

Abstract
G-protein-coupled receptor kinase 2 (GRK2) is a central regulator of G-protein-coupled receptor signaling. We report that Mdm2, an E3-ubiquitin ligase involved in the control of cell growth and apoptosis, plays a key role in GRK2 degradation. Mdm2 and GRK2 association is enhanced by β2-adrenergic receptor stimulation and β-arrestin. Increased Mdm2 expression accelerates GRK2 proteolysis and promotes kinase ubiquitination at defined residues, whereas GRK2 turnover is markedly impaired in Mdm2-deficient cells. Moreover, we find that activation of the PI3K/Akt pathway by insulin-like growth factor-1 alters Mdm2-mediated GRK2 degradation, leading to enhanced GRK2 stability and increased kinase levels. These data put forward a novel mechanism for controlling GRK2 expression in physiological and pathological conditions.
Keywords: degradation, GRK2, IGF-1, Mdm2
Introduction
G-protein-coupled receptor kinases (GRKs) and β-arrestins are critical regulators of a wide variety of biological processes. First, these proteins play a major role in the attenuation of signaling mediated by the large family of G-protein-coupled receptors (GPCRs) (Gainetdinov et al, 2004). Besides promoting activation of heterotrimeric G proteins, agonist stimulation of GPCRs leads to receptor phosphorylation by GRKs (Kohout and Lefkowitz, 2003; Penela et al, 2003). This phosphorylation event facilitates binding of β-arrestins to the receptor, resulting in uncoupling from G proteins. Acting as adaptor proteins for components of the endocytic machinery, arrestins also trigger receptor internalization. Therefore, GRKs and arrestins are involved in GPCR desensitization, endocytosis and resensitization (Ferguson, 2001).
β-Arrestins and GRKs also regulate other membrane receptor families. It has been reported that β-arrestin binds to ligand-stimulated insulin-like growth factor-1 receptor (IGF1-R), thereby promoting receptor internalization and enhancing IGF-dependent mitogenic signaling (Lin et al, 1998). GRK2, a ubiquitously expressed member of the GRK family, also phosphorylates PDGF-Rβ and reduces receptor activity without altering its downregulation (Freedman et al, 2002; Hildreth et al, 2004). In addition, β-arrestins can act as scaffold molecules that bring different signaling molecules into the receptor complex (reviewed by Lefkowitz and Shenoy, 2005). Likewise, GRK2 has been shown to phosphorylate non-receptor substrates and also to interact with a variety of signaling proteins such as PI3Kγ, Gαq or GIT (Penela et al, 2003, 2006, and references therein). GRK2 also inhibits TGF-β-mediated cell growth arrest and apoptosis by inducing Smad phosphorylation (Ho et al, 2005). Overall, these data suggest that GRK2 may have ‘effector' functions beyond receptor desensitization.
Such functional complexity predicts that alterations in GRK2 levels and/or activity may have important effects on cell signaling. Interestingly, several pathological conditions such as congestive heart failure, hypertension and rheumatoid arthritis (RA), among others, display altered GRK2 expression and function (reviewed by Penela et al, 2003, 2006; Metaye et al, 2005). Whereas different regulatory mechanisms of GRK2 activity and subcellular localization have been described (reviewed by Kohout and Lefkowitz, 2003; Penela et al, 2003), the mechanisms that govern GRK2 cellular levels have only recently began to be addressed. We first described that GRK2 is rapidly degraded by the proteasome pathway, and that β2-adrenergic receptor (β2AR) activation enhances GRK2 ubiquitination and turnover (Penela et al, 1998). We have also shown that agonist-dependent binding of β-arrestin to GPCRs supports GRK2 degradation by allowing the recruitment of c-Src and the phosphorylation of GRK2 on critical tyrosine residues (Penela et al, 2001). MAPK-mediated GRK2 phosphorylation also triggers GRK2 degradation in a process that is again dependent on β-arrestin function (Elorza et al, 2003).
Proteasome degradation requires the orchestrated activities of the ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzymes (E2) and ubiquitin ligases (E3) (Pickart, 2001). The specificity of target protein selection is determined by ubiquitin ligases, which interact with their substrates either directly or by means of adaptor molecules. Interestingly, β-arrestins are able to interact with Mdm2, a RING domain-containing E3-ubiquitin ligase involved in the control of tumor suppressor p53 activity (Weissman, 2001). β-Arrestin-mediated recruitment of Mdm2 to several GPCR complexes leads to different β-arrestin ubiquitination patterns, which controls the characteristics of MAPK activation and receptor internalization (Shenoy et al, 2001; Shenoy and Lefkowitz, 2003, 2005; Wang et al, 2003).
In this report, we identify Mdm2 as an E3-ubiquitin ligase for GRK2 that is critically involved in kinase ubiquitination and degradation. Moreover, we put forward a new Mdm2-mediated pathway for the modulation of GRK2 cellular levels by IGF-1. Control of GRK2 protein levels by Mdm2 may represent a versatile mechanism for fine-tuning of the expression of this key regulatory kinase by different extracellular signals.
Results
Mdm2 associates with GRK2 and this process is facilitated by β2AR stimulation and β-arrestin
GRK2 is rapidly degraded by the proteasome pathway in response to β2AR agonist stimulation, whereas the turnover of the GRK2-K220R mutant, which lacks kinase activity, is severely impaired (Penela et al, 1998). We have also reported that GRK2 turnover is dependent on β-arrestin function, and that overexpression of either β-arrestin1 or β-arrestin2 is able to promote GRK2-K220R proteolysis (Penela et al, 2001), thus suggesting that these adaptor molecules may recruit some of the factors needed for GRK2 ubiquitination and proteasome degradation. Interestingly, β-arrestins interact with Mdm2, bringing this protein to the plasma membrane β2AR complex, where Mdm2 triggers ubiquitination of β-arrestins and also auto-ubiquitination (Shenoy et al, 2001; Wang et al, 2003). As GRK2 is also part of activated GPCR complexes, we explored the possibility that Mdm2 could act as a GRK2 ubiquitin ligase. Consistent with this idea, we find that endogenous GRK2 and Mdm2 can associate in MCF7 cells, as shown by co-immunoprecipitation experiments using either anti-GRK2 or anti-Mdm2 antibodies (Supplementary data and Supplementary Figure S1A). This suggests that both proteins can be found in the same molecular complex at steady-sate conditions ‘in situ'.
We have previously shown that agonist stimulation of β2AR decreases GRK2 half-life from 60 min (basal conditions) to ∼30 min (stimulated turnover) (Penela et al, 1998). Therefore, we tested whether Mdm2 association to GRK2 would be modified by receptor activation. Figure 1A and B shows that β2AR stimulation promotes a rapid and transient increase in endogenous GRK2/Mdm2 association in HeLa cells and in HEK-293 cells coexpressing these proteins. It is worth noting that β2AR stimulation also triggers a rapid and transient association of β-arrestins with Mdm2 (Wang et al, 2003). Interestingly, we find that overexpression of β-arrestin1 increases both the extent and kinetics (maximal co-immunoprecipitation detected at 1–2 min) of GRK2/Mdm2 association upon β-agonist challenge (Figure 1B). A similar effect was observed upon expression of β-arrestin2 (data not shown). Consistent with a key mediator role for arrestins in GRK2 turnover, we find that protein decay of endogenous GRK2 (as assessed by chase experiments in the presence of cycloheximide) was severely delayed in mouse embryonic fibroblasts (MEFs) lacking both β-arrestin1 and β-arrestin2 compared to wild-type MEFs (Figure 1C), indicating that expression of β-arrestins is necessary for normal GRK2 degradation in physiological conditions.
Figure 1.
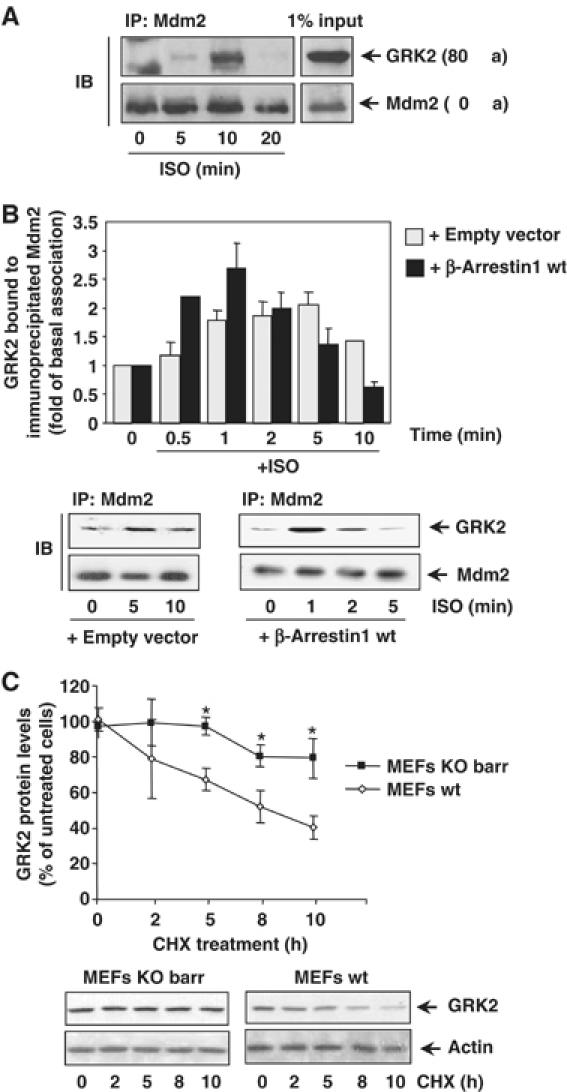
(A) Association of endogenous Mdm2 and GRK2. HeLa cells growing in 150 mm dishes were serum-starved for 2 h and incubated with an inverse β-agonist (betaxolol, 10 μM) for 15 min to lessen basal Mdm2/GRK2 association before challenging with 10 μM isoproterenol for the indicated times. Cells were lysed in buffer A as detailed in Materials and methods and lysates subjected to immunoprecipitation with anti-Mdm2 (SMP14) antibody. Immunoprecipitates were resolved by SDS–PAGE and GRK2 associated to the Mdm2 immunocomplex was detected by Western blot analysis with a specific antibody. 1% of the total lysate used for immunoprecipitation was loaded as input signal. (B) The Mdm2/GRK2 association is enhanced by β2AR stimulation and β-arrestin expression. HEK-293 cells were transiently transfected with plasmids encoding GRK2, β2AR, Mdm2 and β-arrestin1 when indicated. Cells were challenged with 10 μM isoproterenol for different times and GRK2/Mdm2 co-immunoprecipitation assessed as detailed in Materials and methods. GRK2 presence in the non-stimulated Mdm2 immunocomplex was taken as the control condition. Data were normalized by total Mdm2 and depicted as mean±s.e.m. of four independent experiments. A representative gel is shown in the bottom panel. (C) Normal decay of GRK2 protein is impaired upon of β-arrestin1 and β-arrestin2 knockdown. GRK2 protein levels were examined by immunoblot analysis in both wild-type and β-arrestin1/2-deficient MEFs upon treatment with cycloheximide (CHX) for the indicated times. The amount of GRK2 at 0 h was defined as 100%, and data normalized by actin protein levels. In both panels, data are the mean±s.e.m. of four independent experiments (*P<0.05). Representative gels are shown.
Taken together, these results indicate that Mdm2 can interact with GRK2 in an agonist-dependent manner and suggest that β-arrestin could be a pivotal adaptor to position Mdm2 and/or other ligases in the vicinity of GRK2.
Mdm2 promotes GRK2 degradation
We next investigated whether Mdm2 could increase the turnover rate of GRK2. First, the stability of GRK2 was determined by pulse–chase assays in HEK-293 cells transiently coexpressing Mdm2 or an empty vector. As shown in Figure 2A, the levels of GRK2 protein decline more rapidly in the presence of Mdm2 (37±1% of [35S]GRK2 protein remaining at 1 h of chase versus 56±3% in the absence of Mdm2 overexpression). Such Mdm2-induced GRK2 proteolysis is blocked by the proteasome inhibitor MG132 (Supplementary data and Supplementary Figure S1B). We have previously shown that the kinase-dead mutant GRK2-K220R displays a severely impaired degradation, probably as a consequence of reduced β-arrestin recruitment to the receptor complex (Penela et al, 2001). Interestingly, Mdm2 is able to associate with this mutant to an extent similar to the wild-type kinase (Supplementary data and Supplementary Figure S1C) and to rescue its defective turnover (Figure 2A, 60±1% of GRK2-K220R remaining at 1 h of chase compared to 92+5% in the absence of Mdm2 expression). Overall, these data are consistent with a role for Mdm2 in the agonist-induced, β-arrestin-mediated GRK2 degradation.
Figure 2.
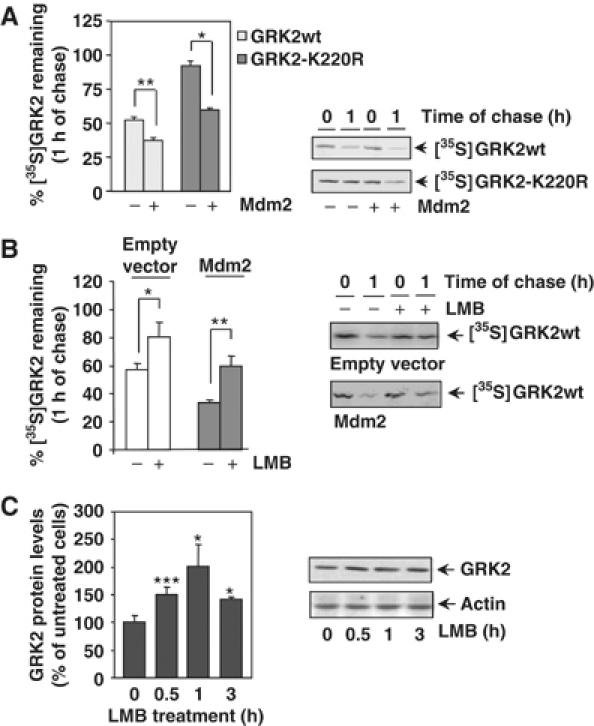
(A) Mdm2 increases degradation of GRK2 and of a slow-turnover GRK2 mutant. HEK-293 cells were transiently transfected with wild-type GRK2 or the GRK2-K220R mutant in the presence or absence of Mdm2, and kinase turnover assessed by pulse–chase experiments as described in Materials and methods. 35S-labeled proteins immunoprecipitated with the anti-GRK2 antibody AbFP2 were resolved by SDS–PAGE followed by fluorography and densitometry. 35S-labeled GRK2 band densities were then normalized to total GRK2 present in the immunoprecipitates, as determined by Western blot analysis. Data are mean±s.e.m. of 3–4 independent experiments performed in duplicate, *P<0.05, **P<0.01. A representative gel autoradiography is shown. (B, C) Effect of LMB treatment on GRK2 turnover. HEK-293 cells transfected with GRK2 and Mdm2 or an empty vector were treated with LMB or vehicle before and during pulse–chase experiments as detailed in Materials and methods, and GRK2 turnover determined as in previous figures. Data are the mean±s.e.m. of three independent experiments performed in duplicate, *P<0.05, **P<0.01. Representative fluorographs are shown. In panel C, endogenous GRK2 expression levels were determined by immunoblot analysis in lysates from MCF7 cells treated for different times with LMB. Data are corrected for actin expression and depicted as percentage of control. *P<0.05, ***P<0.001. A representative blot is shown.
We next analyzed the effect of leptomycin B (LMB) on GRK2 stability. LMB, a drug that blocks nuclear export, has been shown to inhibit Mdm2 nuclear–cytoplasmic shuttling, thus leading to an increased nuclear localization of Mdm2 (Lain et al, 1999). As shown in Figure 2B, the turnover of transiently transfected GRK2 in HEK-293 is significantly retarded in the presence of LMB (80±10% of protein remaining at 1 h of chase compared with 57±5% in control conditions). This treatment does not affect GRK2 localization in our experimental conditions (data not shown), thereby ruling out that this effect is caused by altered kinase distribution. Moreover, the enhanced GRK2 turnover observed upon coexpression of Mdm2 (34±2% of labeled kinase remaining) was not observed in the presence of LMB (60±6% of protein remaining), thus suggesting that the effects of LMB on GRK2 stability are due to the impairment of Mdm2 function and/or localization. In line with this notion, LMB promotes a time-dependent increase (in the range of ≈1.4- to 2-fold) of endogenous GRK2 steady-state levels in MCF7 cells (Figure 2C), consistent with a physiological role for Mdm2 in GRK2 degradation.
Mdm2 modulates GRK2 ubiquitination
We have previously reported that agonist stimulation of β2AR promotes an increase in GRK2 polyubiquitination, consistent with its stimulatory effect on kinase turnover (Penela et al, 1998). As β2AR activation triggers Mdm2/GRK2 association, we next investigated whether Mdm2 would modulate GRK2 ubiquitination. HEK-293 cells were transiently transfected with β2AR, HA-tagged ubiquitin and GRK2 in the presence or absence of Mdm2. Interestingly, both basal and agonist-induced GRK2 polyubiquitination are markedly enhanced upon Mdm2 expression (Figure 3A), supporting a role for Mdm2 as an E3-ubiquitin ligase for GRK2.
Figure 3.
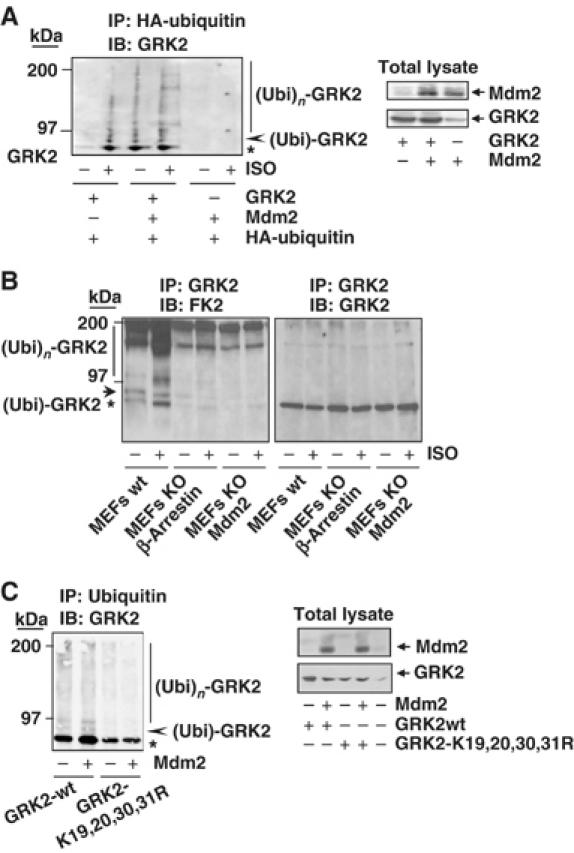
Mdm2 promotes GRK2 ubiquitination. (A) Wild-type GRK2, HA-ubiquitin and β2AR expression plasmids were transfected together with Mdm2 or an empty vector into HEK-293 cells. After 2 h of serum starving, cells were incubated with or without 10 μM isoproterenol for 15 min and lysed for ubiquitination assays as described in Materials and methods. The different GRK2 species present in the ubiquitin immunoprecipitates are indicated. Asterisk and arrowhead indicate mono- and bi-ubiquitinated GRK2 forms (see text for details). Blots depicting expression levels for Mdm2 and GRK2 in total lysates are shown. (B) Both basal and agonist-stimulated ubiquitination of endogenous GRK2 are blocked in either Mdm2- or β-arrestin-deficient cells. ∼50 × 106 wild-type MEFs or MEFs devoid of Mdm2 or β-arrestin proteins were serum-starved and challenged or not with 10 μM isoproterenol for 15 min. Cells were processed as indicated above and ubiquitinated GRK2 species were analyzed by immunoprecipitation of endogenous GRK2 with a specific monoclonal anti-GRK2 antibody (c5/1.1) and detection of endogenous ubiquitin conjugates with anti-ubiquitin antibody FK2. Membranes were stripped and immunoblotted (right panel) with a specific polyclonal anti-GRK2 antibody (C-15) to confirm equal GRK2 loading in each condition. (C) Mutation of critical lysine residues in the N-terminus of GRK2 impairs kinase ubiquitination in the presence of Mdm2. Wild-type GRK2 or the GRK2- K19,20,30,31R mutant was cotransfected with ubiquitin plasmids in the presence or absence of Mdm2 into HEK-293 cells and the kinase ubiquitination pattern was analyzed as above. Expression levels of GRK2 and Mdm2 were assessed in lysates by Western blot analysis. Gels in all panels are representative of 2–3 independent experiments.
The GRK2 ubiquitination patterns induced by receptor stimulation or Mdm2 expression are very similar. The bands of modified kinase migrating at 80 kDa (asterisk) and immediately above (arrowhead) appear to correspond to monoubiquitinated forms of GRK2, whereas the smeared bands in the 97–200 kDa range correspond to polyubiquitinated species of GRK2 (see Supplementary data and Supplementary Figure S2A).
To further substantiate the involvement of Mdm2 as a GRK2 ubiquitin ligase, we compared the ubiquitination of endogenous kinase in wild-type MEFs (expressing endogenous Mdm2 levels) or in MEFs from Mdm2-deficient mice. As shown in Figure 3B, in wild-type MEFs, isoproterenol challenge promotes a marked increase in endogenous GRK2 ubiquitinated species. It must be noted that such basal and agonist-promoted GRK2 ubiquitination pattern is similar to that observed in a heterologous system with overexpressed proteins (see Figure 3A). Interestingly, in Mdm2-deficient MEFs, basal GRK2 ubiquitination was attenuated, whereas the agonist-induced component was severely inhibited, indicating a pivotal role for Mdm2 in this process. Moreover, a similar impairment of GRK2 ubiquitination results also from the absence of β-arrestins, consistent with their role as mediators of the ligase recruitment to GRK2 (Figure 3B).
To explore the specificity of GRK2 ubiquitination by Mdm2, we sought to identify lysine residues in GRK2 critical for supporting Mdm2-dependent ubiquitin attachment. Multiple lysine-to-arginine changes in lysine-rich regions have been reported to result in resistance to proteasomal degradation in several proteins, including p53, the classical Mdm2 target (Rodriguez et al, 2000). As GRK2 lysine residues at positions 19, 21, 30 and 31 are in close proximity to the initial cleavage site on GRK2 by the proteasome (Penela et al, 1998), we tested the ubiquitination and degradation pattern of a GRK2-K19,21,30,31R mutant transiently coexpressed in HEK-293 cells with the β2AR and a tagged HA-ubiquitin construct. Mutation of these lysine residues leads to a clear reduction in the extent and pattern of ubiquitination as compared to wild-type GRK2, both in basal conditions and upon receptor challenge (Supplementary data and Supplementary Figure S2B). Moreover, in contrast to wild-type kinase, the presence of Mdm2 does not modify the extent of GRK2-K19,21,30,31R ubiquitination (Figure 3C), despite the fact that this mutant can associate with Mdm2, as demonstrated by co-immunoprecipitation experiments (Supplementary data and Supplementary Figure S3A). Overall, these results suggest that at least some of these lysine residues in GRK2 are targeted by Mdm2 in an agonist-dependent manner.
Mdm2-mediated ubiquitination of GRK2 lysine residues 19, 21, 30 and 31 is critical for kinase degradation
We next investigated the consequences of impairing Mdm2-dependent ubiquitination of GRK2. The GRK2-K19,21,30,31R mutant was transiently coexpressed in HEK-293 cells with the β2AR, and the kinase turnover was analyzed by pulse–chase assays. As shown in Figure 4A, degradation of this mutant was clearly delayed in basal conditions, with a half-life three-fold longer than that of wild-type GRK2, consistent with its reduced ubiquitination. Moreover, receptor stimulation does not significantly enhance GRK2-K19,21,30,31R degradation (data not shown). It should be noted that this mutant displayed similar subcellular distribution and kinase activity as compared to wild-type GRK2 (Supplementary data and Supplementary Figure S3B and C), thus ruling out the possibility that the altered degradation might arise from changes other than impaired ubiquitination. In this regard, the slower turnover of this mutant was also blocked in the presence of proteasome inhibitors (data not shown), thus confirming that mutation of these lysine residues severely retards but does not change the proteolytic pathway responsible for degrading the protein.
Figure 4.
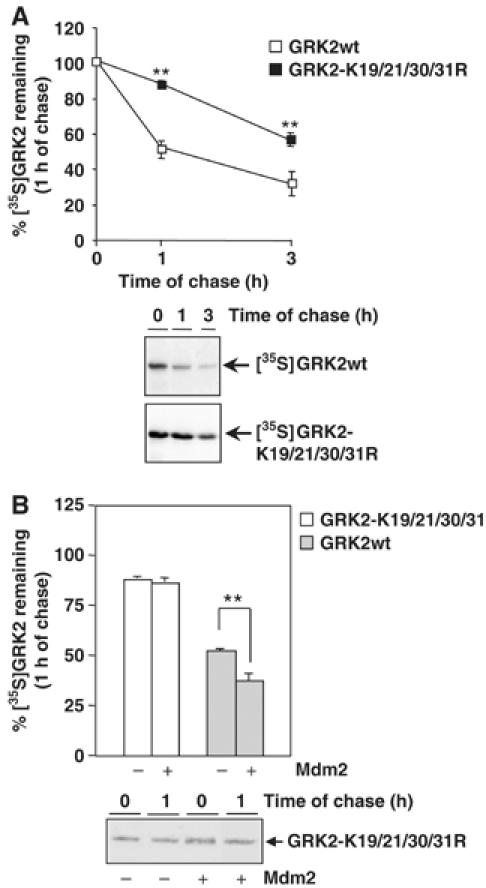
N-terminal lysine residues are critical for Mdm2-mediated GRK2 degradation. (A) Turnover of wild-type GRK2 and GRK2-K19,20,30,31R mutant proteins was assessed by pulse–chase experiments in HEK-293 cells as described in Materials and methods and Figure 2. (B) Similar experiments were performed in cells cotransfected with an Mdm2 construct. In both panels, data are mean±s.e.m. from three to four independent experiments (**P<0.01), and representative fluorographies are shown.
Interestingly, overexpression of Mdm2 is unable to accelerate GRK2-K19,21,30,31R degradation (88±3% of mutant protein remaining in the absence of Mdm2 compared with 86±4% in its presence; Figure 4B). These results indicate that the ubiquitination of defined amino-terminal lysine residues of GRK2 is critical for proper kinase degradation and also strongly suggest that the agonist-dependent increase in GRK2 turnover involves the E3 ligase Mdm2-dependent modification of these residues.
GRK2 turnover rate is markedly decreased by Mdm2 knockdown
In order to substantiate the regulatory role of Mdm2 in GRK2 stability, we next examined whether degradation of ectopically expressed GRK2 is altered in Mdm2-deficient MEFs. Figure 5A shows that the GRK2 protein decay is severely impaired in the absence of Mdm2, with 98±9% of labeled GRK2 remaining after 1 h of chase, which resembles the altered kinase degradation caused by abrogation of its Mdm2-dependent ubiquitination (Figure 4A and B). Interestingly, restoration of Mdm2 expression in the Mdm2-deficient MEFs rescues GRK2 turnover (Figure 5A), leading to a proteolysis pattern close to that observed for GRK2 in different cellular settings with intact Mdm2 expression (Figure 3; Penela et al, 2001). To further confirm that Mdm2 contributes to degradation of GRK2 in physiological conditions, we compared the protein decay of endogenous kinase by chase experiments in the presence of cycloheximide in both wild-type and Mdm2-deficient MEFs. Figure 5B shows that the degradation rate of GRK2 in MEFs lacking Mdm2 is slower than in wild-type MEFs in these conditions. Moreover, the steady-state expression of endogenous GRK2 is higher in Mdm2-deficient MEFs than in Mdm2-intact cells (compare lane 6 versus lane 1 in the lower panel of Figure 5B). Overall, these results indicate that Mdm2 is involved in degradation of GRK2 in endogenous conditions, and that this ligase plays a critical role in maintaining basal GRK2 turnover.
Figure 5.
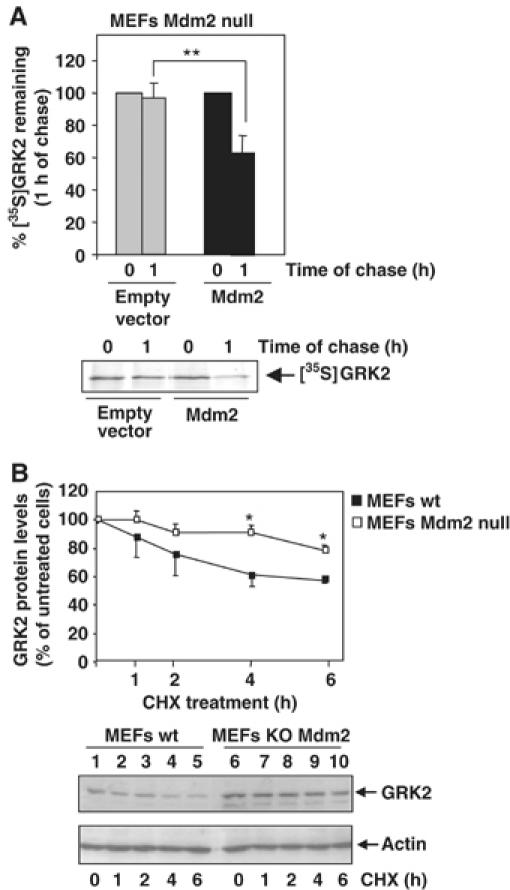
GRK2 turnover is blocked in Mdm2-deficient cells. (A) Mdm2-null MEFs were transfected with wild-type GRK2 in the presence or absence of Mdm2. Degradation of the heterologous GRK2 protein was assessed after 1 h of chase as in previous figures. (B) Stability of endogenous GRK2 is increased in the absence of Mdm2 expression. GRK2 protein levels were examined by immunoblot analysis in both wild-type and Mdm2-deficient MEFs upon treatment with cycloheximide (CHX) for the indicated times. The amount of GRK2 at 0 h was defined as 100%, and data normalized by actin protein levels. In both the panels, data are the mean±s.e.m. of four independent experiments (*P<0.05, **P<0.01). Representative gels are shown.
GRK2 stability is modulated by IGF-1 signaling via Mdm2 and the PI3K/Akt pathway
The experiments shown above demonstrate that GRK2 degradation requires Mdm2-dependent ubiquitination, and suggest that stimulation of GPCRs such as β2AR may enhance GRK2 turnover by facilitating the interaction of GRK2 with a β-arrestin-recruited pool of Mdm2. It has been recently reported that Mdm2 can also be recruited by β-arrestin molecules in response to activation of tyrosine kinase receptors such as the IGF1-R, thus promoting degradation of several targets including the receptor itself (Girnita et al, 2005). Therefore, we asked whether IGF1-R activation might elicit some modulatory effects on GRK2 stability. To this end, the degradation rate of endogenous GRK2 was determined by pulse–chase assays in the breast human carcinoma cell line MCF7, which expresses endogenous IGF1-R. Under normal conditions, GRK2 is degraded in MCF7 cells with a time course similar to that observed for this kinase in other cell types, and GRK2 half-life remains essentially unaltered when chasing MCF7 cells in low serum medium (data not shown), an experimental condition selected to analyze the potential effects of IGF-1 stimulation. Surprisingly, GRK2 degradation was markedly delayed at all chase points examined in cells challenged with IGF-1 as compared to untreated cells (∼1.3-fold of protein decay between 0.5 and 2 h of chase in the presence of IGF-1 versus ∼2-fold decay in its absence) (Figure 6A). Recently, several groups have reported that Akt interacts with and phosphorylates Mdm2 in response to activation of IGF1-R (Mayo and Donner, 2001; Feng et al, 2004) promoting its nuclear shuffling. Therefore, we hypothesized that Akt phosphorylation of the E3 ligase could impair Mdm2-dependent GRK2 degradation. To test this hypothesis, we performed pulse–chase assays in HEK-293 cells expressing GRK2 with a constitutively active mutant of Akt (Akt-myr). As shown in Figure 6B, GRK2 degradation is clearly diminished in the presence of Akt-myr as compared to the turnover of GRK2 alone (74±8% of 35S-labeled kinase remaining at 1 h of chase versus 49±3%, respectively). Interestingly, the expression of Akt-myr causes a dramatic change in the GRK2 proteolysis promoted by Mdm2 (63±15% of protein remaining after 1 h of chase compared to 26±3% in the presence of Mdm2 only) (Figure 6B). These findings suggest that activation of Akt by IGF-1 signaling can prevent the effect of Mdm2 on GRK2 turnover, thus leading to kinase stabilization.
Figure 6.
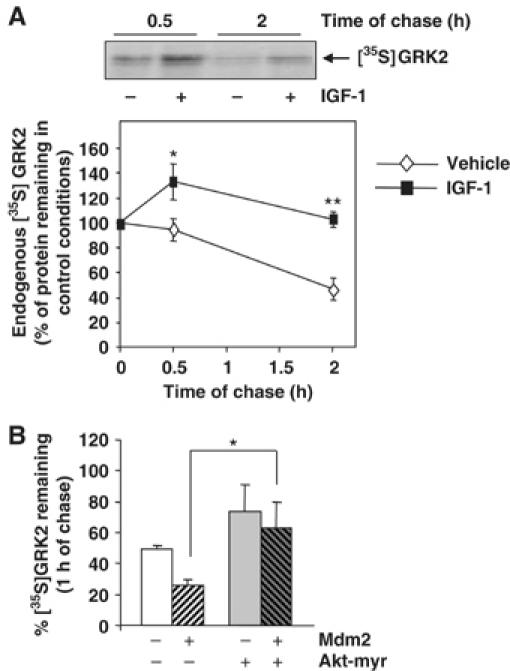
IGF receptor activation and Akt activity promote GRK2 stabilization. (A) Turnover of endogenous GRK2 was analyzed in MCF7 cells as detailed in Materials and methods in the presence of 50 ng/ml of IGF-1 or vehicle. Labeled proteins were immunoprecipitated with a specific anti-GRK2 antibody (C5/1.1). The density of the 35S-labeled GRK2 band after the pulse period was taken as 100%. Data are mean±s.e.m. of 3–5 independent experiments. *P<0.05; **P<0.01. A representative fluorography is shown. (B) HEK-293 cells were transfected with different combinations of GRK2, Mdm2 and a constitutively active Akt mutant (Akt-myr) as indicated. GRK2 degradation rate was determined by pulse–chase analysis. Results (mean±s.e.m.) from three independent experiments are shown. *P<0.05.
In line with these data, steady-state expression levels of endogenous GRK2 increase progressively in MCF7 cells upon treatment with IGF-1 as assessed by immunoblot analysis (Figure 7A). The increase was apparent after 4 h of incubation and attained a value of ≈two-fold at 24 h. Interestingly, the peak activation of Akt upon IGF-1 challenge precedes the burst in GRK2 levels (Figure 7A, lower panel). Moreover, Akt-dependent phosphorylation of Mdm2, as detected with specific phospho-antibodies, correlates with the timing of GRK2 accumulation (Figure 7A, lower panel). This suggests that modification of Mdm2 by the PI3K/Akt pathway abrogates ligase activity towards GRK2 and therefore leads to protein accumulation. Consistent with this notion, IGF-1-mediated GRK2 upregulation was impeded in the presence of a PI3K inhibitor (Figure 7A), which also abolishes Akt activation and Mdm2 phosphorylation upon IGF-1 challenge (data not shown). Furthermore, IGF1-mediated GRK2 protein increase was not observed in MCF7 cells when an Mdm2 mutant unable to be modulated by the AKT/PI3K pathway (Mdm2 S166, 186A) was overexpressed (Figure 7B). Consistent with the idea that Akt phosphorylation of Mdm2 alters its ability to induce GRK2 degradation, the association of GRK2 with the Mdm2 mutant unable to be phosphorylated by Akt is notably increased, whereas that with an Mdm2 mutant that mimics such phosphorylation (Mdm2 S166, 186D) is severely impaired (Supplementary data and Supplementary Figure S4). Overall, these results indicate that modulation of Mdm2 by AKT plays a pivotal role in GRK2 stabilization.
Figure 7.
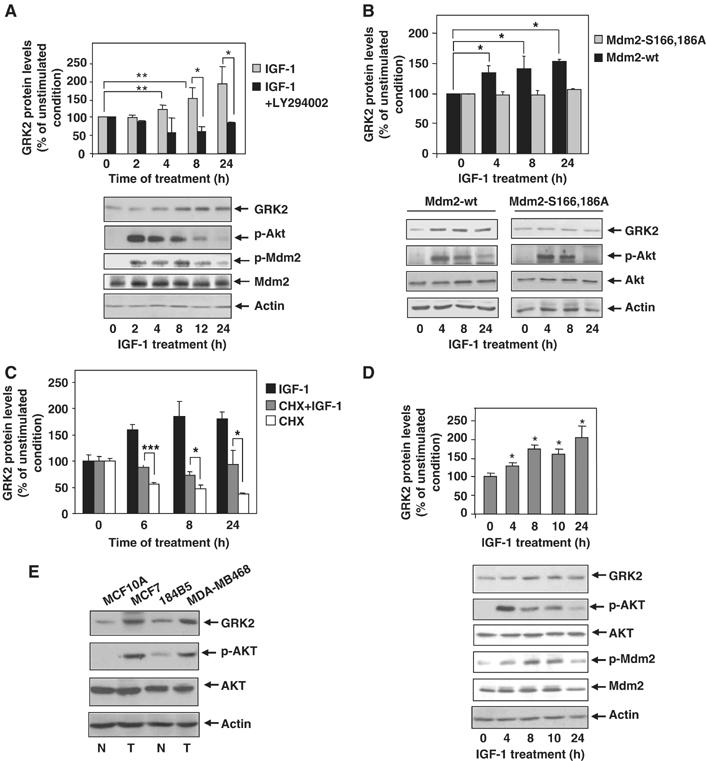
IGF-1 stimulation induces endogenous GRK2 protein accumulation in a PI3K-dependent manner. (A) MCF7 cells were serum-starved for 12 h and challenged with IGF-1 (50 ng/ml) for different times in the presence or absence of the PI3K inhibitor LY294002 (10 μM). Cells were subjected to immunoblot analysis with anti-GRK2, anti-Akt-pSer473, anti-Mdm2-pSer166 and anti-Mdm2 antibodies. GRK2 band densities were normalized by actin protein levels and expressed as percentage of GRK2 level in unstimulated conditions. Data are mean±s.e.m. of five independent experiments performed in duplicate. *P<0.05, **P<0.01. (B) IFG-1-dependent upregulation of GRK2 is prevented by expression of an Mdm2 mutant defective in Akt-mediated phosphorylation. MCF7 cells transiently overexpressing either wild-type Mdm2 or the ligase mutant Mdm2 S166, 186A were serum-starved as above and stimulated with IGF-1 (50 ng/ml) for the indicated times. Expression levels of GRK2, Akt-pSer473, Akt and actin were determined by immunoblotting. Data were analyzed and depicted as in panel A. Representative blots are shown. (C) IGF-1 increases GRK2 protein by decreasing its degradation rate without altering its protein synthesis. Endogenous GRK2 protein levels were determined by immunoblot analysis in MCF7 cells stimulated with IGF-1 as above in the presence or absence of cycloheximide and compared with cycloheximide treatment alone. Data are mean±s.e.m. of three independent experiments. *P<0.05; ***P<0.001. (D) GRK2 protein accumulation is induced by IGF-1 challenge, in the non-transformed mammary cell line MCF10A. Cells were treated as above and levels of GRK2, Akt-pSer473, Akt, Mdm2-pSer166, Mdm2 and actin immunodetected with specific antibodies. The patterns of Akt activation, Mdm2 phosphorylation and GRK2 protein increase are similar to those observed in MCF7 cells. (E) Transformed mammary cells display higher levels of Akt activity and GRK2 protein. Both malignant transformed (T, MDA-MB486 and MCF7) and non-transformed cells (N, MCF10A and 184B5) were lysed in RIPA buffer and expression levels of GRK2, Akt-pSer473, Akt and actin were determined as above. Representative blots from three independent experiments are displayed.
In order to rule out that IGF1-R activation could enhance transcription and/or GRK2 protein synthesis by a PI3K/Akt-dependent pathway, we measured protein kinase levels in the presence of IGF-1 plus cycloheximide, a well-known protein synthesis inhibitor. As shown in Figure 7C, treatment of MCF7 cells in the presence of this compound alone leads to a reduction in GRK2 protein levels, which is detected as earlier as 6 h of incubation, as it would be expected for a short-lived protein. Interestingly, IGF-1 challenge can relieve the protein decay of GRK2 due to the cycloheximide-dependent inhibition of protein synthesis, thereby confirming that modulation of GRK2 stability is the main process targeted by IGF-1-dependent activation of Akt. Moreover, these findings also indicate that modulation of GRK2 turnover by IGF-1 stimulation does not require synthesis of any protein factor.
Confirming the physiological relevance of the modulation of GRK2 expression levels by the PI3K/AKT/Mdm2 axis observed in the MCF7 breast cancer cell line, GRK2 was similarly upregulated upon IGF-1 challenge in several other transformed cells such as the breast epithelial MDA-MB468 and the melanoma B16F10 cell lines (Supplementary data and Supplementary Figure S5). Interestingly, GRK2 protein accumulation is also triggered by IGF1-R activation in MCF10A cells, a non-transformed mammary epithelial cell line in which the pattern of Akt activation and of downstream phosphorylation of Mdm2 also correlate with GRK2 upregulation (Figure 7D). Moreover, silencing of Akt activity by addition of a pharmacological inhibitor completely prevents the increase in GRK2 protein triggered by IGF-1 both in transformed (MB468) and non-transformed (MCF10A) cell lines (Supplementary data and Supplementary Figure S6A and B). Cycloheximide-dependent downregulation of GRK2 expression was also prevented in the presence of IGF-1 in the non-transformed cell line (Supplementary Figure S6B).
Taken together, these results indicate that the IGF-1/Akt/Mdm2 regulatory axis of GRK2 turnover operates in different cell types, and suggest that upregulation of GRK2 by this novel modulatory mechanism might take place in some cellular contexts of malignant transformation. In this regard, we find that mammary transformed cells show higher cellular levels of GRK2 when compared to normal cells, in parallel with higher activation of AKT (Figure 7E).
Finally, to further establish the involvement of Mdm2 in the stabilization of GRK2 promoted by the IGF-1/PI3K/Akt activation axis, we compared the effect of IGF-1 challenge on endogenous GRK2 levels in both wild-type and Mdm2-deficient MEFs (Figure 8). We find that GRK2 protein is accumulated in response to IGF-1 in MEFs expressing Mdm2 (panel A) with a time course and to an extent similar to those observed in MCF7 cells. Remarkably, in MEFs isolated from Mdm2-knockout mice, the absence of the ligase precludes any significant change in GRK2 expression upon IGF-1 treatment (panel B), despite the fact that these cells display an IGF1-R-dependent activation of the PI3K/Akt pathway similar to that detected in normal MEFs (data not shown). Overall, these results demonstrate that IGF1-R stimulation modulates GRK2 cellular levels by a mechanism involving the PI3K/Akt pathway and the Mdm2 E3 ligase.
Figure 8.
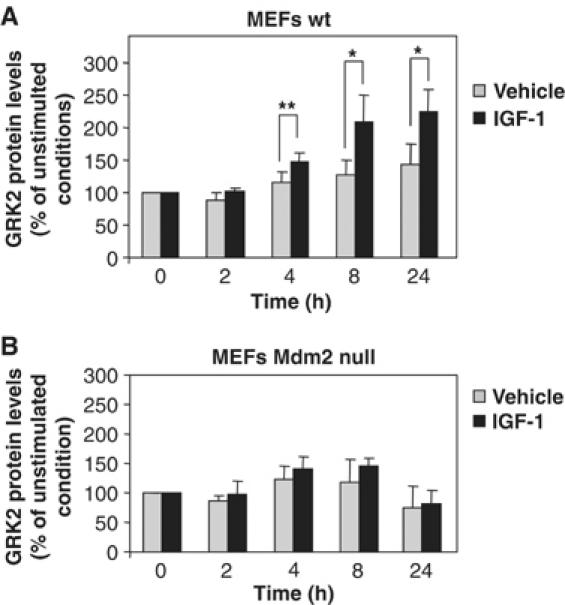
Knockdown of Mdm2 prevents endogenous GRK2 protein accumulation in response to IGF-1.Wild-type (A) and Mdm2-deficient MEFs (B) were treated with IGF-1 or vehicle for the indicated times as in Figure 7. GRK2 protein levels were determined by Western blot analysis and normalized by actin expression. Data are mean±s.e.m. of four experiments performed in triplicate, *P<0.05, **P<0.01.
Discussion
The Mdm2 oncoprotein is an E3-ubiquitin ligase best known by its role in controlling p53 degradation and transcriptional activity (Yang et al, 2004; Bond et al, 2005). Growing evidence indicates that other proteins interact with Mdm2 and can be regulated by this ligase (Lin et al, 2002; Girnita et al, 2003; Iwakuma and Lozano, 2003; Jin et al, 2003; Pan and Chen, 2003; Uchida et al, 2005). In this report, we identify GRK2 as a new Mdm2 target.
Several lines of evidence support the notion that Mdm2 serves as an E3 ligase for GRK2 ubiquitination and is critical for modulating its degradation and cellular levels. First, a pool of endogenous GRK2 and Mdm2 can be found in the same molecular complex, as indicated by co-immunoprecipitation of both proteins under basal conditions. Moreover, stimulation of the β2AR promotes a marked and transient increase in the endogenous GRK2/ Mdm2 association, suggesting that this is a modulated and specific process. Second, overexpression of Mdm2 markedly increases the extent of ‘in situ' GRK2 ubiquitination in both basal and agonist-stimulated conditions, rescues the defective degradation of the GRK2 mutant GRK2-K220R and enhances GRK2 proteolysis to an extent similar to that promoted by receptor stimulation, supporting the idea that Mdm2 is a key ligase mediating agonist-dependent GRK2 ubiquitination and subsequent proteasome degradation. Third, GRK2 ubiquitination and turnover is significantly impaired in Mdm2-deficient MEFs compared to wild-type cells, which results in increased endogenous GRK2 protein levels. Restoration of ligase expression in Mdm2-knockout cells rescues GRK2 degradation, leading to a turnover rate similar to that observed in different cell types with native Mdm2 expression. Finally, we find that modulation of Mdm2 by the PI3K/Akt pathway upon IGF1-R stimulation alters GRK2 degradation and augments kinase cellular levels, putting forward a new mechanism for controlling GRK2 expression.
Mdm2 has been shown to control ubiquitination and stability of different molecules in response to membrane receptor occupancy (Shenoy et al, 2001; Girnita et al, 2005). β2AR stimulation leads to the recruitment of Mdm2 to the receptor complex in a β-arrestin-dependent manner. Both β-arrestin1 and β-arrestin2 are able to interact with Mdm2 (Wang et al, 2003) and therefore to bring this ligase to the vicinity of activated receptors. We have found that β-arrestin1 also plays a role in GRK2 degradation by facilitating the β-agonist-triggered GRK2/Mdm2 association. Consistently, GRK2 protein decay and ubiquitination pattern are also markedly altered in β-arrestin1/2-knockout MEFs. These findings are compatible with a sequence of events in which Mdm2 is recruited to the receptor complex at the plasma membrane, where the ligase would sequentially ubiquitinate several targets in a β-arrestin-dependent manner.
As Mdm2 preferentially associates to β-arrestins in their basal receptor-unbound conformation (Song et al, 2006), it is feasible that β-arrestin might deliver Mdm2 to the receptor complex in an active form that is suitable to interact with and promote GRK2 ubiquitination, as has been reported for other targets (Shenoy et al, 2001; Wang et al, 2003). Mdm2 shuttles back and forth between the nucleus and the cytoplasm as a result of its interaction with different proteins. In this regard, it has been reported that β-arrestin2 promotes Mdm2 cytoplasmic relocation (Wang et al, 2003), whereas β-arrestin1 might cooperate with β-arrestin2 in the maintenance of an accessible cytosolic pool of Mdm2 for degradation of targets such as GRK2 in response to specific stimuli. In line with this idea, disruption of Mdm2 shuttling by promoting its nuclear retention with LMB, which also alters the balance between nuclear and cytoplasmic pools of β-arrestin2 (Scott et al, 2002; Wang et al, 2003), markedly affects GRK2 stability, retarding kinase degradation and increasing the steady-state levels of endogenous GRK2.
Although Mdm2 emerges as a crucial ligase involved in GRK2 degradation, the contribution of other ligase activities to GRK2 ubiquitination cannot be ruled out. β-Arrestin has been suggested to recruit additional ubiquitin ligases to the β2AR complex (Shenoy et al, 2001). On the other hand, it has been described that Mdm2 mediates multiple monoubiquitination attachment to its prototypical target p53, whereas p300 mediates subsequent polyubiquitination acting as an E4 ligase (Lai et al, 2001; Grossman et al, 2003). Although a comprehensive map of GRK2 sites involved in ubiquitination awaits further research, our results point to lysines 19, 21, 30 and 31 as critical residues involved in both mono- and polyubiquitination of GRK2 (see also Supplementary data). Our results are compatible with Mdm2 mediating directly either types of ubiquitination or, alternatively, multiple or single monoubiquitination that would prime subsequent polyubiquitination by another ligase in a β-arrestin-mediated manner.
The dependence of GRK2 degradation on Mdm2 function opens the possibility that factors affecting the activity as well as subcellular localization of this E3 ligase may modulate GRK2 turnover (Meek and Knippschild, 2003). In this regard, several groups have reported the ability of Akt to phosphorylate Mdm2 at residues 166 and 186 or 188 (Mayo and Donner, 2001; Ashcroft et al, 2002; Feng et al, 2004; Milne et al, 2004). In response to the IGF-1, the Akt-dependent phosphorylation of Mdm2 stimulates translocation of the ligase from the cytoplasm to the nucleus (Mayo and Donner, 2001; Zhou et al, 2001) and its stabilization (Feng et al, 2004). Although this scenario favors the downregulation of p53 by Mdm2, it may hamper the Mdm2-dependent regulation of other cellular targets. In keeping with this idea, we show that IGF-1 stimulation promotes GRK2 protein accumulation in different cell types by diminishing its degradation rate. The stabilization of GRK2 promoted by IGF-1 challenge does not depend on protein synthesis, and requires both the activation of the PI3K/Akt pathway and Mdm2 expression, as IGF-1 effect is abolished in the presence of PI3K inhibitors as well as in Mdm2-knockout cells. Upregulation of GRK2 by IGF-1 in MCF7 cells is prevented upon overexpression of an Mdm2 mutant unable to be phosphorylated by Akt, in line with the notion that Akt-mediated phosphorylation of Mdm2 is required for the effects on GRK2 stability.
The fact that GRK2 turnover is retarded by IGF1-R activation but enhanced by GPCR stimulation might be explained by differences in the composition or sequential assembly of the receptor multimolecular complexes, particularly regarding GRK2 recruitment. It is also possible that IGF1-R activation modulates phosphorylation of β-arrestins, as recently reported for insulin receptor activation (Hupfeld et al, 2005), thus impairing their function as scaffolds in the GRK2 degradation process. Alternatively, or in addition, IGF-1 signaling could trigger modifications of either Mdm2 or β-arrestins that would alter their capacity to promote efficient GRK2 degradation. Consistently, GRK2 displays differential association with Mdm2 mutants that either mimic or prevent Akt phosphorylation; however, whether Akt-dependent phosphorylation of Mdm2 hampers the ligase-mediated degradation of GRK2 by altering direct protein–protein interactions between Mdm2 and GRK2 or by keeping away these proteins in the cell, or both, remains to be determined.
Altered expression of GRK2 has been associated to several human diseases and reported in animal models of different pathologies. GRK2 protein levels were found to be downregulated in RA (Lombardi et al, 1999), in multiple sclerosis (Vroon et al, 2005) and in models of ischemic heart damage (Girnita et al, 2005). GRK2 is upregulated in other pathological settings, such as human congestive heart failure or hypertension (Metaye et al, 2005; Penela et al, 2006). It is worth noting that modification of kinase stability emerges as an important process that may trigger aberrant GRK2 expression in different situations. Increased GRK2 degradation has been reported in RA (Lombardi et al, 1999). Interestingly, the expression of Mdm2 has been found to be upregulated in samples from RA patients (Taranto et al, 2005), thus raising the intriguing possibility that Mdm2 might contribute to the observed decay in GRK2 protein in these patients. Whether Mdm2 might be altered functionality in other diseases characterized by changes in GRK2 expression levels deserves to be explored.
On the other hand, as our data indicate that control of Mdm2 protein levels, activity or localization can influence GRK2 protein stability, it is conceivable that dysfunction of the PI3K/Akt pathway might result in altered GRK2 regulation in different cell types. Interestingly, a significant proportion of primary human breast tumors displays increased Akt activity and Akt-dependent Mdm2 phosphorylation (Zhou et al, 2001), which are critical markers for tumor progression. In those samples positive for Neu/Akt, Mdm2 was mainly localized in the nucleus, in contrast to the mixed nuclear and cytoplasmic distribution of the ligase in the Neu/Akt-negative breast tumors. Although the expression of GRK2 in tumor tissues has not been addressed in detail, it is tempting to suggest that deregulated Akt activity, by relieving the Mdm2-dependent degradation of GRK2, could stabilize the kinase and play a role in GRK2 upregulation reported by others in some human tumor malignancies (Metaye et al, 2002; King et al, 2003). Further studies will be needed to explore the functional interplay of the PI3K/Akt/ Mdm2/GRK2 axis and their relevance in physiological and malignant cell growth.
Materials and methods
Materials and plasmids
See Supplementary data.
Site-directed mutagenesis
Cell culture and transfection
HEK-293, MCF-7, wild-type and Mdm2/p53 double-null MEFs were cultured in Dulbecco's modified Eagle's medium plus 10% (v/v) fetal bovine serum at 37°C in a humidified 5% atmosphere. Cells grown to 80% confluence in 60 mm dishes were transfected with the indicated plasmids (5 μg of total DNA) by using the LipofectAmine (HEK-293) or the FuGENE6 (MCF7 and MEFs) reagents. Empty vector was added as needed to keep the total amount of DNA transfected constant.
Cell treatment and metabolic labeling
See Supplementary data.
Immunoprecipitation and Western blot analysis
Cells were lysed in buffer A (20 mM Tris–HCl, pH 7.5, 100 mM NaCl, 1 mM EDTA, 0.5% NP-40, 10% glycerol, with a cocktail of protease inhibitors) for 1 h at 4°C and then centrifuged (15 000 g, 10 min). Supernatants were incubated with a specific agarose-conjugated anti-Mdm2 monoclonal antibody (SMP14, Santa Cruz Biotechnology) or with anti-GRK2 monoclonal antibody (clone C5/1.1, Upstate). Immune complexes were resolved in 7.5% SDS–PAGE and transferred to nitrocellulose membranes. Blots were probed with a specific GRK2 polyclonal antibody AbFP2 (Murga et al, 1996) or with an Mdm2 polyclonal antibody (N-20, Santa Cruz Biotechnology). After membrane stripping, the same blots were re-probed with the corresponding antibodies directed against the immunoprecipitated proteins. Blots were developed using a chemiluminescent method (ECL, Amersham). Band density was quantitated by laser densitometric analysis.
Ubiquitination assays
HEK-293 cells seeded in 100 mm dishes were transfected with different combinations of expression vectors for HA-ubiquitin, β2AR, Mdm2 and wild-type GRK2 or the GRK2-K19,21,30,31R mutant. After 2 h of serum starving, cells were either left untreated or stimulated with 10 μM isoproterenol for the indicated times. Cells that were not challenged with any agonist were maintained in the presence of serum. Subsequently, cells were lysed in 1 ml of 20 mM Tris–HCl (pH 7.4), 0.3 M NaCl, 1% Triton-X100 and 0.5% sodium deoxycholate supplemented with 5 mM N-ethyl-maleimide. Ubiquitin conjugates were purified by immunoprecipitation with a monoclonal anti-HA antibody (12CA5, Roche Molecular Biochemicals) or with a specific monoclonal anti-ubiquitin antibody that does not crossreact with free ubiquitin molecules (FK2, Affinity). The presence of ubiquitinated GRK2 was then analyzed by Western blot analysis as described (Penela et al, 1998). Similar GRK2 and ubiquitin expression levels were confirmed in cell lysates by dot blot and Western blot using their specific antibodies. For analysis of endogenous ubiquitination, MEF cells were grown at confluency in 150 mm dishes and treated as above. Ubiquitin conjugates of GRK2 were detected by immunoprecipitation of GRK2 protein with a monoclonal antibody (clone C5/1.1, Upstate) and immunoblotting with the FK2 antibody.
Supplementary Material
Supplementary Information
Acknowledgments
We thank Susana Rojo and Lucía Horrillo for skillful technical and secretarial assistance, respectively, and Drs G Lozano, L Mayo, RJ Lefko Witz, M Hung and M Serrano for experimental tools. This work was supported by grants from Ministerio de Educación y Ciencia (SAF2002-0408 and SAF2005-03053), Fundación Ramón Areces and the MAIN European Network (LSHG-CT-2003-502935). PP is a recipient of a Ramón y Cajal contract.
References
- Ashcroft M, Ludwig RL, Woods DB, Copeland TD, Weber HO, MacRae EJ, Vousden KH (2002) Phosphorylation of HDM2 by Akt. Oncogene 21: 1955–1962 [DOI] [PubMed] [Google Scholar]
- Bond GL, Hu W, Levine AJ (2005) MDM2 is a central node in the p53 pathway: 12 years and counting. Curr Cancer Drug Targets 5: 3–8 [DOI] [PubMed] [Google Scholar]
- Elorza A, Penela P, Sarnago S, Mayor F Jr (2003) MAPK-dependent degradation of G protein-coupled receptor kinase 2. J Biol Chem 278: 29164–29173 [DOI] [PubMed] [Google Scholar]
- Feng J, Tamaskovic R, Yang Z, Brazil DP, Merlo A, Hess D, Hemmings BA (2004) Stabilization of Mdm2 via decreased ubiquitination is mediated by protein kinase B/Akt-dependent phosphorylation. J Biol Chem 279: 35510–35517 [DOI] [PubMed] [Google Scholar]
- Ferguson SS (2001) Evolving concepts in G protein-coupled receptor endocytosis: the role in receptor desensitization and signaling. Pharmacol Rev 53: 1–24 [PubMed] [Google Scholar]
- Freedman NJ, Kim LK, Murray JP, Exum ST, Brian L, Wu JH, Peppel K (2002) Phosphorylation of the platelet-derived growth factor receptor-beta and epidermal growth factor receptor by G protein-coupled receptor kinase-2. Mechanisms for selectivity of desensitization. J Biol Chem 277: 48261–48269 [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR, Premont RT, Bohn LM, Lefkowitz RJ, Caron MG (2004) Desensitization of G protein-coupled receptors and neuronal functions. Annu Rev Neurosci 27: 107–144 [DOI] [PubMed] [Google Scholar]
- Girnita L, Girnita A, Larsson O (2003) Mdm2-dependent ubiquitination and degradation of the insulin-like growth factor 1 receptor. Proc Natl Acad Sci USA 100: 8247–8252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girnita L, Shenoy SK, Sehat B, Vasilcanu R, Girnita A, Lefkowitz RJ, Larsson O (2005) {beta}Arrestin is crucial for ubiquitination and down-regulation of the insulin-like growth factor-1 receptor by acting as adaptor for the MDM2 E3 ligase. J Biol Chem 280: 24412–24419 [DOI] [PubMed] [Google Scholar]
- Grossman SR, Deato ME, Brignone C, Chan HM, Kung AL, Tagami H, Nakatani Y, Livingston DM (2003) Polyubiquitination of p53 by a ubiquitin ligase activity of p300. Science 300: 342–344 [DOI] [PubMed] [Google Scholar]
- Hildreth KL, Wu JH, Barak LS, Exum ST, Kim LK, Peppel K, Freedman NJ (2004) Phosphorylation of the platelet-derived growth factor receptor-beta by G protein-coupled receptor kinase-2 reduces receptor signaling and interaction with the Na(+)/H(+) exchanger regulatory factor. J Biol Chem 279: 41775–41782 [DOI] [PubMed] [Google Scholar]
- Ho J, Cocolakis E, Dumas VM, Posner BI, Laporte SA, Lebrun JJ (2005) The G protein-coupled receptor kinase-2 is a TGFbeta-inducible antagonist of TGFbeta signal transduction. EMBO J 24: 3247–3258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hupfeld CJ, Resnik JL, Ugi S, Olefsky JM (2005) Insulin-induced beta-arrestin1 Ser-412 phosphorylation is a mechanism for desensitization of ERK activation by Galphai-coupled receptors. J Biol Chem 280: 1016–1023 [DOI] [PubMed] [Google Scholar]
- Iwakuma T, Lozano G (2003) MDM2, an introduction. Mol Cancer Res 1: 993–1000 [PubMed] [Google Scholar]
- Jin Y, Lee H, Zeng SX, Dai MS, Lu H (2003) MDM2 promotes p21waf1/cip1 proteasomal turnover independently of ubiquitylation. EMBO J 22: 6365–6377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King DW, Steinmetz R, Wagoner HA, Hannon TS, Chen LY, Eugster EA, Pescovitz OH (2003) Differential expression of GRK isoforms in nonmalignant and malignant human granulosa cells. Endocrine 22: 135–142 [DOI] [PubMed] [Google Scholar]
- Kohout TA, Lefkowitz RJ (2003) Regulation of G protein-coupled receptor kinases and arrestins during receptor desensitization. Mol Pharmacol 63: 9–18 [DOI] [PubMed] [Google Scholar]
- Lai Z, Ferry KV, Diamond MA, Wee KE, Kim YB, Ma J, Yang T, Benfield PA, Copeland RA, Auger KR (2001) Human mdm2 mediates multiple mono-ubiquitination of p53 by a mechanism requiring enzyme isomerization. J Biol Chem 276: 31357–31367 [DOI] [PubMed] [Google Scholar]
- Lain S, Midgley C, Sparks A, Lane EB, Lane DP (1999) An inhibitor of nuclear export activates the p53 response and induces the localization of HDM2 and p53 to U1A-positive nuclear bodies associated with the PODs. Exp Cell Res 248: 457–472 [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ, Shenoy SK (2005) Transduction of receptor signals by beta-arrestins. Science 308: 512–517 [DOI] [PubMed] [Google Scholar]
- Lin FT, Chen W, Shenoy S, Cong M, Exum ST, Lefkowitz RJ (2002) Phosphorylation of beta-arrestin2 regulates its function in internalization of beta(2)-adrenergic receptors. Biochemistry 41: 10692–10699 [DOI] [PubMed] [Google Scholar]
- Lin FT, Daaka Y, Lefkowitz RJ (1998) beta-arrestins regulate mitogenic signaling and clathrin-mediated endocytosis of the insulin-like growth factor I receptor. J Biol Chem 273: 31640–31643 [DOI] [PubMed] [Google Scholar]
- Lombardi MS, Kavelaars A, Schedlowski M, Bijlsma JW, Okihara KL, Van de Pol M, Ochsmann S, Pawlak C, Schmidt RE, Heijnen CJ (1999) Decreased expression and activity of G-protein-coupled receptor kinases in peripheral blood mononuclear cells of patients with rheumatoid arthritis. FASEB J 13: 715–725 [DOI] [PubMed] [Google Scholar]
- Mayo LD, Donner DB (2001) A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc Natl Acad Sci USA 98: 11598–11603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meek DW, Knippschild U (2003) Posttranslational modification of MDM2. Mol Cancer Res 1: 1017–1026 [PubMed] [Google Scholar]
- Metaye T, Gibelin H, Perdrisot R, Kraimps JL (2005) Pathophysiological roles of G-protein-coupled receptor kinases. Cell Signal 17: 917–928 [DOI] [PubMed] [Google Scholar]
- Metaye T, Menet E, Guilhot J, Kraimps JL (2002) Expression and activity of g protein-coupled receptor kinases in differentiated thyroid carcinoma. J Clin Endocrinol Metab 87: 3279–3286 [DOI] [PubMed] [Google Scholar]
- Milne D, Kampanis P, Nicol S, Dias S, Campbell DG, Fuller-Pace F, Meek D (2004) A novel site of AKT-mediated phosphorylation in the human MDM2 onco-protein. FEBS Lett 577: 270–276 [DOI] [PubMed] [Google Scholar]
- Murga C, Ruiz-Gomez A, Garcia-Higuera I, Kim CM, Benovic JL, Mayor F Jr (1996) High affinity binding of beta-adrenergic receptor kinase to microsomal membranes. Modulation of the activity of bound kinase by heterotrimeric G protein activation. J Biol Chem 271: 985–994 [DOI] [PubMed] [Google Scholar]
- Pan Y, Chen J (2003) MDM2 promotes ubiquitination and degradation of MDMX. Mol Cell Biol 23: 5113–5121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penela P, Elorza A, Sarnago S, Mayor F Jr (2001) Beta-arrestin- and c-Src-dependent degradation of G-protein-coupled receptor kinase 2. EMBO J 20: 5129–5138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penela P, Murga C, Ribas C, Tutor AS, Peregrin S, Mayor F Jr (2006) Mechanisms of regulation of G-protein-coupled receptor kinases (GRKs) and cardiovascular disease. Cardiovasc Res 69: 46–56 [DOI] [PubMed] [Google Scholar]
- Penela P, Ribas C, Mayor F Jr (2003) Mechanisms of regulation of the expression and function of G protein-coupled receptor kinases. Cell Signal 15: 973–981 [DOI] [PubMed] [Google Scholar]
- Penela P, Ruiz-Gomez A, Castano JG, Mayor F Jr (1998) Degradation of the G protein-coupled receptor kinase 2 by the proteasome pathway. J Biol Chem 273: 35238–35244 [DOI] [PubMed] [Google Scholar]
- Pickart CM (2001) Mechanisms underlying ubiquitination. Annu Rev Biochem 70: 503–533 [DOI] [PubMed] [Google Scholar]
- Rodriguez MS, Desterro JM, Lain S, Lane DP, Hay RT (2000) Multiple C-terminal lysine residues target p53 for ubiquitin–proteasome-mediated degradation. Mol Cell Biol 20: 8458–8467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott MG, Le Rouzic E, Perianin A, Pierotti V, Enslen H, Benichou S, Marullo S, Benmerah A (2002) Differential nucleocytoplasmic shuttling of beta-arrestins. Characterization of a leucine-rich nuclear export signal in beta-arrestin2. J Biol Chem 277: 37693–37701 [DOI] [PubMed] [Google Scholar]
- Shenoy SK, Lefkowitz RJ (2003) Multifaceted roles of beta-arrestins in the regulation of seven-membrane-spanning receptor trafficking and signalling. Biochem J 375: 503–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy SK, Lefkowitz RJ (2005) Receptor-specific ubiquitination of beta-arrestin directs assembly and targeting of seven-transmembrane receptor signalosomes. J Biol Chem 280: 15315–15324 [DOI] [PubMed] [Google Scholar]
- Shenoy SK, McDonald PH, Kohout TA, Lefkowitz RJ (2001) Regulation of receptor fate by ubiquitination of activated beta 2-adrenergic receptor and beta-arrestin. Science 294: 1307–1313 [DOI] [PubMed] [Google Scholar]
- Song X, Raman D, Gurevich EV, Vishnivetskiy SA, Gurevich VV (2006) Visual and both non-visual arrestins in their inactive conformation bind JNK3 and Mdm2 and relocalize them from the nucleus to the cytoplasm. J Biol Chem 281: 21491–21499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taranto E, Xue JR, Lacey D, Hutchinson P, Smith M, Morand EF, Leech M (2005) Detection of the p53 regulator murine double-minute protein 2 in rheumatoid arthritis. J Rheumatol 32: 424–429 [PubMed] [Google Scholar]
- Uchida C, Miwa S, Kitagawa K, Hattori T, Isobe T, Otani S, Oda T, Sugimura H, Kamijo T, Ookawa K, Yasuda H, Kitagawa M (2005) Enhanced Mdm2 activity inhibits pRB function via ubiquitin-dependent degradation. EMBO J 24: 160–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vroon A, Kavelaars A, Limmroth V, Lombardi MS, Goebel MU, Van Dam AM, Caron MG, Schedlowski M, Heijnen CJ (2005) G protein-coupled receptor kinase 2 in multiple sclerosis and experimental autoimmune encephalomyelitis. J Immunol 174: 4400–4406 [DOI] [PubMed] [Google Scholar]
- Wang P, Gao H, Ni Y, Wang B, Wu Y, Ji L, Qin L, Ma L, Pei G (2003) Beta-arrestin 2 functions as a G-protein-coupled receptor-activated regulator of oncoprotein Mdm2. J Biol Chem 278: 6363–6370 [DOI] [PubMed] [Google Scholar]
- Weissman AM (2001) Themes and variations on ubiquitylation. Nat Rev Mol Cell Biol 2: 169–178 [DOI] [PubMed] [Google Scholar]
- Yang Y, Li CC, Weissman AM (2004) Regulating the p53 system through ubiquitination. Oncogene 23: 2096–2106 [DOI] [PubMed] [Google Scholar]
- Zhou BP, Liao Y, Xia W, Zou Y, Spohn B, Hung MC (2001) HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2 phosphorylation. Nat Cell Biol 3: 973–982 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information