

Abstract
Genetic evidence indicates that Drosophila defense against Gram-positive bacteria is mediated by two putative pattern recognition receptors acting upstream of Toll, namely Gram-negative binding protein 1 (GNBP1) and peptidoglycan recognition protein SA (PGRP-SA). Until now however, the molecular recognition proceedings for sensing of Gram-positive pathogens were not known. In the present, we report the physical interaction between GNBP1 and PGRP-SA using recombinant proteins. GNBP1 was able to hydrolyze Gram-positive peptidoglycan (PG), while PGRP-SA bound highly purified PG fragments (muropeptides). Interaction between these proteins was enhanced in the presence of PG or muropeptides. PGRP-SA binding depended on the polymerization status of the muropeptides, pointing to constraints in the number of PGRP-SA molecules bound for signaling initiation. We propose a model whereby GNBP1 presents a processed form of PG for sensing by PGRP-SA and that a tripartite interaction between these proteins and PG is essential for downstream signaling.
Keywords: Drosophila , innate immunity, non-self-recognition
Introduction
The ability to detect pathogens is an essential component of the immune system. The first line of sensing is stimulated by the recognition of conserved components of microorganisms called pathogen-associated molecular patterns (PAMPs; Janeway and Medzhitov, 2002). These PAMPs consist of molecules not found in the host, including bacterial cell wall peptidoglycan (PG) and lipopolysaccharide. Recognition of PAMPs by specific proteins called pattern recognition receptors (PRRs) activates a broad specificity inflammatory response characterized by the recruitment and activation of phagocytes and the production of antimicrobial compounds. These types of innate defenses are well conserved from invertebrates to mammals (Hoffmann et al, 1999).
Some of the most prevalent and severe infections known to medicine are caused by Gram-positive bacteria, the cell wall of which presents multiple layers of two insoluble, networked carbohydrates, namely, teichoic acid and PG (Sriskandan and Cohen, 1999; Brown, 2004). The latter is composed of β-1,4 carbohydrate chains, alternating N-acetylglucosamine and N-acetyl muramic acid sugars crosslinked by short peptide chains called stem peptides (Weber et al, 2003b). The third residue of the stem peptide is commonly a lysine (Lys-type PG) although some Bacilli contain diaminopimelic acid (DAP), a residue usually found in Gram-negative PG (Weber et al, 2003b). There is considerable variation in the modes of crosslinking between stem peptides as well as the residues included in the inter-peptide bridges. This heterogeneity in even the basic cell wall composition has confounded efforts to fit the Gram-positive innate immune response to the elegant concept of pattern recognition. It is thought that recognition of these pathogens is accomplished by several different strategies, some of which are among the most ancient and conserved elements of the innate immune response (Weber et al, 2003b).
To address the question of PG recognition by the host, we have used Drosophila as a genetically tractable model, which relies solely on innate defenses to fight off infection. These defenses are centered around two pathways, the Toll signaling pathway, which is triggered following fungal or Gram-positive bacterial infection and the IMD (for immune deficiency) signaling pathway activated in Gram-negative sepsis. These cascades culminate in NF-κB-dependent regulation of many target genes including those coding for potent antimicrobial peptides (AMPs; reviewed in Leclerc and Reichhart, 2004; Wang and Ligoxygakis, 2006).
Genetic screens have identified two putative PRRs as necessary for activating Toll following Gram-positive bacterial infection. PG recognition protein SA (PGRP-SA) and Gram-negative binding protein 1 (GNBP1) are blood-circulating proteins that were shown to be essential components of signaling upstream of Toll (Michel et al, 2001; Gobert et al, 2003). Point mutations in PGRP-SA and RNAi-mediated knock down of GNPB1 or deletions of its open reading frame led to severe defects in AMP expression and rendered defective flies susceptible to infection (Michel et al, 2001; Gobert et al, 2003; Pili-Floury et al, 2004). Concomitant overexpression of both proteins resulted in challenge-independent AMP activation (Gobert et al, 2003). In addition, structural studies of PGRP-SA have identified a binding domain for muropeptides and a protein–protein interaction surface that could serve as a platform for a potential interface with GNBP1 (Chang et al, 2004; Reiser et al, 2004). Finally, two recent papers describing the complex of the human PGRP-Iα with a muramyl peptapeptide (MPP) from Gram-positive bacteria have highlighted the induced structural rearrangements in the PG-binding domain that are essential for entry of the MPP and at the same time the way in which PGRPs could discriminate between Lys and DAP types of PG (Guan et al, 2006; Swaminathan et al, 2006).
We have previously determined that injection of intact Lys-PG in PGRP-SA- or GNBP1-deficient flies does not result in Toll activation (Filipe et al, 2005). In contrast, injections of highly purified fragments of Gram-positive Lys-PG were able to trigger the Toll pathway in a GNBP1 but not in a PGRP-SA mutant background (Filipe et al, 2005). In addition, hemolymph (the insect analog of the mammalian blood) from wild-type flies had a hydrolyzing activity against Lys-PG, which was missing from GNBP1 mutant blood (Filipe et al, 2005). Taken together, these data formulated the hypothesis that GNBP1 could present Lys-PG to PGRP-SA while an interaction involving the two proteins and PG could be envisaged (Filipe et al, 2005).
To clarify the molecular recognition mechanism of Gram-positive bacteria by the Drosophila host, we produced recombinant GNBP1 and PGRP-SA in a baculovirus expression system. Here, we show that rGNBP1 has a hydrolyzing activity against Lys-PG, which is enhanced in the presence of rPGRP-SA. We analyzed by mass spectrometry the majority of the products of this hydrolysis, which were released in the supernatant. These were identified as muropeptides demonstrating the muramidase-like activity of GNBP1. Moreover, when injected, the hydrolysis products were able to activate AMP expression in wild-type but not in PGRP-SA mutant flies. Additionally, the two proteins physically interact and this interaction is enhanced in the presence of GNBP1-hydrolyzed PG or highly purified PG fragments. These PG fragments are bound by rPGRP-SA and binding depends on the state of muropeptide polymerization and the presence of the nonreducing end of the terminal muramic acid. The above put forward a model whereby pattern recognition of Gram-positive PG is achieved by a GNBP1/PGRP-SA complex.
Results
To characterize the molecular events during Gram-positive PG sensing, we expressed and purified PGRP-SA and GNBP1 in a baculovirus insect cell culture system. Optimal expression conditions were determined for both proteins (see Materials and methods for details). Using a two-step chromatographic procedure, namely Ni-NTA affinity chromatography and gel filtration, rPGRP-SA and rGNBP1 were purified. The recombinant proteins were subjected to N-terminal protein sequencing and the identity of a mature form of purified rPGRP-SA lacking the signal peptide was confirmed (Figure 1A, lane 1). Similarly, the recombinant GNBP1 was established to be the mature protein with the signal peptide removed (Figure 1A, lane 2). Characterization of the purified proteins by analytical ultracentrifugation (AUC; see Materials and methods) showed that, in solution, rPGRP-SA was present as a monomer. Interestingly, rGNBP1 was able to form homodimers (Figure 1B).
Figure 1.
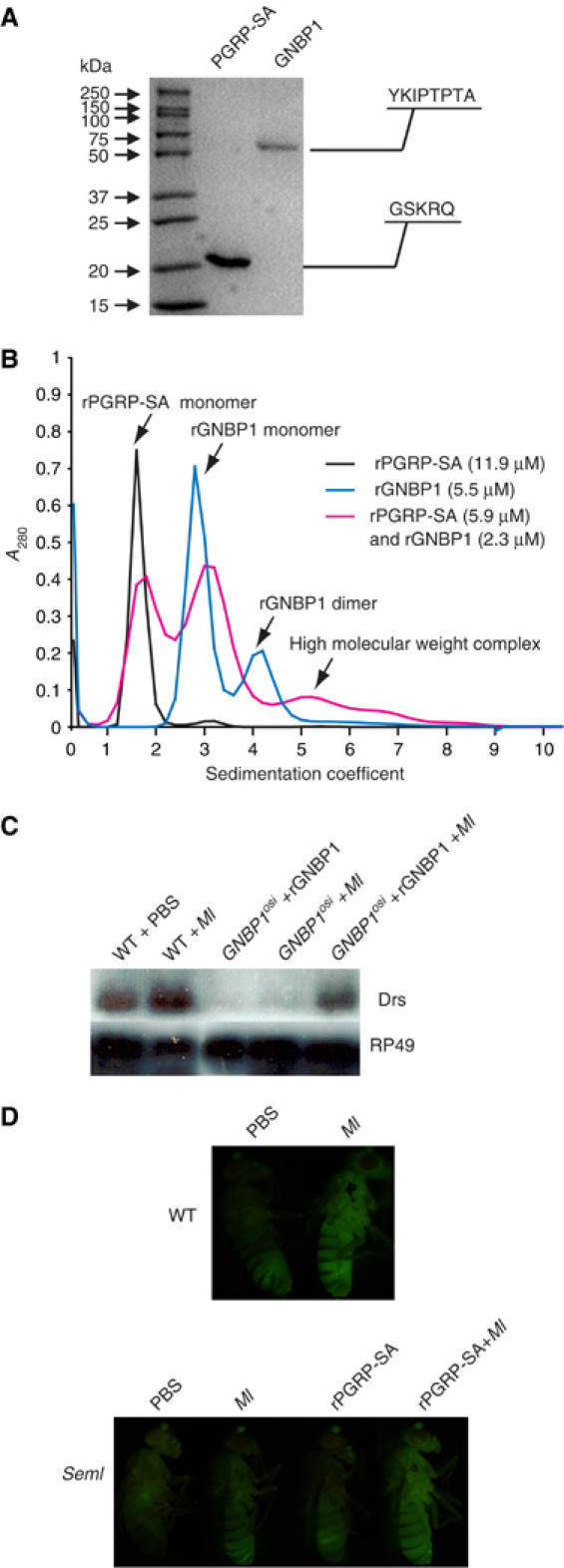
Protein expression, functional assays and AUC analysis. (A) Purified rPGRP-SA (predicted MW 20 kDa) and rGNBP1 (predicted MW 60 kDa) were analyzed on a 12% reducing SDS–PAGE gel. The identity of both proteins was verified by N-terminal protein sequencing (protein and nucleic acid chemistry service facility, Department of Biochemistry, University of Cambridge). As shown, the ∼20 amino-acid signal peptide was cleaved in both cases and the mature, secreted product detected. rGNBP1 and rPGRP-SA are biologically functional in vivo. (B) Analytical ultracentrifugation (AUC) analysis. Majority of rPGRP-SA appeared in a peak with a sedimentation coefficient (S) of 1.6, indicating PGRP-SA to be monomeric in solution. rGNBP1 showed two peaks at 2.8 and 4.2, respectively. The higher S species corresponds to a potential protein dimer of GNBP1 at higher concentrations in solutions. AUC of two proteins mixed showed a broad shoulder peak at an even higher S, suggesting a weak protein–protein interaction between the two proteins. The AUC curves for rPGRP-SA, rGNBP1 and the protein mixtures are shown in black, blue and purple, respectively. (C) Injection of 20 ng rGNBP1 is able to restore Drosomycin expression as assayed by Northern blot after Ml challenge in GNBP1 mutant flies. Injection of rGNBP1 does not activate any response in the absence of challenge. Wild-type (WT) flies and GNBP1osi flies infected with Ml were used as control. PBS injections were performed in WT flies as injection controls. (D) 10 ng injection of rPGRP-SA restores the GFP fluorescence in PGRP-SA mutant flies carrying a Drosomycin-GFP reporter gene after Ml challenge. Wild-type flies carrying the same reporter gene were used as controls. Again injection of the recombinant protein without challenge does not trigger Drs-GFP.
We tested the in vivo activity of the purified recombinant proteins by injecting them into the respective mutant flies. We used as an assay the expression of the AMP gene drosomycin (drs), a transcriptional target of the Toll pathway (Filipe et al, 2005). Expression of drs was restored in GNBPosi mutant flies that were injected with 20 ng or rGNBP1 protein and subsequently infected by the Gram-positive bacterium Micrococcus luteus (Ml; Figure 1C). The recombinant PGRP-SA was injected into homozygous PGRP-SAseml mutant flies carrying a drs-GFP transgene. Injection of 10 ng rPGRP-SA into the mutant flies restored GFP expression following infection by Ml (Figure 1D). Taken together, these results indicate that both recombinant proteins are biologically functional.
GNBP1 binds to and hydrolyzes Lys-type PG
We have previously speculated that GNBP1 is able to act enzymatically on Lys-type PG, as hemolymph from GNBP1 mutant flies is not able to provoke a reduction in OD measurements of a blood/PG mixture, in contrast to wild-type flies where PG was hydrolyzed (Filipe et al, 2005). Reintroduction of GNBP1 through the expression of a UAS-transgene restored the observed activity (Filipe et al, 2005). Based on these results, we tested whether rGNBP1 was able to hydrolyze Lys-type PG. Recombinant GNBP1 (at 0.5 μM) was indeed able to hydrolyze PG from Ml and Enterococcus faecalis but not Staphylococcus aureus or Bacillus subtilis (Figure 2A and data not shown). As shown in Figure 2A, this activity is enhanced in the presence of rPGRP-SA (at 0.5 μM). These results indicate the cooperation of the two proteins and underscore the enzymatic ability of GNBP1 against certain types of Lys-PG. This selectiveness of GNBP1 as an enzyme correlates with the ability of rGNBP1 to bind some Lys-PG molecules and not others as denoted in Figure 2B. In addition, a limited degradation of PG in the presence of PGRP-SA was also observed (Figure 2A). It is interesting to note here that PGRP-SA exhibited a broader capacity of PG binding, as it could also bind DAP-type PG from Bacillus subtilis (Figure 2B).
Figure 2.
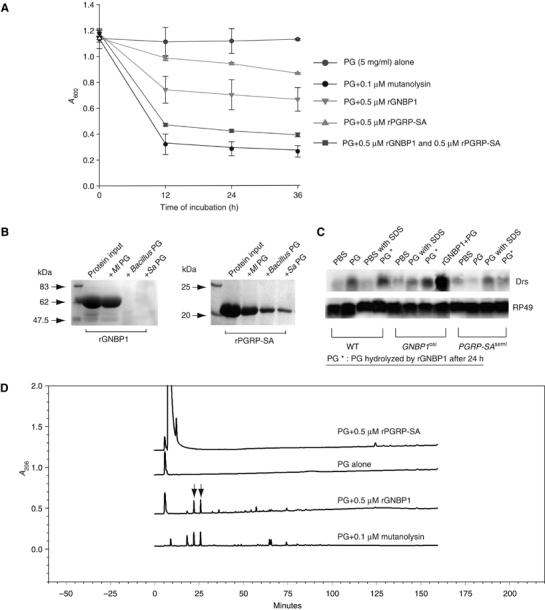
rGNBP1 binds and hydrolyzes PG with rPGRP-SA, generating an immunostimulatory PG in GNBP1 mutant flies, but not PGRP-SA mutant flies. (A) The hydrolytical assay was performed with rGNBP1 and/or rPGRP-SA in a sodium phosphate buffer (pH5.5) at 37°C. OD absorbance at 600 nm was taken at 0, 12, 24 and 36 h. In a 1:1 molar ratio, PG from Ml can be degraded more efficiently in the presence of both proteins than rGNBP1 alone. (B) Both rGNBP1 and rPGRP-SA bind to PG. In all, 20 μg of rGNBP1 or rPGRP-SA was used as input for the binding assay (see Materials and methods). Briefly, after incubation with the recombinant proteins, PG from different bacterial strains was removed from the reaction by centrifugation, washed and the association with recombinant proteins analyzed by SDS–PAGE. Recombinant GNBP1 and rPGRP-SA both bind to Ml PG (Lys-type), whereas rPGRP-SA exhibited significant binding affinity for Bacillus PG (DAP-type) and S. aureus PG (Lys-type). GNBP1 did not bind to these PGs. (C) GNBP1 but not PGRP-SA mutant flies respond to the rGNBP1-hydrolyzed PG (denoted as PG*). Intact PG or PG* were injected into flies at 5 mg/ml. Wild-type or mutant flies injected with PBS, PBS with 1% SDS (used for denaturing rGNBP1), intact PG and intact PG in 1% SDS were performed as controls. No difference was observed in the expected results when intact PG or intact PG in 1% SDS was injected to wild-type or mutant flies (this figure and data not shown). Injection of rGNBP1 in GNBP1-deficient flies before PG injection restores drosomycin expression to wild-type levels (lane rGNBP1+PG). Measurements of drosomycin expression were carried out 24 h following injection. Upper panel: Drosomycin probe (Drs). Lower panel: RP49 probe as internal RNA loading control. (D) Recombinant GNBP1 but not rPGRP-SA was able to release small PG fragments comparable to mutanolysin. Chromatography of the hydrolytically products after HPLC from top to bottom: PG after PGRP-SA treatment, PG alone, PG after rGNBP1 digestion and PG after mutanolysin digestion as control. Released small PG fragments that had their molecular weight determined by MALDI-MS are indicated by arrows.
To test for the inflammatory properties of the reaction products following the GNBP1-mediated hydrolysis, we injected the supernatant (after precipitation of the remaining PG and heat inactivation of GNBP1) to adult flies. We monitored activation by analyzing the expression of drs. As shown in Figure 2C, induction of drs is achieved in both wild-type and GNBP1osi mutants but not in PGRP-SA-deficient flies. These data indicate that the products of the reaction have inflammatory properties and that the limiting factor for the step following GNBP1-mediated PG hydrolysis is recognition by PGRP-SA. Moreover, injection of Ml PG digested by GNBP1 to various extends resulted in a correlation between the level of hydrolysis and drs expression (Supplementary Figure S1). Max levels of drs were observed when OD of PG was reduced at 80% something that happens between 24 and 36 h. The products that resulted from PG hydrolysis by GNBP1 were as capable to promote drs induction as purified polymerized muropeptides (Filipe et al, 2005). We asked therefore, whether GNBP1 could be releasing muropeptides into the supernatant when incubated with PG. To this end, we analyzed the supernatant of the reaction mixture by high-pressure liquid chromatography (HPLC) (Figure 2D). Two major peaks could be identified eluting at 22 and 26 min, similarly to what was observed when PG was digested by mutanolysin (arrows in Figure 2D). We collected the fractions eluting at 22 and 26 min and determined their molecular weight by Mass Spectrometry (MS). For both GNBP1 and mutanolysin digests, the obtained molecular weights corresponded to a monomeric muropeptide with only one terminating D-Ala residue (m/z 955) or with two terminating D-Ala residues (m/z 1026) (SRF, unpublished results). The ability of GNBP1 to release monomeric muropeptides from PG, similarly to mutanolysin, thus confirmed a muramidase-like activity for GNBP1.
However, this activity was less efficient than that of mutanolysin, as only monomeric muropeptides were released indicating that GNBP1 attacks bonds not as frequently or does it at specific sites of Ml's PG. The released monomeric muropeptides should not be active because we have shown that monomeric muropeptides are unable to trigger an immune response (Filipe et al, 2005). Instead, it is conceivable that the processed remaining PG should have an increased number of reducing ends that result from the infrequent cuts of GNBP1. The specific site where GNBP1 cleaves and the structural requirements for an efficient cut are under current investigation (SRF and PL, in preparation).
PG enhances the physical interaction between GNBP1 and PGRP-SA
Data from AUC pointed to the formation of a higher molecular weight complex between the two proteins. Briefly, when rPGRP-SA and rGNBP1 were mixed at 5.9 and 2.3 μM, respectively, we observed the formation of a higher molecular weight complex while the rGNBP1 dimer peak disappeared (compare the blue and purple lines in Figure 1B). Given this and the results described previously, we sought to determine whether we could quantify this interaction in a purified system. We addressed the question using the technique of surface plasmon resonance (Biacore). For these experiments, we immobilized PGRP-SA on a sensor chip and measured the relative response following injection of GNBP1. A direct interaction between PGRP-SA and GNBP1 could be reproducibly detected (Figure 3A). By using serial dilutions of pure rGNBP1 injections, we were able to quantify this interaction (Figure 3B). We confirmed binding by doing the reciprocal experiment with immobilized GNBP1 and injection of rPGRP-SA (Figure 3B). These results indicate a physical interaction between the two proteins with a KD in the low micromolar range. This interaction was enhanced if before injection GNBP1 was mixed with Methylobacter luteus PG or S. aureus muropeptides and then passed over the PGRP-SA sensor chip (Figure 3C left panel for PG and right panel for muropeptides). A summary of the dissociation constants measured for all interactions is shown in Figure 3B. Specifically, mixing different dilutions of a tetrameric muropeptide (7.5–100 nM) with GNBP1 and subsequent passing through the PGRP-SA chip augmented the interaction between the two proteins from a calculated KD of 21 μM (PGRP-SA/GNBP1) to that of 15 μM (PRGP-SA/GNBP1/Muropeptide tetramer; Figure 3D). The reverse, namely mixing PGRP-SA with Ml PG and passing over the GNBP1 sensor chip did not have the same effect (data not shown). This points towards the existence of a tripartite complex as well as a specific mode of sensing involving the two proteins and PG, whereby GNBP1 interacts with PG facilitating the binding of PGRP-SA (see discussion). The structure of the tetrameric muropeptide is shown in Figure 4A.
Figure 3.
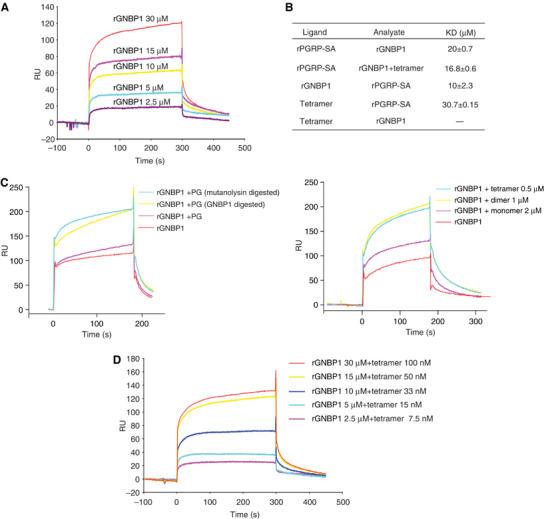
rGNBP1 and rPGRP-SA interact and hydrolyzed PG or muropeptides can enhance the strength of this interaction. (A) Surface plasmon resonance (Biacore) was used to study the interaction between rGNBP1 and rPGRP-SA. (A) rPGRP-SA was immobilized on a CM5 sensor chip and serial dilutions of rGNBP1 were injected over the sensor chip. With the increase of protein concentration, the read-out in response units (RU) increased accordingly. The reverse experiment namely immobilization of rGNBP1 on a CM5 sensor chip and injection of serial dilutions of rPGRP-SA gave similar results (data not shown). Dissociation constants (KDs) for both experiments are indicated in panel (B). (B) A summary of KDs based on a series of binding curves for the Biacore experiments described in this figure as well as in Figure 4 (PGRP-SA/muropeptide interaction). All KDs were calculated using the BIAevulation software (see Materials and methods). Three binding curves were used for each KD measurement. (C) Hydrolyzed PG (left panel) or muropeptides (right panel) enhanced the binding between rGNBP1 and rPGRP-SA. In this experiment, rPGRP-SA was coated on the sensor chip. (D) Quantification of the interaction of rPGRP-SA and rGNBP1 in the presence of muropeptides, using serial dilutions of a highly purified tetrameric muropeptide from S. aureus. As in (C), rPGRP-SA was coated on the sensor chip. The calculated KD is presented in (B). Note that from a KD of 20 μM for the interaction of rPGRP-SA/rGNBP1, there is a decrease in the calculated KD (16 μM) and therefore an enhancement of interaction when before injection rGNBP1 is mixed with the tetrameric muropeptide.
Figure 4.
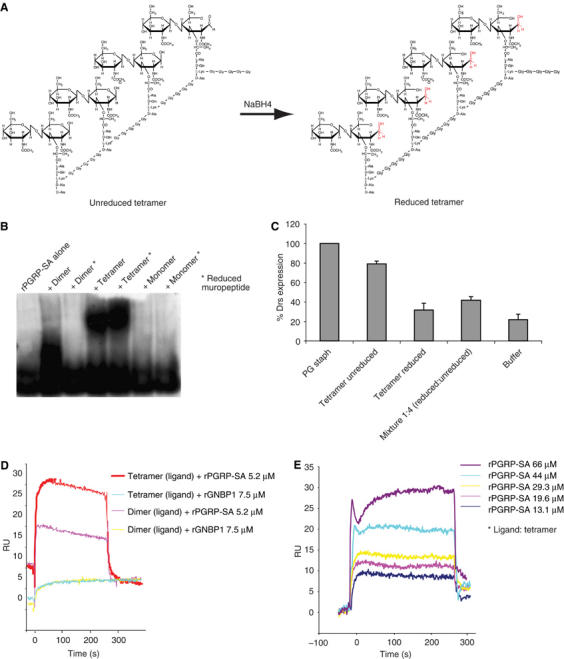
Polymerized muropeptides are able to shift rPGRP-SA to a higher molecular weight and rPGRP-SA is the protein that responds to muropeptide but not rGNBP1. (A) Chemical structure of unreduced and reduced muropeptide tetramer from S. aureus. (B) Polymerized muropeptides shifted rPGRP-SA to a higher molecular weight on a native gel. In each lane, 10 μg of rPGRP-SA were loaded and incubated with 25 μl of different biotinylated muropeptides (0.1 mg/ml) for 3 h at room temperature before loading onto the gel. After separation, proteins were transferred to PVDF as described in Materials and methods and probed with a streptavidin–peroxidase conjugate. Muropeptides with the muramic acid reduced (shown in red in A) were also investigated for their ability to bind to rPGRP-SA. Whereas dimeric muropeptides only bind rPGRP-SA in an unreduced state, muropeptide tetramers bind rPGRP-SA in both reduced and unreduced states. Monomeric muropeptides are unable to bind to the protein. (C) Although both reduced and unreduced tetramers bind to PGRP-SA, only the latter induce immune response confirming the significance of the free reducing end of the muramyl acid for recognition (Filipe et al, 2005). Moreover, mixing of reduced tetramers with their unreduced counterparts at a ratio of 1 reduced to 4 unreduced molecules had a severe effect in drs expression levels. This result indicates that reduced tetramers act as competitors for recognition by binding to PRGP-SA and thus diluting the elicitor effect of unreduced tetramers. (D) Biotinylated tetrameric and dimeric muropeptides were immobilized on a streptavidin-coated Biacore sensor chip and rGNBP1 or rPGRP-SA injected. Whereas rGNBP1 does not bind to either muropeptide, rPGRP-SA interacts weakly with both dimer and tetramer. (E) Quantification of the interaction shown in (D) using a tetrameric muropeptide coated on the sensor chip and injecting a dilution series of rPGRP-SA. The calculated KD is shown in Figure 3B.
PGRP-SA binds to polymerized muropeptides
The data presented so far indicate that GNBP1 is able to present PG to PGRP-SA in a processed form. To study the binding requirements of PGRP-SA and its affinity for fragments of Lys-PG, we sought to investigate whether PGRP-SA was able to bind highly purified muropeptides from S. aureus. We tested this by mixing the protein with different, biotin labeled, muropeptides and then visualized the interaction on native protein gels followed by immunoblotting (see Materials and methods). We had previously identified that a muropeptide dimer as the minimal structure activating the Toll pathway when injected to wild-type adults (Filipe et al, 2005). However, this structure could not trigger AMP gene expression in a PGRP-SAseml mutant background (Filipe et al, 2005). In agreement with our previous results, we found that rPGRP-SA is indeed able to bind dimeric muropeptides but not monomers. Moreover, the free end of the muramic acid is important for binding as reducing it prevents binding of PGRP-SA (Figure 4B) and it renders the dimer unable to activate the Toll pathway in flies (Filipe et al, 2005). As shown in Figure 4B, PGRP-SA also exhibits a binding affinity for tetrameric muropeptides. It was surprising that the reduced tetrameric muropeptides were able to interact with PGRP-SA, as we have previously shown that a reduced tetramer is unable to trigger an inflammatory response in Drosophila (Filipe et al, 2005). However, this binding did not lead to signaling. Mixing of reduced and unreduced tetramers in a ratio of 1:4 molecules significantly reduced drs activation when injected to wild-type flies (Figure 4C; see Figure 4A for the structural differences between unreduced/reduced tetramers).
This indicates that the reduced and unreduced tetrameric muropeptides are competing for the same receptor and that the free reducing end is still important for an inflammatory response. In addition, this points to different requirements for binding between dimeric and tetrameric muropeptides (as reduced tetramers bind PGRP-SA but reduced dimers do not). These results along with the recent 3D structural data for PGRP-SA (Chang et al, 2004; Reiser et al, 2004) suggest that the stoichiometry of the interaction is one PGRP-SA molecule for every muropeptide dimer moiety and that as muropeptide polymerization increases more protein molecules are recruited. Cooperative binding of PGRP-SA molecules to the tetrameric structure may facilitate the interaction between the receptor and this muropeptideas opposed to the dimereven in a reduced state. Co-crystallization studies of rPGRP-SA and reduced or unreduced dimer and tetramer will eventually provide a definite answer (NJ Gay unpublished). Finally, the interaction between rPGRP-SA and muropeptides was further confirmed using Biacore, where different muropeptides were passed through a PGRP-SA-coated chip (Figure 4D). This interaction was quantified with a KD of 30 μM using the tetrameric muropeptide from S. aureus (Figure 4E). Of note is the fact that GNBP1 was never seen to interact with any of these structures.
Discussion
In recent years, genetic screens have revealed that in Drosophila, the Toll pathway is activated by distinct PRRs sensing PG upstream of the receptor itself (Michel et al, 2001; Gobert et al, 2003; Leulier et al, 2003; Pili-Floury et al, 2004) Until now however, the molecular recognition events that define the mechanism of Gram-positive-PG recognition remained elusive. In the present, we have analyzed the mode of sensing of two PRRs (PGRP-SA and GNBP1) needed in response to some classes of Gram-positive bacteria. Our results show that GNBP1 functions as an enzyme, which recognizes β-1,4 N-glucosaminoglycans such as PG and is able to hydrolyze and present PG to PGRP-SA. GNBP1 is part of the GNBP/β-1,3-Glucan recognition family of proteins, which comprises members from several insects (Zhang et al, 2003). These proteins contain an N-terminal glucan-binding domain and a C-terminal domain similar to β-1,3- and β-1,4-bacterial glucanases and a β-1,3-glucanase from the sea urchin Strongylocentrotus purpuratus (Yahata et al, 1990; Bachman and McClay, 1996). Although critical residues in the active sites of bacterial glucanases have been replaced with other amino acids in GNBP1, there is PG degrading activity in the hemolymph of wild-type flies, which is lost in the GNBP1 mutants (Filipe et al, 2005).
Recombinant GNBP1 is only able to bind to and hydrolyze Lys-type PG from some Gram-positive bacteria as opposed to rPGRP-SA, which binds to both (Lys and DAP) types of PG (Chang et al, 2004 and Figure 2A). It is tempting to speculate therefore that it is GNBP1, which provides the specificity of the response to Gram-positive bacteria in the GNBP1/PGRP-SA complex. It is possible that GNBP1 can only bind to poorly crosslinked PG or PG in which there are several unsubstituted muramic acid residues as is the case for M. luteus (Schleifer and Kandler, 1972). Ongoing genetic screening by chemical mutagenesis for new null alleles of GNBP1 has produced point mutations in the glucanase-like domain indicating the importance of this domain for GNBP1 function (PL, unpublished data).
In addition, analysis by HPLC of the supernatant resulting from the incubation of PG with GNBP1 revealed the release of specific products. Further analysis of these products by MALDI-MS showed they are monomeric muropeptides. This indicates that GNBP1 is able to produce muropeptide monomers as is the case for mutanolysin. The most likely way for this to happen is if GNBP1 cleaves the β(1–4) linkage of the MurNAc and GlcNAc of two successive muropeptides (which would correspond to a muramidase-like activity). As only 25% of Ml PG is crosslinked (Schleifer and Kandler, 1972; Swaminathan et al, 2006), this would allow the release of monomeric muropeptides. Moreover, it would reduce the size of the glycan chains while unmasking more free reducing ends available for PGRP-SA recognition and binding on the remaining polymeric structures (especially if cleavage takes place in the middle of a glycan chain). Our results indicate that the more the level of processing (as exemplified by the reduction in OD), the higher the inflammatory activity (Supplementary Figure S1). We therefore believe that the formation of new free reducing ends through the activity of GNBP1 in time may account for the higher level of challenge observed.
Finally, the fact that the hydrolysis products generated by GNBP1 are able to activate the immune response in GNBP1 but not PGRP-SA mutant flies shows that, GNBP1 is needed to produce and present a processed form of PG for recognition by PGRP-SA. Although both proteins bind PG, it is only PGRP-SA that binds to purified PG fragments. A muropeptide dimer (with the free reducing end of the muramic acid intact) is the minimal PG moiety recognized by PGRP-SA (Filipe et al, 2005). It is interesting to note here that based on the 3D structure of PGRP-SA, it has been hypothesized that the binding of a PG compound onto the surface groove of the protein could create a new molecular surface that would allow the interaction with other PRRs such as GNBP1 (Chang et al, 2004). This correlates with the enhanced interaction between PGRP-SA and GNBP1 seen in the presence of PG or PG fragments and brings forward a model for pattern recognition of Gram-positive bacteria involving a PG/GNBP1/PGRP-SA tripartite interaction (Figure 5).
Figure 5.
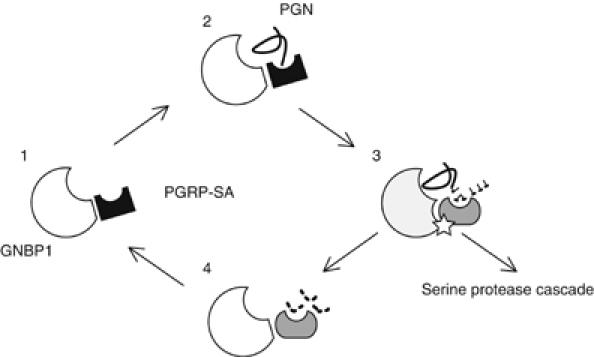
The molecular events involving GNBP1 and PGRP-SA in PG sensing and downstream signal transduction. Without immune challenge, GNBP1 and PGRP-SA form a protein complex by a very weak and dynamic protein–protein interaction, which is unable to generate a signal (Gobert et al, 2003; this study; stage 1). When Gram-positive PG (solid line) appears in the hemolymph, it is rapidly and efficiently recognized by and bound to the GNBP1/PGRP-SA protein complex. At this point, a tripartite GNBP1/PGRP-SA/PG complex is formed (stage 2). With the aid of PGRP-SA, activated GNBP1 breaks down PG releasing monomeric muropeptides, and at the same time producing free reducing ends in glycan chains of smaller sizes, an important feature for PGRP-SA binding (Filipe et al, 2005; this study). While GNBP1 continues to hydrolyze PG, PGRP-SA may change its conformation to allow the binding to small fragments (dashed line) of PG (stage 3). At this stage, the complex is able to relay the signal to the serine protease cascade, culminating in the cleavage of Spätzle and binding to Toll (Weber et al, 2003a). When the polymerized PG is converted into smaller fragments to which only PGRP-SA is able to respond (stage 4), the signal is gradually reduced as the interaction between PGRP-SA and muropeptides alone is relatively weak. Finally, GNBP1 and PGRP-SA form a static protein complex again ready to capture any PG present in the hemolymph.
The recognition signal generated by this complex is amplified through a serine protease cascade, which culminates in the cleavage and activation of substantial amounts of the protein ligand Spätzle (Spz). The 106 C-terminal amino acids of Spz constitute the active domain of the protein, which binds to Toll with an affinity of 80 nM (Weber et al, 2003a, 2005). The observed physical interaction between PGRP-SA and GNBP1 was in the order of low μM (approximately a thousand times weaker) indicating a more transient and dynamic association, which rapidly leads to a strong signaling step on the cell surface. The association of the two PRRs in the blood and in the absence of PG (Gobert et al, 2003; this study) as a 1:1 complex (Figure 1D), the enhancement of their interaction by PG and the amplification step by a serine protease cascade are probably adaptations of the particular physiology of insects. The lack of a circulatory system makes all tissues accessible to hemolymph. In turn, this means that any strong association between the circulating GNBP1 and PGRP-SA could lead to an autoactivation of signaling in the absence of an immune challenge. Stabilizing this interaction only in the presence of PAMPs like PG would avoid such a precarious possibility.
The next step in the elucidation of how Drosophila senses infection is to visualize the complexes of PRRs and PAMPs in vivo and in real time. Experiments underway, with fluorescent-tagged versions of PGRP-SA and GNBP1 will pinpoint whether these complexes are created directly on cell walls of Gram-positive bacteria or on PG fragments shed by these pathogens.
Materials and methods
Protein expression and purification
Full-length PGRP-SA cDNA was obtained from the Drosophila genomics research center (http://dgrc.cgb.indiana.edu/). GNBP1 full-length cDNA was obtained from Dr Won-Jae Lee. Primers encoding a C-terminal rTEV (tobacco etch virus) protease cleavage site and a His6-tag sequence were used in amplification of both GNBP1 and PGRP-SA. PCR products were subcloned into the vector pFastBac1 (Invitrogen) and sequenced, and recombinant baculoviruses were generated using the Bac-to-Bac system (Invitrogen). Viruses were amplified by infection of SF9 cell suspension cultures with 2% FBS, and the best protein expression condition were confirmed by Western blotting using Tetra anti-his tag antibody conjugated with horseradish peroxidase (Qiagen). Protein expression for GNBP1 was from a 10-l suspension culture of SF9 cells infected at multiplicity of infection (MOI) of 1.0 72 h post infection. For PGRP-SA, a 10-l suspension culture was infected at multiplicity of 10 and harvested after 72 h. After centrifugation, supernatant with the desired proteins were concentrated on a Centramate tangential flow system (Pall Filtron) with buffer exchanged to binding buffer (150 mM NaCl and 20 mM Tris–HCl, pH 7.5, 10 mM Imidazole). Following the addition of protease inhibitors (Roche), both His6-tagged proteins were purified by Qiagen Superflow NiNTA agarose and gel filtration on Superdex 75 or 200 columns (GE Healthcare) on an FPLC system (GE Healthcare).
Analytical ultracentrifugation of rGNBP1 and rPGRP-SA
All analytical ultracentrifugation experiments were performed on an Optima XLA/I (Beckman Coulter) centrifuge equipped with a four-hole titanium rotor, double-sector centerpieces and an interference optical system for data acquisition. Sedimentation velocity runs were performed at 55 000 r.p.m., with 3 min between scans of A280, at 10°C. The sample volume was 400 μl. Data were analyzed using Sedfit software. The partial specific volumes, buffer density and viscosity were estimated using SEDINTERP (Laue et al, 1992). Protein concentrations were 5 μM in 25 mM phosphate buffer pH 7.2, with 50 mM NaCl.
Fly strains, functional assays, PG hydrolysis and infections
Oregon-R and wN flies were used as wild-type controls. PGRP-SAseml and GNBP1osi mutants were previously described (Michel et al, 2001; Gobert et al, 2003). For functional assays of rPGRP-SA, 2–4 days old female flies carrying the homozygous mutant for PGRP-SAseml were collected and 9.2 nl (10 ng) of purified proteins was injected using thin glass capillaries mounted on a Nanoject II micro-injector (Drummond Scientific). Drosomycin-GFP expression was monitored and visualized directly under a fluorescence stereoscope. The GNBP1 functional assay was performed using 2–4 days old GNBP1 mutant flies injected with 9.2 nl (20 ng) of purified proteins. After a 1-h recovery from the injection, the flies were infected with M. luteus and collected 24 h later for Northern blotting. For the GNBP1-hydrolyzed PG studies, an rGNBP1-PG reaction was centrifuged after 24 h incubation (see below) and the supernatant was treated by boiling with 1% SDS for 5 min to inactivate the remaining enzymatic activity of rGNBP1. Subsequently, 30 nl of 5 mg/ml of intact PG or rGNBP1 hydrolyzed PG was injected into the GNBP1 and PGRP-SA mutant flies, respectively. Flies were collected 24 h after injection. For the infections in different time points, PG was injected to flies (wild type or GNBP1osi) following incubation with GNBP1 to different extends (30, 50 and 80% hydrolysis as measured by the drop in the OD—see Figure 2A). Treatment of samples before injection was as above. For Northern blotting, total RNA was prepared and was performed as previously described (Filipe et al, 2005). PBS buffer or PBS containing 1% SDS (used to inactivate rGNBP1) was used in all the injection experiments as negative control. The basal level of drs expression was monitored using non-challenged wild-type flies.
Enzymatic and binding activity of rGNBP1/rPGRP-SA and HPLC
PG from M. luteus was purified as previously reported (Filipe et al, 2005). rGNBP1 and/or rPGRP-SA was added into 500 μl of 5 mg/ml PG at 0.5 M. Absorbance readings at 600 nm were taken at 0, 12, 24 and 36 h in a Beckman DU 500 UV/Vis Spectrophotometer (Beckman-Coulter). The reaction was performed at 37°C with gentle agitation. PG treated with mutanolysin (Sigma) was used as positive control. The supernatant obtained from the incubation of M. luteus PG with GNBP1 for 12 h at 37°C was collected. It was boiled for 5 min to allow precipitation of proteins and centrifuged at 16 000 g for 5 min. The supernatant was then mixed with an equal volume of 0.5 M borate buffer, pH 9.0, reduced by the addition of sodium borohydride and analyzed as previously described (De Jonge et al, 1992). Fractions eluting at 22 and 26 min were desalted and analyzed by MALDI-MS using cyano-4-hydroxycinnamic acid as the MALDI matrix at the MS Service in ITQB, Lisbon. The binding activity of the proteins to PG was studied by incubating 20 μg each protein with 200 μg of PG in a 300 μl reaction with binding buffer (20 mM Tris–HCl, pH 8.0; 300 mM NaCl). After 3 h incubation on a shaking platform, PG was spun down at 16 000 g for 5 min. PG pellet was then washed three times with 1 ml washing buffer (100 mM Tris–HCl, pH 8.0; 500 mM NaCl and 0.02% Tween-20). Subsequently, the PG pellet was dissolved directly in 20 μl SDS sample buffer and subjected to a SDS-reducing PAGE gel directly.
Surface plasmon resonance of rGNBP1/rPGRP-SA/PG/muropeptides
To study the interaction of rGNBP1 and rPGRP-SA and its kinetics, surface plasmon resonance experiments were carried out on a Biacore 2000 instrument using CM5 sensor chips (BIAcore AB). His6-tagged rGNBP1 and rPGRP-SA were immobilized onto the chips by amine coupling. Concentrations of rGNBP1 at 7.5 μM and rPGRP-SA at 5.2 μM were used. With rPGRP-SA immobilized on the chip, a serial dilution of rGNBP1 (2.5, 5, 10 and 20 μM) was injected at a flow rate of 20 μl/min using the KINJECT mode for 5 min and dissociation was carried over a period of 6 min. Recombinant GNBP1 incubated with PG, treated PG or muropeptides were injected at the same protein concentration and settings were as that for the protein alone. Namely, 2 μM monomer, 1 μM dimer and 0.5 μM tetramer were coinjected with 7.5 μM of rGNBP1.
Muropeptides from S. aureus used in this study were purified as previously described (Filipe et al, 2005). The interaction between proteins and muropeptides was investigated by using biotinylated muropeptides (see below) on a PGRP-SA sensor chip. The biotinylated muropeptides (6 μM) were coated on the chip by the method recommended by the manufacturer (Biacore AB). For each muropeptide, 200 RU were coated and rPGRP-SA of different concentration was injected at a flow rate of 5 μl/min for 5 min and dissociation was carried over a period of 4 min. Evaluation of binding kinetic was performed by using the BIAevaluation software, version 3.1 (Biacore AB). All binding curves were subtracted by a mock reference flow cell to remove bulk refractive index contribution and mass transfer. Namely, the average equilibrium response was determined for each ligand concentration and plotted on a graph of the response versus concentration. A nonlinear fit of the 1:1 Langmuir binding to the data determined the equilibrium dissociation constant (KD).
Biotinylation of muropeptides and native blot
Muropeptides were biotinylated using EZ-link Biotin-PEO-Amine (Pierce Biotechnology) to couple the biotin moiety to the free COOH-termini within the peptide chain. Biotinylation reactions were set up as follows: 40 μl muropeptide solution (at 30 μM in H2O) were mixed with 1.2 μl EZ-link Biotin-PEO-Amine (50 mM in MES buffer) and 0.4 μl EDC (10 mM Stock in DMSO; EDC is required to activate COOH groups), and the reaction incubated at RT for 2 h. Subsequently, uncoupled biotinylation reagent was removed by dialysis against 2 lt PBS overnight at 4°C using a 500 MWCO dialysis membrane (Spectrapor). After dialysis, the volume for each reaction was adjusted to 150 μl with buffer, yielding an approximate concentration of 6 μM.
To check for binding to rPGRP-SA, 25 μl biotinylated muropeptides were incubated with 5 μl rPGRP-SA (10 μg) in a 50 μl reaction for 3 h. Reaction mixtures were separated on a 4–16% Novex Tris–Glycine gel (Invitrogen) under native conditions. Before transfer to PVDF membrane (GE Healthcare) using standard semidry blotting procedures, the gel was incubated in 0.1% SDS-containing buffer for 30 min to promote protein transfer. Subsequently, the blot was blocked in PBS with 0.1% Tween-20, probed with a streptavidin-POD-conjugate (Roche) and subjected to ECL detection (GE Healthcare).
Supplementary Material
Supplementary Figure S1
Acknowledgments
PL would like to dedicate this paper to the loving memory of our dear friend and colleague Ahmet Kilinc (1975–2005). He will be sadly missed.
We thank Dr Won-Jae Lee for the GNBP1 cDNA and the DGRC for the PGRP-SA cDNA. We acknowledge the Mass Spectrometry Service at the ITQB, Universidade Nova de Lisboa. LW was supported by a Dorothy Hodgkin Postgraduate Award from the BBSRC and Hutchison Whampoa. Part of this work was supported by grant POCI/SAU-IMI/56501/2004 from the Fundação para a Ciência e a Tecnologia (to SRF) and by a Portugal–UK joint project grant from the Royal Society (to SRF and PL). Work in the UK was supported by the Medical Research Council, through a Program Grant (to NJG) and a Career Establishment Grant (to PL).
References
- Bachman ES, McClay DR (1996) Molecular cloning of the first metazoan beta-1,3 glucanase from eggs of the sea urchin Strongylocentrotus purpuratus. Proc Natl Acad Sci USA 93: 6808–6813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown EJ (2004) The molecular basis of streptococcal toxic syndrome. N Engl J Med 350: 2093–2094 [DOI] [PubMed] [Google Scholar]
- Chang CI, Pili-Floury S, Herve M, Parquet C, Chelliah Y, Lemaitre B, Mengin-Lecreulx D, Deisenhofer JA (2004) Drosophila pattern recognition receptor contains a peptidoglycan docking groove and unusual L,D-carboxypeptidase activity. PLoS Biol 2: E277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jonge BL, Chang YS, Gage D, Tomasz A (1992) Peptidoglycan composition of a highly methicillin-resistant Staphylococcus aureus strain. The role of penicillin binding protein 2A. J Biol Chem 267: 11248–11254 [PubMed] [Google Scholar]
- Filipe SR, Tomasz A, Ligoxygakis P (2005) Requirements of peptidoglycan structure that allow detection by the Drosophila Toll pathway. EMBO Rep 6: 327–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobert V, Gottar M, Matskevich AA, Rutschmann S, Royet J, Belvin M, Hoffmann JA, Ferrandon D (2003) Dual activation of the Drosophila Toll pathway by two pattern recognition receptors. Science 302: 2126–2130 [DOI] [PubMed] [Google Scholar]
- Guan R, Brown RH, Swaminathan CP, Roychowdhury A, Boons GJ, Mariuzza RA (2006) Crystal structure of human peptidoglycan recognition protein I alpha bound to a muramyl pentapeptide from Gram-positive bacteria. Protein Sci 15: 1199–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann JA, Kafatos FC, Janeway CA, Ezekowitz RA (1999) Phylogenetic perspectives in innate immunity. Science 284: 1313–1318 [DOI] [PubMed] [Google Scholar]
- Janeway CA Jr, Medzhitov R (2002) Innate immune recognition. Annu Rev Immunol 20: 197–216 [DOI] [PubMed] [Google Scholar]
- Laue TM, Shah B, Ridgeway TM, Pelletier SL (1992) In Analytical Ultracentrifugation in Biochemistry and Polymer Science, Harding SE, Rowe AJ and Horton JC (eds) pp 90–125. Publisher: R Soc Chem, Cambridge: UK [Google Scholar]
- Leclerc V, Reichhart J-M (2004) The immune response of Drosophila melanogaster. Immunol Rev 198: 59–71 [DOI] [PubMed] [Google Scholar]
- Leulier F, Parquet C, Pili-Floury S, Ryu JH, Caroff M, Lee WJ, Mengin-Lecreulx D, Lemaitre B (2003) The Drosophila immune system detects bacteria through specific peptidoglycan recognition. Nat Immunol 4: 478–484 [DOI] [PubMed] [Google Scholar]
- Michel T, Reichhart JM, Hoffmann JA, Royet J (2001) Drosophila Toll is activated by Gram-positive bacteria through a circulating peptidoglycan recognition protein. Nature 414: 756–759 [DOI] [PubMed] [Google Scholar]
- Pili-Floury S, Leulier F, Takahashi K, Saigo K, Samain E, Ueda R, Lemaitre B (2004) In vivo RNA interference analysis reveals an unexpected role for GNBP1 in the defence against Gram-positive bacterial infection in Drosophila adults. J Biol Chem 279: 12848–12853 [DOI] [PubMed] [Google Scholar]
- Reiser J-B, Teyton L, Wilson IA (2004) Crystal structure of the Drosophila peptidoglycan recognition protein SA (PGRP-SA) at 1.56A resolution. J Mol Biol 340: 909–917 [DOI] [PubMed] [Google Scholar]
- Schleifer KH, Kandler O (1972) Peptidoglycan types of bacterial cell walls and their taxonomic implications. Bacteriol Rev 36: 407–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriskandan S, Cohen J (1999) Gram-positive sepsis: mechanisms and differences from gram-negative sepsis. Inf Dis Clin N Am 13: 397–412 [DOI] [PubMed] [Google Scholar]
- Swaminathan CP, Brown PH, Roychowdhury A, Wang Q, Guan R, Silverman N, Goldman WE, Boons GJ, Mariuzza RA (2006) Dual strategies for peptidoglycan discrimination by peptidoglycan recognition proteins. Proc Natl Acad Sci USA 103: 684–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Ligoxygakis P (2006) Pathogen recognition and signaling in the Drosophila innate immune response. Immunobiol 211: 251–261 [DOI] [PubMed] [Google Scholar]
- Weber AN, Tauszig-Delamasure S, Hoffmann JA, Lelievre E, Gascan H, Ray KP, Morse MA, Imler JL, Gay NJ (2003a) Binding of the Drosophila cytokine Spatzle to Toll is direct and establishes signalling. Nat Immunol 4: 794–800 [DOI] [PubMed] [Google Scholar]
- Weber AN, Moncrieffe MC, Gangloff M, Imler JL, Gay NJ (2005) Ligand–receptor and receptor–receptor interactions act in concert to activate 23 signaling in the Drosophila toll pathway. J Biol Chem 280: 22793–22799 [DOI] [PubMed] [Google Scholar]
- Weber RW, Moreillon P, Tuomanen E (2003b) Innate sensors of Gram-positive bacteria. Curr Opin Immunol 15: 408–415 [DOI] [PubMed] [Google Scholar]
- Yahata N, Watanabe T, Nakamura Y, Yamamoto Y, Kamimiya S, Tanaka H (1990) Structure of the gene encoding beta-1,3-glucanase A1 of Bacillus circulans WL-12. Gene 86: 113–117 [DOI] [PubMed] [Google Scholar]
- Zhang R, Cho HY, Kim HS, Ma YG, Osaki T, Kawabata S, Sönderhäll K, Lee BL (2003) Characterisation and properties of a 1,3-£]-D-glucan pattern recognition protein of Tenebrio molitor larvae that is specifically degraded by serine protease during prophenoloxydase cascade. J Biol Chem 278: 42072–42079 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1