated apoptotic response in other experimental settings, we analyzed several other types of cell that have been shown to undergo p53-mediated apoptosis.
Figure 6.
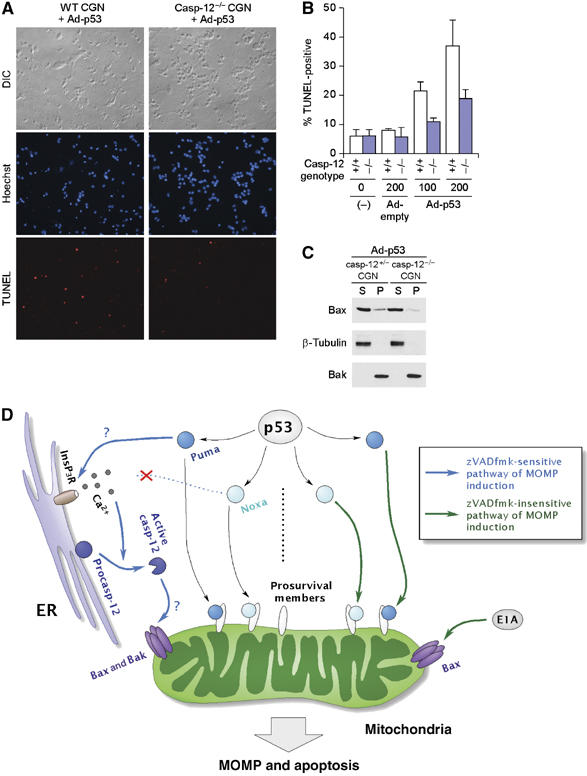
Requirement for caspase-12 in p53-induced apoptosis of CGNs. (A, B) Apoptosis induced by ectopic expression of p53 in wild-type (WT) (+/+) and caspase-12-deficient (casp-12−/−) CGNs. WT and casp-12−/− CGNs were prepared from the same litter and infected with adenovirus expressing p53 (Ad-p53) or control adenovirus; apoptosis was then analyzed by TUNEL. Representative images of CGNs infected with p53-expressing adenovirus at a multiplicity of infection (m.o.i.) of 200 are shown in panel A. Quantitation of TUNEL positivity is shown in panel B, wherein values shown are means±s.d. from four experiments, each using an independent pair of clones. (C) p53-induced Bax membrane insertion in caspase-12-heterozygous (casp-12+/−) and casp-12−/− CGNs. casp-12+/− and casp-12−/− CGNs were prepared from the same litter, and infected with the adenovirus expressing p53 at 200 m.o.i. (D) Dual regulation of mitochondrial apoptotic pathway by p53 targets, Puma and Noxa. In certain types of cells such as normal fibroblasts, thymocytes and CGNs, MOMP is induced at least partly through the zVADfmk-sensitive pathway (left, blue arrow), which is activated by Puma but not Noxa. This zVADfmk-sensitive pathway involves ER events, that is, calcium release through InsP3Rs, which in turn activates calpain. These ER events lead to the activation of caspase-12 and presumably other members of the caspase family; the activation of this pathway, together with the direct inhibition of prosurvival Bcl-2 family proteins by the BH3-only proteins on mitochondria, induces efficient activation of both Bax and Bak, and MOMP induction. Under certain conditions, such as fibroblasts expressing the E1A oncoprotein, a zVADfmk-insensitive MOMP-inducing pathway (right, green arrow), which can be activated by both Puma and Noxa, becomes effective. E1A presumably facilitates the operation of this pathway through the partial activation of Bax. Further studies are required to confirm the validity of this model, particularly regarding the role of Puma in the modulation of InsP3R function, and regarding the role of caspase-12 and other caspases in the activation of Bax and Bak.
The apoptosis of thymocytes caused by X-ray irradiation has been well studied and it proceeds exclusively in a p53-dependent manner (Clarke et al, 1993; Lowe et al, 1993b). It has also been shown that Puma deficiency but not Noxa deficiency confers significant resistance to this type of apoptosis (Shibue et al, 2003; Villunger et al, 2003). Treatment with zVADfmk revealed that not only cell death but also MOMP-related apoptotic events including Bax membrane insertion and cytochrome c release were clearly impaired by the inhibition of caspases in the X-ray-irradiation-induced apoptosis of thymocytes (Supplementary Figure 7A–C). However, caspase-12 deficiency did not confer resistance to this form of apoptosis (Supplementary Figure 7D), suggesting the redundant contribution of other members of the caspase family.
We then examined primarily cultured cerebellar granule neurons (CGNs), which also undergo p53-induced apoptosis (Slack et al, 1996), an event simulating the death of neurons caused by brain-damaging stresses such as ischemia and hypoxia. Indeed, this type of apoptosis is mediated by Puma (Cregan et al, 2004). As shown in Figure 6A and B, caspase-12-deficient CGNs showed a higher resistance to apoptosis induced by the adenovirus-mediated expression of p53 than the littermate control cells. Moreover, the membrane insertion of Bax was also impaired by the caspase-12 deficiency (Figure 6C). These observations of thymocytes and CGNs further emphasize the crucial role of the zVADfmk-sensitive MOMP induction pathway in certain forms of the p53-mediated apoptotic response. Moreover, observation of thymocytes offers a good explanation of why Puma but not Noxa is involved in the p53-dependent apoptosis of these cells (Shibue et al, 2003; Villunger et al, 2003).
Discussion
Our study suggests the operation of two pathways involved in the p53-mediated apoptotic response, the Ca2+/calpain-dependent (zVADfmk-sensitive) and -independent (zVADfmk-insensitive) pathways of MOMP induction, in which the former pathway is activated by Puma and the latter by both Puma and Noxa (Figure 6D). This, in turn, suggests that the differential activation of these two pathways should affect the fate of cells upon p53 activation. Indeed, the induction of Puma and Noxa mRNAs upon genotoxic stress was found to differ significantly among different types of cell: expression of Noxa mRNA, rather than that of Puma mRNA, is highly induced upon DNA damage in fibroblasts, which is the opposite in thymocytes (Supplementary Figure 8). In view of the inability of Noxa to activate the zVADfmk-sensitive MOMP pathway (Figure 6D), the strong induction of Noxa and the weak induction of Puma in fibroblasts are consistent with the observation that they undergo cell cycle arrest rather than apoptosis upon DNA damage, unless they express oncoproteins such as E1A.
A strong induction of Puma mRNA in thymocytes upon X-ray-irradiation (Supplementary Figure 8) also offers an explanation to the long-known observation that these irradiated cells readily undergo apoptosis regardless of oncoprotein expression: a high-level Puma induction would effectively trigger the Ca2+/calpain-dependent, zVADfmk-sensitive MOMP pathway. Indeed, this notion is consistent with our experimental data showing that zVADfmk treatment inhibits MOMP-related events on thymocytes induced by X-ray irradiation (Supplementary Figure 7B and C). As thymocyte apoptosis is suppressed by calpain inhibitors (Squier et al, 1994) but occurs normally in caspase-12-deficient thymocytes (Supplementary Figure 7D), we infer the involvement of alternative, Ca2+/calpain-dependent caspases function in these cells (Blomgren et al, 2001). Previous studies have shown several conditions wherein caspases play a role in the apoptotic pathway upstream of MOMP (Lassus et al, 2002; Marsden et al, 2002), but the mechanism underlying the activation of each caspase, which should be independent of MOMP, remained largely unclear. Our study now showed that a BH3-only protein, Puma, can activate caspase-12 (and presumably other members of the caspase family) by inducing calcium release from ER. Activated caspase-12, presumably in cooperation with other caspases, contributes to the induction of MOMP in certain forms of p53-mediated apoptosis (Figure 6D). It is worth noting that Puma activates both Ca2+/calpain-dependent and -independent pathways of MOMP induction: although the details regarding the roles of these two pathways, such as their relative contribution in p53-mediated apoptosis and their inter-relationship, remain largely unclear, we infer that operation of these two pathways ensures efficient progression of p53-mediated apoptosis.
The mechanism underlying the Puma-induced calcium release from InsP3Rs remains to be studied further (Figure 6D). Immunoprecipitation analysis indicated that the interaction between InsP3Rs and Bcl-2 or Bcl-XL is attenuated by Puma but not Noxa (Figure 4E), which is consistent with the result that Puma but not Noxa binds to these two prosurvival members (Supplementary Figure 9): Puma may compete with InsP3Rs for binding to Bcl-2 and Bcl-XL. In this regard, several reports point to the role of Bcl-2 family proteins in the regulation of ER calcium: either Bcl-2 overexpression or double deficiency in Bax and Bak causes a reduction in steady-state calcium concentration in ER (Pinton et al, 2000; Scorrano et al, 2003), whereas Bax and Bak activation on ER membrane induces calcium release from ER (Zong et al, 2003; Mathai et al, 2005). Moreover, modulation of InsP3Rs calcium channel activity by Bcl-2 or Bcl-XL through the direct interaction has also been reported (Chen et al, 2004; White et al, 2005). In light of these reports, it is an interesting possibility that Puma affects the function of InsP3Rs indirectly by interfering with the interaction between InsP3Rs and Bcl-2 or Bcl-XL; however, whether and how this leads to calcium release awaits future investigations. We also performed subcellular fractionation analysis using Puma-expressing fibroblasts; the vast majority of the Puma protein was detected in the mitochondrial fraction, whereas it was rarely detectable in the ER fraction (Supplementary Figure 10), suggesting another possibility that Puma induces calcium release from ER indirectly through its effect on mitochondria. An intriguing hypothesis that is congruent with this idea is the involvement of cytochrome c in this process. It has been reported that in staurosporin-treated HeLa cells, a small amount of cytochrome c is released early in apoptosis, which subsequently binds to InsP3Rs to elicit calcium release from ER (Boehning et al, 2003). This observation supports the idea that Puma first induces a minimal release of cytochrome c from mitochondria, which in turn binds to InsP3Rs on ER and induces calcium release. Testing each of these possibilities will be helpful for fully illustrating the Puma-mediated, calcium/calpain-dependent pathway of MOMP induction.
Puma belongs to the BH3-only subgroup of the Bcl-2 family, whose members have been implicated in the initiation of most forms of apoptosis (Cory and Adams, 2002). It is therefore interesting to determine whether other BH3-only members also exert some effects other than directly affecting the permeability of the mitochondrial outer membrane. Several studies have been performed to compare the proapoptotic activities of BH3-only members. Most of them are based on an experimental system in which isolated mitochondria or some analogues are treated with peptides resembling the BH3-domain sequence of BH3-only proteins (Letai et al, 2002; Kuwana et al, 2005). Through these studies, both Puma and Noxa have been identified to be ineffective by themselves to trigger MOMP (Kuwana et al, 2005). However, when expressed in intact cells, Puma but not Noxa can trigger MOMP as a result of its extramitochondrial effect to activate caspase-12 through calcium release from ER (Figure 6D). It should therefore be important to identify the extramitochondrial effects of BH3-only molecules as well, to determine how they trigger apoptotic response.
In contrast to Puma, Noxa does not induce the extramitochondrial activation of caspase-12; therefore, it is not effective in inducing the apoptosis of NIH3T3 cells unless they express E1A. How E1A induces the oligomerization of Bax but not Bak, thereby sensitizing cells to Noxa-induced apoptosis, remains to be studied further. As Noxa induces the apoptosis of cells deficient in Bcl-XL, one plausible model is the downregulation of Bcl-XL by E1A, but this turned out not to be the case (Supplementary Figure 1C). It has also been reported that Noxa shows a characteristic binding affinity to prosurvival members of the Bcl-2 family: it strongly binds to Mcl-1 but not to Bcl-2 or Bcl-XL (Supplementary Figure 9) (Chen et al, 2005). Accordingly, Noxa can initiate the apoptotic response only in limited situations. Whatever the mechanism, the limited function of Noxa may be an important aspect in balancing the p53-induced cell fate, preventing unnecessary cell death induced by p53 activation.
Between the two branching pathways of p53-mediated apoptosis, the zVADfmk-sensitive MOMP pathway seems to be activated regardless of cell condition, whereas the zVADfmk-insensitive pathway operates more effectively in cells that are ready to undergo apoptosis than in normal healthy cells (Figure 6D). As such, the differential activation of these two pathways, which can be achieved by changing the induction of Puma and Noxa, for example, should allow p53 to induce apoptosis only under appropriate conditions. It should be noted that the p53-mediated apoptotic response is an essential inherent mechanism for tumor suppression, and many cancer therapeutic agents exert their effects via this mechanism (Brown and Attardi, 2005). In this context, our results indicate that the selective activation of the zVADfmk-insensitive MOMP pathway might be beneficial for effectively eliminating cells expressing certain oncoproteins (Figure 6D). Further investigation of the role of these two pathways will help us design a method of manipulating the p53-mediated apoptotic response for the purpose of selectively damaging cancerous cells.
Materials and methods
Detection of apoptosis
In the quantitation of cell death by the flow cytometric detection of Annexin V, cells were collected 24 h after the pMx-derived retroviral infection of NIH3T3 cells and MEFs, and 12 h after the X-ray irradiation of thymocytes. Subsequently, the cells were stained with Annexin V-Cy3 or -FITC (MBL) and analyzed by flow cytometry, according to the manufacturer's protocol. The apoptosis of CGNs was detected by the TUNEL reaction using In Situ Cell Death Detection Kit, TMR Red (Roche Diagnostics), followed by microscopic examination. To obtain results shown in Figure 6B, at least 200 cells were microscopically examined for TUNEL positivity in each sample.
Supplementary Material
Supplementary Figures 1 and 2
Supplementary Figures 3 and 4
Supplementary Figures 5 and 6
Supplementary Figures 7 and 8
Supplementary Figures 9 and 10
Supplementary Methods
Acknowledgments
We are most grateful to RA Weinberg for his continuous support and encouragement throughout this work. We also thank N Tanaka and E Oda-Sato for Noxa cDNA, pBabe-puro-E2F1 and helpful advice; T Nakagawa for the anti-caspase-12 rat monoclonal antibody and discussion; J Yuan and A Steele for caspase-12-deficient mice; T Nakajima for adenovirus 2 E1A 12S cDNA; T Kiyono for pLR-hygro-E7; A Miyawaki for pcDNA3-YC3.60; N Fujita and T Tsuruo for pFLAG-CMV-2-mBcl-2 and -mBcl-XL; A Umezawa and J Hata for mouse Mcl-1 cDNA; T Yamashita and T Tokino for adenovirus expressing p53; Q Chang for technical help in primary neuron culture; J Yang and T Onder for the critical reading of the manuscript; and K Honda, H Tanaka and H Tamiya for invaluable advise. This work was supported in part by Kakenhi (one of the grants for Advanced Research on Cancer and a Grant-In-Aid for Scientific Research on Priority Areas from the Ministry of Education, Culture, Sports, Science and Technology of Japan) and Research Grants from the Princess Takamatsu Cancer Research Fund and Amgen Inc.
References
- Blomgren K, Zhu C, Wang X, Karlsson JO, Leverin AL, Bahr BA, Mallard C, Hagberg H (2001) Synergistic activation of caspase-3 by m-calpain after neonatal hypoxia–ischemia: a mechanism of ‘pathological apoptosis'? J Biol Chem 276: 10191–10198 [DOI] [PubMed] [Google Scholar]
- Boehning D, Patterson RL, Sedaghat L, Glebova NO, Kurosaki T, Snyder SH (2003) Cytochrome c binds to inositol (1,4,5) trisphosphate receptors, amplifying calcium-dependent apoptosis. Nat Cell Biol 5: 1051–1061 [DOI] [PubMed] [Google Scholar]
- Breckenridge DG, Germain M, Mathai JP, Nguyen M, Shore GC (2003) Regulation of apoptosis by endoplasmic reticulum pathways. Oncogene 22: 8608–8618 [DOI] [PubMed] [Google Scholar]
- Brown JM, Attardi LD (2005) The role of apoptosis in cancer development and treatment response. Nat Rev Cancer 5: 231–237 [DOI] [PubMed] [Google Scholar]
- Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM, Huang DC (2005) Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell 17: 393–403 [DOI] [PubMed] [Google Scholar]
- Chen R, Valencia I, Zhong F, McColl KS, Roderick HL, Bootman MD, Berridge MJ, Conway SJ, Holmes AB, Mignery GA, Velez P, Distelhorst CW (2004) Bcl-2 functionally interacts with inositol 1,4,5-trisphosphate receptors to regulate calcium release from the ER in response to inositol 1,4,5-trisphosphate. J Cell Biol 166: 193–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke AR, Purdie CA, Harrison DJ, Morris RG, Bird CC, Hooper ML, Wyllie AH (1993) Thymocyte apoptosis induced by p53-dependent and independent pathways. Nature 362: 849–852 [DOI] [PubMed] [Google Scholar]
- Cory S, Adams JM (2002) The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer 2: 647–656 [DOI] [PubMed] [Google Scholar]
- Cregan SP, Arbour NA, Maclaurin JG, Callaghan SM, Fortin A, Cheung EC, Guberman DS, Park DS, Slack RS (2004) p53 activation domain 1 is essential for PUMA upregulation and p53-mediated neuronal cell death. J Neurosci 24: 10003–10012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degterev A, Boyce M, Yuan J (2003) A decade of caspases. Oncogene 22: 8543–8567 [DOI] [PubMed] [Google Scholar]
- Ekert PG, Read SH, Silke J, Marsden VS, Kaufmann H, Hawkins CJ, Gerl R, Kumar S, Vaux DL (2004) Apaf-1 and caspase-9 accelerate apoptosis, but do not determine whether factor-deprived or drug-treated cells die. J Cell Biol 165: 835–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Calvo M, Peterson EP, Leiting B, Ruel R, Nicholson DW, Thornberry NA (1998) Inhibition of human caspases by peptide-based and macromolecular inhibitors. J Biol Chem 273: 32608–32613 [DOI] [PubMed] [Google Scholar]
- Goping IS, Gross A, Lavoie JN, Nguyen M, Jemmerson R, Roth K, Korsmeyer SJ, Shore GC (1998) Regulated targeting of BAX to mitochondria. J Cell Biol 143: 207–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DR, Kroemer G (2004) The pathophysiology of mitochondrial cell death. Science 305: 626–629 [DOI] [PubMed] [Google Scholar]
- Hsu YT, Youle RJ (1997) Nonionic detergents induce dimerization among members of the Bcl-2 family. J Biol Chem 272: 13829–13834 [DOI] [PubMed] [Google Scholar]
- Kang SJ, Wang S, Hara H, Peterson EP, Namura S, Amin-Hanjani S, Huang Z, Srinivasan A, Tomaselli KJ, Thornberry NA, Moskowitz MA, Yuan J (2000) Dual role of caspase-11 in mediating activation of caspase-1 and caspase-3 under pathological conditions. J Cell Biol 149: 613–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko LJ, Prives C (1996) p53: puzzle and paradigm. Genes Dev 10: 1054–1072 [DOI] [PubMed] [Google Scholar]
- Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR, Newmeyer DD (2005) BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell 17: 525–535 [DOI] [PubMed] [Google Scholar]
- Lassus P, Opitz-Araya X, Lazebnik Y (2002) Requirement for caspase-2 in stress-induced apoptosis before mitochondrial permeabilization. Science 297: 1352–1354 [DOI] [PubMed] [Google Scholar]
- Lavia P, Mileo AM, Giordano A, Paggi MG (2003) Emerging roles of DNA tumor viruses in cell proliferation: new insights into genomic instability. Oncogene 22: 6508–6516 [DOI] [PubMed] [Google Scholar]
- Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ (2002) Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2: 183–192 [DOI] [PubMed] [Google Scholar]
- Li P, Allen H, Banerjee S, Franklin S, Herzog L, Johnston C, McDowell J, Paskind M, Rodman L, Salfeld J, Towne E, Tracey D, Wardwell S, Wei F-Y, Wong W, Kamen R, Seshadri T (1995) Mice deficient in IL-1 beta-converting enzyme are defective in production of mature IL-1 beta and resistant to endotoxic shock. Cell 80: 401–411 [DOI] [PubMed] [Google Scholar]
- Liu X, Kim CN, Yang J, Jemmerson R, Wang X (1996) Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 86: 147–157 [DOI] [PubMed] [Google Scholar]
- Lowe SW, Ruley HE, Jacks T, Housman DE (1993a) p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell 74: 957–967 [DOI] [PubMed] [Google Scholar]
- Lowe SW, Schmitt EM, Smith SW, Osborne BA, Jacks T (1993b) p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature 362: 847–849 [DOI] [PubMed] [Google Scholar]
- Marsden VS, O'Connor L, O'Reilly LA, Silke J, Metcalf D, Ekert PG, Huang DC, Cecconi F, Kuida K, Tomaselli KJ, Roy S, Nicholson DW, Vaux DL, Bouillet P, Adams JM, Strasser A (2002) Apoptosis initiated by Bcl-2-regulated caspase activation independently of the cytochrome c/Apaf-1/caspase-9 apoptosome. Nature 419: 634–637 [DOI] [PubMed] [Google Scholar]
- Mathai JP, Germain M, Shore GC (2005) BH3-only BIK regulates BAX,BAK-dependent release of Ca2+ from endoplasmic reticulum stores and mitochondrial apoptosis during stress-induced cell death. J Biol Chem 280: 23829–23836 [DOI] [PubMed] [Google Scholar]
- Merritt AJ, Potten CS, Kemp CJ, Hickman JA, Balmain A, Lane DP, Hall PA (1994) The role of p53 in spontaneous and radiation-induced apoptosis in the gastrointestinal tract of normal and p53-deficient mice. Cancer Res 54: 614–617 [PubMed] [Google Scholar]
- Nagai T, Yamada S, Tominaga T, Ichikawa M, Miyawaki A (2004) Expanded dynamic range of fluorescent indicators for Ca(2+) by circularly permuted yellow fluorescent proteins. Proc Natl Acad Sci USA 101: 10554–10559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Yuan J (2000) Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis. J Cell Biol 150: 887–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J (2000) Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 403: 98–103 [DOI] [PubMed] [Google Scholar]
- Nakano K, Vousden KH (2001) PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell 7: 683–694 [DOI] [PubMed] [Google Scholar]
- Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T, Tanaka N (2000) Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 288: 1053–1058 [DOI] [PubMed] [Google Scholar]
- Oren M (2003) Decision making by p53: life, death and cancer. Cell Death Differ 10: 431–442 [DOI] [PubMed] [Google Scholar]
- Orrenius S, Zhivotovsky B, Nicotera P (2003) Regulation of cell death: the calcium–apoptosis link. Nat Rev Mol Cell Biol 4: 552–565 [DOI] [PubMed] [Google Scholar]
- Pinton P, Ferrari D, Magalhaes P, Schulze-Osthoff K, Di Virgilio F, Pozzan T, Rizzuto R (2000) Reduced loading of intracellular Ca(2+) stores and downregulation of capacitative Ca(2+) influx in Bcl-2-overexpressing cells. J Cell Biol 148: 857–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, Korsmeyer SJ (2003) BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science 300: 135–139 [DOI] [PubMed] [Google Scholar]
- Shibue T, Takeda K, Oda E, Tanaka H, Murasawa H, Takaoka A, Morishita Y, Akira S, Taniguchi T, Tanaka N (2003) Integral role of Noxa in p53-mediated apoptotic response. Genes Dev 17: 2233–2238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack RS, Belliveau DJ, Rosenberg M, Atwal J, Lochmuller H, Aloyz R, Haghighi A, Lach B, Seth P, Cooper E, Miller FD (1996) Adenovirus-mediated gene transfer of the tumor suppressor, p53, induces apoptosis in postmitotic neurons. J Cell Biol 135: 1085–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squier MK, Miller AC, Malkinson AM, Cohen JJ (1994) Calpain activation in apoptosis. J Cell Physiol 159: 229–237 [DOI] [PubMed] [Google Scholar]
- Suzuki K, Hata S, Kawabata Y, Sorimachi H (2004) Structure, activation, and biology of calpain. Diabetes 53 (Suppl 1): S12–S18 [DOI] [PubMed] [Google Scholar]
- Thornberry NA, Rano TA, Peterson EP, Rasper DM, Timkey T, Garcia-Calvo M, Houtzager VM, Nordstrom PA, Roy S, Vaillancourt JP, Chapman KT, Nicholson DW (1997) A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J Biol Chem 272: 17907–17911 [DOI] [PubMed] [Google Scholar]
- Villunger A, Michalak EM, Coultas L, Mullauer F, Bock G, Ausserlechner MJ, Adams JM, Strasser A (2003) p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science 302: 1036–1038 [DOI] [PubMed] [Google Scholar]
- Vousden KH, Lu X (2002) Live or let die: the cell's response to p53. Nat Rev Cancer 2: 594–604 [DOI] [PubMed] [Google Scholar]
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ (2001) Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292: 727–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White C, Li C, Yang J, Petrenko NB, Madesh M, Thompson CB, Foskett JK (2005) The endoplasmic reticulum gateway to apoptosis by Bcl-X(L) modulation of the InsP3R. Nat Cell Biol 7: 1021–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Kong YY, Yoshida R, Elia AJ, Hakem A, Hakem R, Penninger JM, Mak TW (1998) Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell 94: 739–750 [DOI] [PubMed] [Google Scholar]
- Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B (2001) PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell 7: 673–682 [DOI] [PubMed] [Google Scholar]
- Zamzami N, Kroemer G (2005) p53 in apoptosis control: an introduction. Biochem Biophys Res Commun 331: 685–687 [DOI] [PubMed] [Google Scholar]
- Zong WX, Li C, Hatzivassiliou G, Lindsten T, Yu QC, Yuan J, Thompson CB (2003) Bax and Bak can localize to the endoplasmic reticulum to initiate apoptosis. J Cell Biol 162: 59–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures 1 and 2
Supplementary Figures 3 and 4
Supplementary Figures 5 and 6
Supplementary Figures 7 and 8
Supplementary Figures 9 and 10
Supplementary Methods