

Abstract
Several of the inhibitor of apoptosis protein (IAP) family members regulate apoptosis in response to various cellular assaults. Some members are also involved in cell signalling, mitosis and targeting proteins to the ubiquitin-proteasome degradation machinery. The most intensively studied family member, X-linked IAP (XIAP), is a potent inhibitor of caspase activity; hence, it is generally assumed that direct caspase inhibition is an important conserved function of most members of the family. Biochemical and structural studies have precisely mapped the elements of XIAP required for caspase inhibition. Intriguingly, these elements are not conserved among IAPs. Here, we review current knowledge of the caspase-inhibitory potential of the human IAPs and show that XIAP is probably the only bona fide caspase inhibitor, suggesting that the other family members never gained the ability to directly inhibit caspase activity.
Keywords: apoptosis, BIR, caspase, exosite, protease
Introduction
The importance of proteolytic signalling pathways in human physiology was first appreciated after the characterization of the coagulation pathway. Proteolytic enzymes transmit signals by hydrolysing the peptide bond between two amino acids. This proteolysis can either initiate signalling by activating a protein, or halt it by inactivating a crucial participant in a given pathway. Unlike kinase and phosphatase pathways, protease signalling is irreversible. For this reason, many points of regulation have evolved to keep proteases silent until their activity is required. For example, in the coagulation pathway, the latent inactive protease zymogens of blood-clotting factors are proteolytically activated in a cascade-like manner. This pathway can also be suppressed at various points by specific protease inhibitors.
It took more than 50 years to identify and characterize the coagulation pathway. Due to massive technological advances in the post-genomic era, it has taken just over 10 years to gain an equivalent understanding of the proteolytic cascade that dismantles cells during apoptosis, through which damaged or unwanted cells are neatly packaged into small membrane-bound fragments for engulfment and disposal by neighbouring phagocytes.
At the heart of the apoptosis pathway are the caspases (formally designated as members of the C14 family of clan CD cysteine proteases), which are so named because of their stringent specificity for cleaving substrates after aspartic-acid residues (Alnemri et al, 1996). There are 11 human caspases, and those that have a clear role in apoptosis can be divided into those involved in the initiation (caspase-2, caspase-8, caspase-9 and caspase-10) or the execution (caspase-3, caspase-6 and caspase-7) phase of apoptosis (Fuentes-Prior & Salvesen, 2004).
The initiator caspases are the first to be activated, translating a death-inducing signal into proteolytic activity. Similar to the coagulation cascade, this initial activation event is not proteolysis itself, but rather the recruitment of a caspase to an activating protein, or platform of proteins. On these activation platforms, the initiator caspase zymogens are activated through dimerization, and subsequent autocatalytic processing is required for stabilization of the catalytically competent initiator caspase dimer (Boatright et al, 2003).
In humans, there are two main pathways to initiator caspase activation. The ‘intrinsic pathway' senses intracellular damage and growth factor or cytokine availability, and activates caspase-9 at the apoptosome (Jiang & Wang, 2004). The ‘extrinsic pathway' is a sensor for extracellular signals, and activates caspase-8 through the ligation of tumour necrosis factor transmembrane-receptor family members to form the death-inducing signalling complex (Debatin & Krammer, 2004). Although activated by different cues, caspase-8 and caspase-9 converge on the executioner caspases, caspase-3 and caspase-7, which exist as inactive dimeric zymogens in resting cells (Fuentes-Prior & Salvesen, 2004). Using a mechanism reminiscent of classical protease activation, the executioner caspase zymogens are cleaved in their interchain linker and, once active, are responsible for most of the proteolytic events that destroy the cell (Fischer et al, 2003).
Viruses have learned that interfering with apoptotic caspases is a good way to keep the host cell alive long enough for viral replication to occur. For example, p35 and p49 from baculovirus are potent inhibitors of most caspases (Bump et al, 1995; Jabbour et al, 2002; Ryan et al, 2002; Zhou et al, 1998). Orthopox viruses contain a serpin, cytokine response-modifier A (CrmA), which also directly inhibits several caspases (Kamada et al, 1997; Zhou et al, 1997). Although they have no structural similarity, CrmA and p35 both inactivate their cognate proteases in a mechanism-based manner (Stennicke et al, 2002). The reactive-site loop binds to the active site of the protease and is cleaved, thereby inducing a conformational change that irreversibly locks the protease in an inactive conformation.
In principle, there are two ways to terminate the activity of a caspase: first, remove it from the cell by using the ubiquitin-targeted proteasome degradation machinery; or second, directly inhibit its enzymatic activity. Intriguingly, there is evidence that members of the inhibitor of apoptosis protein (IAP) family (Fig 1) are capable of both functions (Riedl & Shi, 2004; Salvesen & Duckett, 2002; Vaux & Silke, 2005a). The evolutionarily conserved mechanism seems to be caspase removal. The ubiquitin-targeting function of Drosophila melanogaster IAP (DIAP1) is essential for apoptosis inhibition in vivo, whereas in vitro it only weakly inhibits D. melanogaster caspase activity (Goyal et al, 2000; Hawkins et al, 1999; Lisi et al, 2000; Yan et al, 2004). By contrast, the most studied of the mammalian IAPs, X-linked IAP (XIAP), has evolved to potently inhibit the enzymatic activity of mammalian caspases at both the initiation phase (caspase-9) and the execution phase (caspase-3 and caspase-7) of apoptosis (Deveraux et al, 1997). There is some evidence that XIAP might participate in caspase polyubiquitination and monoubiquitination in vitro; however, this function is yet to be confirmed in vivo with endogenous proteins. Moreover, XIAP-directed caspase ubiquitination might not result in degradation by the proteasome (Creagh et al, 2004; Hao et al, 2004; Morizane et al, 2005; Suzuki et al, 2001). In this review, we discuss recent structural and mechanistic studies indicating that XIAP is the only mammalian IAP that has adopted the ability to directly inhibit the enzymatic activity of caspases.
Figure 1.
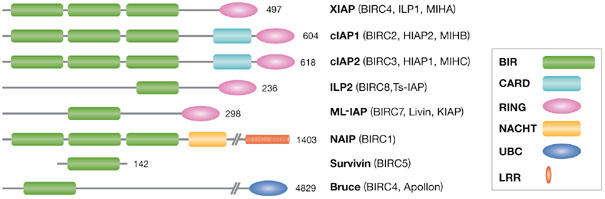
Schematic representation of the human inhibitor of apoptosis protein family. In addition to at least one baculoviral IAP repeat (BIR) domain, most IAPs have other distinct functional domains. The really interesting new gene (RING) domain found in many IAPs is an E3 ligase that presumably directs targets to the ubiquitin-proteasome degradation system. Caspase-recruitment domains (CARDs) can mediate homotypic protein–protein interactions, although the binding partners for the cellular IAP (cIAP) CARDs have not yet been elucidated. Bruce has a ubiquitin-conjugation (UBC) domain that is found in many ubiquitin-conjugating enzymes. The NACHT domain of neuronal apoptosis-inhibitory protein (NAIP) resembles a nucleotide-oligomerization domain related to the AAA+ NTPases, whereas the leucine-rich repeats (LRRs) are similar to those of the Toll-like receptors that function as pathogen sensors. BIRC, baculoviral IAP-repeat-containing; HIAP, human IAP; IAP, inhibitor of apoptosis protein; ILP, IAP-like protein; KIAP, kidney IAP; MIH, mammalian IAP homologue; ML-IAP, melanoma IAP; NACHT, domain found in NAIP, CIITA, HET-E and TP-1; Ts-IAP, testicular IAP; XIAP, X-linked IAP.
XIAP: a potent natural caspase inhibitor
Biochemical and structural analyses have precisely mapped the elements in XIAP that are required for the inhibition of caspase activity. Initial biochemical studies determined that a region encompassing the second baculoviral IAP repeat (BIR) domain (BIR2) inhibits caspase-3 and caspase-7, whereas the third BIR domain (BIR3) inhibits caspase-9 (Deveraux et al, 1999). Although these BIR domains share close to 40% identity, structural analyses revealed what initially seemed to be strikingly different methods for inhibiting proteases of the same mechanistic family. However, further biochemical dissection has shown that there are, in fact, some shared elements.
Both BIR domains use a two-site binding mechanism for potent caspase inhibition. One of these sites is a conserved surface groove found in BIR2 and BIR3 of XIAP, and in other BIR domains (Fig 2A,D). We have defined this site as the IAP-binding motif (IBM)-interacting groove, because of its specificity for proteins containing an IBM at their immediate amino terminus (Scott et al, 2005). These include the pro-apoptotic mammalian IAP antagonists Smac/DIABLO and HtrA2, and the Drosophila proteins Hid, Grim and Reaper (Vaux & Silke, 2003). Although no structures are known for the BIR1 domain of XIAP, sequence constraints indicate that the IBM-interacting groove is absent from this domain (Fig 2A).
Figure 2.
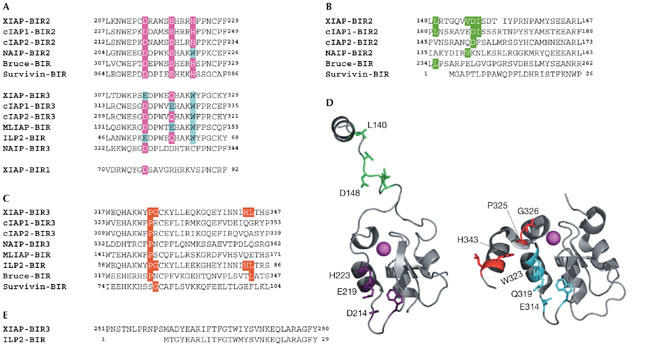
The caspase-binding elements of human inhibitor of apoptosis proteins. Alignment of baculoviral IAP repeat (BIR) domains with the highest identity. (A) IAP-binding motif (IBM)-interacting groove exosite. The residues that line this groove are highlighted; those identical to XIAP BIR2 or XIAP BIR3 are shown in purple and cyan, respectively. The BIR3 of cIAPs, the BIR2 of neuronal apoptosis-inhibitory protein (NAIP) and the single BIR of ML-IAP share some features of both BIR2 and BIR3 of XIAP. (B) Executioner caspase-inhibitory peptide strand of XIAP that lies at the amino terminus of the BIR2 domain. Crucial residues are shown in green. (C) The helix distal to XIAP BIR3 targets the dimer interface of caspase-9. Crucial residues are shown in red. (D) Crystal structure of BIR2 (right, from Protein Data Bank (PDB) 1I3O) and BIR3 (left, from PDB 1NW9) from XIAP. The executioner caspase-inhibitory element is shown in green, whereas the caspase-9-inhibitory distal helix is shown in red. The IBM-interacting grooves of BIR2 and BIR3 are shown in purple and cyan, respectively. (E) Alignment of XIAP BIR3 with ILP2 showing the truncated amino terminus of the latter. cIAP, cellular IAP; IAP, inhibitor of apoptosis protein; ILP2, IAP-like protein 2; ML-IAP, melanoma IAP; XIAP, X-linked IAP.
On activation, caspases are proteolytically processed between their large and small subunits as a result of either autocatalytic processing following dimerization or the activity of upstream proteases. For caspase-3, caspase-7 and caspase-9, the newly generated small-subunit amino terminus constitutes an IBM. To achieve potent inhibition, the BIR domain binds to at least the newly generated IBM. For the caspase-9 and BIR3 complex, this interaction was first suggested by biochemical studies (Srinivasula et al, 2001) and later observed in a crystal structure of the complex (Shiozaki et al, 2003). However, although it was appreciated that the BIR2 domain itself contributed to caspase-3 and caspase-7 inhibition (Huang et al, 2001; Sun et al, 1999), three independently derived crystal structures of the caspase-7/BIR2 complex failed to reveal any electron density for the BIR2 domain (Chai et al, 2001; Huang et al, 2001). By contrast, the caspase-3/BIR2 complex not only showed the entire BIR2 domain, but also revealed that the caspase small-subunit amino terminus docks in the IBM-interacting groove (Riedl et al, 2001). Although this interaction probably results from crystal packing, it is nevertheless required for potent caspase-3 and caspase-7 inhibition in solution (Scott et al, 2005). Therefore, the IBM-interacting groove of the BIR domain functions as an exosite, which is a term used for a functionally important region outside the active site of the enzyme. In a similar manner to the carboxy-terminal tail of hirudin binding to exosite I of the coagulation protease thrombin, the IBM-interacting groove acts as an anchor, strengthening inhibitor binding (Fig 3; Rydel et al, 1990).
Figure 3.
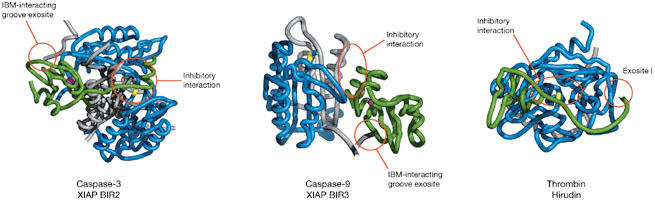
Two-site binding mechanisms for protease inhibition. The colour coding is as follows: the inhibitors are shown in green with the co-ordinating zinc in purple, the protease large subunit is shown in blue with the catalytic residue in yellow and the small subunit is shown in grey. (A) X-linked IAP (XIAP) baculoviral IAP repeat 2 (BIR2) complex with caspase-3 (Protein Data Bank (PDB) 1I3O). The amino-terminal peptide strand preceding the BIR2 domain binds across the substrate-binding site, whereas the IAP-binding motif (IBM) of a caspase-3 small subunit, in an adjacent asymmetric unit, docks in the IBM-interacting groove. The mechanism of inhibition is steric occlusion of the active site. (B) XIAP BIR3 complex with a caspase-9 monomer (PDB 1NW9). The IBM of caspase-9 docks into the IBM-interacting groove of BIR3, facilitating interactions of the carboxy-terminal BIR3 helix with the dimer interface of caspase-9. This prevents dimerization or induces monomerization of caspase-9 with concomitant collapse of the active site. (C) Hirudin complex with thrombin (PDB 1HRT). The substrate-binding site is blocked, whereas the carboxy terminus docks to exosite I of thrombin.
Although this anchoring interaction in BIR2 and BIR3 of XIAP is conserved, the mechanism of enzyme inhibition is not. Early biochemical studies identified residues in XIAP that are crucial for caspase-3 inhibition (Fig 2B,D). Surprisingly, these residues are not in the BIR2 domain, but rather in the preceding peptide strand, with Leu 141 and Asp 148 being important for tight inhibition (Sun et al, 1999). After structural data became available, the importance of these residues was clear. All structures of a BIR2/caspase complex show that this peptide strand (residues 140–156) stretches across the catalytic-binding cleft of the enzyme (Fig 3). Asp 148 forms hydrogen bonds, whereas Leu 141 makes hydrophobic contacts with the caspase. Thus, tight inhibition of the executioner caspases, caspase-3 and caspase-7, by XIAP requires two surfaces: an exosite-anchoring motif and an active site-directed interaction.
Although targeting the active site is reminiscent of a standard ‘lock and key' inhibitor mechanism, this is not the case for BIR2 and executioner caspases. If we compare caspase-3 inhibition by BIR2 with synthetic peptidyl inhibitors, such as DEVD-CHO, there are significant differences. Peptidyl inhibitors bind similarly to a substrate, occupying the specificity recognition sites of the protease. They exploit the catalytic function of the protease to trap the bound state, and act as covalent modifiers or transition-state analogues. Although the BIR2 peptide strand binds across the substrate-binding cleft, and Asp 148 occupies the S4-binding pocket, the S1, S2 and S3 pockets of the caspase remain unoccupied. In addition, all potential carbonyls are too far from the catalytic cysteine for nucleophilic attack. Equally important, the peptide strand binds in a ‘back to front' orientation. This unusual reverse-binding mechanism has been observed for members of the papain family of proteases (clan CA, family C1A). Here, the zymogen has a fully formed active site, but the propeptide of the enzyme spans the substrate-binding cleft in the reverse direction, keeping the enzyme silent until it reaches the lysosome (Coulombe et al, 1996). Cytotoxic T-lymphocyte antigen 2, which has sequence similarity with the propeptides of some papain-like enzymes, might inhibit cathepsin L through the same mechanism (Kurata et al, 2003).
As opposed to targeting the enzyme active site directly, the BIR3 domain of XIAP abolishes activity using a fundamentally different mechanism. The functional inhibitory surface—separate from the IBM-interacting groove—is a helix found immediately after the BIR3 domain (Fig 2C,D). Mutagenesis identified His 343 as crucial for caspase-9 inhibition (Sun et al, 2000). Structural analysis of a BIR3/caspase-9 complex (Fig 3) showed that this distal helix packs against the dimer interface and monomerizes caspase-9, forcing the protease into an inactive conformation with a collapsed active site (Shiozaki et al, 2003). Active caspases are obligate dimers, so this reverses the mechanism of caspase-9 activation. Another possibility is that BIR3 binds to monomeric caspase-9 that has been adventitiously cleaved, but not activated, through its proteolytically generated small-subunit IBM. This would facilitate the interaction at the BIR3 distal helix/caspase-9 interface, preventing dimerization and activation. Nonetheless, XIAP is the first example of a natural protease inhibitor that uses this kind of allosteric mechanism.
Are the other mammalian IAPs caspase inhibitors?
As a result of these studies on XIAP, IAPs have become attractive targets for drug design for the treatment of chronic and acute diseases that have a cell-death component (Schimmer & Dalili, 2005). So, are all IAPs caspase inhibitors?
Cellular IAP1 (cIAP1) and cIAP2 consist of three BIR domains and a really interesting new gene (RING) domain, and are the closest paralogues of XIAP (Fig 1). Similar to XIAP, overexpression of cIAPs protects cells from apoptosis. Together with their high sequence and domain conservation, it has been assumed that cIAPs are also caspase inhibitors and are redundant with XIAP. Indeed, one study found that mice deficient in XIAP showed elevated levels of cIAP1 and cIAP2 proteins (Harlin et al, 2001), although another independently derived line did not (Olayioye et al, 2005).
The BIR domains of cIAPs contain IBM-interacting grooves that are highly conserved with those in XIAP (Fig 2A). Furthermore, the peptide strand preceding BIR2 of cIAP1 has some of the residues that are crucial for XIAP-mediated inhibition of the executioner caspases. cIAP2 has the crucial Asp 153, but other residues in this peptide strand are less conserved (Fig 2B). Experimentally, recombinant cIAP BIR2 fragments that encompass the preceding peptide strand have no executioner caspase-inhibitory activity (Eckelman & Salvesen, 2005; Table 1). However, replacing the cIAP1 strand with that of XIAP can restore potent executioner caspase inhibition. Regarding caspase-9, the BIR3 domain of both cIAPs has only one of four dimer interface-interacting residues (Fig 2C), and so neither cIAP can demolish caspase-9 activity. It should be noted that cIAPs can bind caspases in vitro, in an active-site-independent manner, although the significance of this has yet to be established (Eckelman & Salvesen, 2005; Tenev et al, 2005). Consequently, although the cIAPs contain a caspase-binding scaffold, they either lost or never gained the specific residues required for caspase inhibition.
Table 1.
Ki values for mammalian baculoviral inhibitor of apoptosis repeat domains against human caspases
| Ki (nM) | Caspase-3 | Caspase-7 | Caspase-9 |
|---|---|---|---|
| XIAP full length | <0.8a,b | <0.07a | 210b |
| XIAP BIR2c | 0.7 | 0.2 | NI |
| XIAP BIR3d | NI | NI | 10 |
| GST-cIAP1 full lengthe | 108 | 42 | ND |
| cIAP1 full lengthf | >2,000 (NI)b,g | >2,000 | >2,000 |
| cIAP1 BIR2f | >10,000 | >10,000 | NI |
| X-cIAP1-BIR2f,h | 19 | <0.2 | NI |
| cIAP1 BIR3f | NI | NI | >5,000 |
| cIAP1 BIR3 (R342G/Q349H/G350L)f,i | NI | NI | <80 |
| GST-cIAP2 full lengthe | 35 | 29 | ND |
| cIAP2 full lengthb,g | NI | ND | ND |
| cIAP2 BIR2f | >5,000 | >5,000 | NI |
| cIAP2 BIR3f | NI | NI | >5,000 |
| ML-IAP BIRj | ND | ND | 3,200 |
| ML-IAP-Q (S150G/Q167H/E168L)j,k | ND | ND | <0.01 |
| GST-ILP2 full lengthl | NI | NI | 752 |
| ILP2 BIRl | NI | NI | 472 |
| GST-NAIP BIR123m | 14 | 50 | ND |
| GST-NAIP BIR3 | 185m | ND | 33n |
| NAIP BIR2o | NI | NI | NI |
ND, not determined; NI, not inhibited.
aScott et al, 2005.
bHalf-maximal inhibitory concentration (IC50; Silke et al, 2001).
dIC50 (Sun et al, 2000).
fKi apparent (Eckelman & Salvesen, 2005).
gIn vitro toxicity assay in yeast.
hcIAP1 BIR2 domain with the caspase-3 and caspase-7 binding elements of XIAP fused to the amino terminus.
iCrucial residues are mutated to those found in XIAP BIR3 IAP-binding motif (IBM)-interacting groove.
jKi apparent (Vucic et al, 2005).
kML-IAP truncated at the carboxy terminus to Gln 172 with mutations in the IBM-interacting groove to those found in XIAP BIR3.
oB.P.E., unpublished results. All recombinant proteins that did not contain a glutathione-S-transferase (GST) tag contained a histidine tag or no tag.
BIR, baculoviral IAP repeat domains; cIAP, cellular IAP; IAP, inhibitor of apoptosis; ILP2, IAP-like protein 2; ML-IAP, melanoma IAP; NAIP, neuronal apoptosis-inhibitory protein; XIAP, X-linked IAP.
On the basis of structural requirements, a BIR must contain both of the caspase-binding elements that are found in XIAP—the IBM-interacting groove exosite and an inhibitory element—to inhibit caspase activity. We therefore predict that the other mammalian IAPs are not caspase inhibitors (Fig 2). Vucic and colleagues showed that melanoma IAP (ML-IAP) is not a tight inhibitor of caspase-9 (Table 1), and elegantly demonstrated that the mutation of three residues in the distal helix to those found in XIAP endowed ML-IAP with potent caspase-9-inhibitory function (Vucic et al, 2005). For neuronal apoptosis-inhibitory protein (NAIP) BIR2, Lys 143 replaces Asp 148 of XIAP (Fig 2B). This drastic substitution should preclude the inhibition of executioner caspases, and, on the basis of sequence-conservation arguments, the third BIR domain might not even contain an IBM-interacting groove. Shin and colleagues have shown that although IAP-like protein 2 (ILP2) contains all of the surface elements required for caspase-9 inhibition, the BIR domain is an intrinsically unstable domain owing to a natural amino-terminal truncation (Fig 2E) and is not a caspase inhibitor (Shin et al, 2005). There is still a possibility that ILP2 requires a binding partner to stabilize the polypeptide and to promote caspase inhibition. However, ILP2 messenger RNA has only been detected in testis, and the ILP2 protein has yet to be detected in vivo. Survivin does not directly inhibit caspases; however, together with hepatitis B X-interacting protein (HBXIP), this IAP might prevent caspase-9 activation at the apoptosome (Marusawa et al, 2003). Bruce might also prevent caspase-9 activation, although the mechanism is unclear (Qiu & Goldberg, 2005).
The problem with glutathione-S-transferase tags
On the basis of structural requirements, it seems clear that no other human IAP contains all the elements required for inhibition of caspase activity. However, in addition to the studies discussed above, others have suggested that some IAPs can inhibit caspases. A possible explanation for these conflicting results is the method used to purify the respective IAP protein. A recent study investigated the effect of the glutathione-S-transferase (GST)-purification tag on the caspase-inhibitory potential of XIAP. A chimeric protein containing the peptide strand of XIAP (residues 124–168) attached to green fluorescent protein in place of the BIR domain was fused to either a GST or a hexahistidine tag (Scott et al, 2005). As this chimaera has only the active site-occluding surface, and lacks the anchoring exosite interaction, this protein should be a poor caspase inhibitor. The chimaeras showed markedly different inhibitory properties depending on their oligomeric state. The hexahistidine-tagged chimaera was a monomer displaying weak inhibition of caspase-3, whereas the GST-tagged chimaera was a mixture of oligomeric species displaying 100-fold enhancement of inhibitory function. GST-XIAP BIR2 is also a mixture of oligomeric species, whereas XIAP BIR2 with a hexahistidine tag is a monomer in solution (F.L.S., unpublished data). Initial studies with GST-tagged cIAPs revealed a similar problem, with apparent inhibition of caspase-3 and caspase-7, albeit two to three orders of magnitude weaker than XIAP (Table 1). GST-tagged BIR domains from ML-IAP and NAIP were suggested to be potent caspase inhibitors (Davoodi et al, 2004; Maier et al, 2002; Vucic et al, 2000); however, more recent studies with hexahistidine-tagged BIRs have produced results that agree with structural and biochemical predictions. We eagerly await kinetic analyses of non-oligomeric versions of NAIP BIR domains to test their inhibitory properties, with preliminary results indicating that they are incompetent (Table 1). Consequently, the artefactual inhibition observed in IAPs that are fused to GST can be attributed to the generation of oligomeric species, warning against using GST constructs to characterize enzyme inhibition. There is a possibility that IAPs might form larger oligomeric complexes that could mimic the artefactual effect of GST on caspase inhibition, although we are unaware of any evidence for this. There is some evidence that IAPs can form heterocomplexes in vitro, but these seem to influence the susceptibility of IAPs to proteasomal degradation, rather than caspase inhibition (Dohi et al, 2004; Silke et al, 2005).
Conclusion
The structural and biochemical requirements of caspase inhibition by XIAP have been thoroughly established. On the basis of these, it is probable that other human IAPs are not direct caspase inhibitors, unless they use a distinctly different mechanism. Earlier studies concluding that IAPs other than XIAP were direct caspase inhibitors have largely been revised, and it seems that XIAP is the only mammalian IAP that directly inhibits caspase activity. The mechanism by which other mammalian IAPs attenuate apoptosis is less clear and remains under intense investigation. One possibility is that IAPs bind proapoptotic molecules (including caspases and Smac/DIABLO, which is an IAP antagonist) through their IBM-interacting grooves, and target them for degradation by the ubiquitin-proteasome pathway (Vaux & Silke, 2005a, b). Targeting could occur through their own RING domains or by forming complexes with other RING-containing IAPs. There is also some shared topography between the IBM-interacting grooves of different BIR domains in different IAPs, suggesting some overlap in binding specificity. Finally, the formation of IAP heterocomplexes might influence their stability and, hence, their ability to regulate apoptosis (Silke et al, 2005).
From an evolutionary perspective, it seems that XIAP has diverged from the other IAPs and acquired the ability to inhibit caspase activity directly. The implications are that XIAP is the first line of defence against inappropriate caspase activity, placing it at an important node in the mammalian apoptotic pathway. However, this assignment is contentious considering the fact that the XIAP-null mice have yet to display a significant phenotype (Harlin et al, 2001; Olayioye et al, 2005). It is therefore tempting to suggest that there is an evolutionarily conserved ‘back-up system' that takes over, at least during development. This back-up might be IAP-mediated targeting of activated caspases for proteasomal degradation. It is far from clear which mammalian IAP could perform this function. cIAP2 can monoubiquitinate executioner caspases in vitro, but this modification is not normally associated with proteasome targeting and degradation (Huang et al, 2000). Monoubiquitination might sequester active caspases away from death substrates, but this has yet to be shown. There is also some evidence that Bruce (Apollon) can bind caspase-3, caspase-7 and caspase-9, and can polyubiquitinate caspase-9, when ectopically expressed in vitro (Bartke et al, 2004; Hao et al, 2004; Qiu & Goldberg, 2005).
Given the recent interest in the development of therapeutic small molecules for targeting IAPs in disease, and the evidence that XIAP is probably the only bona fide caspase inhibitor, we look forward to studies that delineate the mechanisms used by other IAPs to protect cells from apoptosis both in vitro and in vivo (Schimmer & Dalili, 2005).

Brendan P. Eckelman, Fiona L. Scott & Guy S. Salvesen
Acknowledgments
We thank all members of the Salvesen laboratory and collaborators, both past and present, for insightful discussion and support. We apologize to our colleagues whose work we could not cite due to space limitations. This work was supported by National Institute of Health grants R01AG15402 and T32CA77109.
References
- Alnemri ES, Livingston DJ, Nicholson DW, Salvesen G, Thornberry NA, Wong WW, Yuan J (1996) Human ICE/CED-3 protease nomenclature. Cell 87: 171. [DOI] [PubMed] [Google Scholar]
- Bartke T, Pohl C, Pyrowolakis G, Jentsch S (2004) Dual role of BRUCE as an antiapoptotic IAP and a chimeric E2/E3 ubiquitin ligase. Mol Cell 14: 801–811 [DOI] [PubMed] [Google Scholar]
- Boatright KM et al. (2003) A unified model for apical caspase activation. Mol Cell 11: 529–541 [DOI] [PubMed] [Google Scholar]
- Bump NJ et al. (1995) Inhibition of ICE family proteases by baculovirus antiapoptotic protein p35. Science 269: 1885–1888 [DOI] [PubMed] [Google Scholar]
- Chai J, Shiozaki E, Srinivasula SM, Wu Q, Datta P, Alnemri ES, Shi Y (2001) Structural basis of caspase-7 inhibition by XIAP. Cell 104: 769–780 [DOI] [PubMed] [Google Scholar]
- Coulombe R, Grochulski P, Sivaraman J, Menard R, Mort JS, Cygler M (1996) Structure of human procathepsin L reveals the molecular basis of inhibition by the prosegment. EMBO J 15: 5492–5503 [PMC free article] [PubMed] [Google Scholar]
- Creagh EM, Murphy BM, Duriez PJ, Duckett CS, Martin SJ (2004) Smac/Diablo antagonizes ubiquitin ligase activity of inhibitor of apoptosis proteins. J Biol Chem 279: 26906–26914 [DOI] [PubMed] [Google Scholar]
- Davoodi J, Lin L, Kelly J, Liston P, MacKenzie AE (2004) Neuronal apoptosis-inhibitory protein does not interact with Smac and requires ATP to bind caspase-9. J Biol Chem 279: 40622–40628 [DOI] [PubMed] [Google Scholar]
- Debatin KM, Krammer PH (2004) Death receptors in chemotherapy and cancer. Oncogene 23: 2950–2966 [DOI] [PubMed] [Google Scholar]
- Deveraux Q, Takahashi R, Salvesen GS, Reed JC (1997) X-linked IAP is a direct inhibitor of cell death proteases. Nature 388: 300–304 [DOI] [PubMed] [Google Scholar]
- Deveraux QL, Leo E, Stennicke HR, Welsh K, Salvesen GS, Reed JC (1999) Cleavage of human inhibitor of apoptosis protein XIAP results in fragments with distinct specificities for caspases. EMBO J 18: 5242–5251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohi T et al. (2004) An IAP–IAP complex inhibits apoptosis. J Biol Chem 279: 34087–34090 [DOI] [PubMed] [Google Scholar]
- Eckelman BP, Salvesen GS (2006) The human anti-apoptotic proteins, cIAP1 and cIAP2 bind but do not inhibit caspases. J Biol Chem 281: 3254–3260 [DOI] [PubMed] [Google Scholar]
- Fischer U, Janicke RU, Schulze-Osthoff K (2003) Many cuts to ruin: a comprehensive update of caspase substrates. Cell Death Differ 10: 76–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentes-Prior P, Salvesen GS (2004) The protein structures that shape caspase activity, specificity, activation and inhibition. Biochem J 384: 201–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal L, McCall K, Agapite J, Hartwieg E, Steller H (2000) Induction of apoptosis by Drosophila Reaper, Hid and Grim through inhibition of IAP function. EMBO J 19: 589–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y et al. (2004) Apollon ubiquitinates SMAC and caspase-9, and has an essential cytoprotection function. Nat Cell Biol 6: 849–860 [DOI] [PubMed] [Google Scholar]
- Harlin H, Reffey SB, Duckett CS, Lindsten T, Thompson CB (2001) Characterization of XIAP-deficient mice. Mol Cell Biol 21: 3604–3608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins CJ, Wang SL, Hay BA (1999) A cloning method to identify caspases and their regulators in yeast: identification of Drosophila IAP1 as an inhibitor of the Drosophila caspase DCP-1. Proc Natl Acad Sci USA 96: 2885–2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Joazeiro CA, Bonfoco E, Kamada S, Leverson JD, Hunter T (2000) The inhibitor of apoptosis, cIAP2, functions as a ubiquitin-protein ligase and promotes in vitro monoubiquitination of caspases 3 and 7. J Biol Chem 275: 26661–26664 [DOI] [PubMed] [Google Scholar]
- Huang Y, Park YC, Rich RL, Segal D, Myszka DG, Wu H (2001) Structural basis of caspase inhibition by XIAP: differential roles of the linker versus the BIR domain. Cell 104: 781–790 [PubMed] [Google Scholar]
- Jabbour AM, Ekert PG, Coulson EJ, Knight MJ, Ashley DM, Hawkins CJ (2002) The p35 relative, p49, inhibits mammalian and Drosophila caspases including DRONC and protects against apoptosis. Cell Death Differ 9: 1311–1320 [DOI] [PubMed] [Google Scholar]
- Jiang X, Wang X (2004) Cytochrome C-mediated apoptosis. Annu Rev Biochem 73: 87–106 [DOI] [PubMed] [Google Scholar]
- Kamada S, Funahashi Y, Tsujimoto Y (1997) Caspase-4 and caspase-5, members of the ICE/CED-3 family of cysteine proteases, are CrmA-inhibitable proteases. Cell Death Differ 4: 473–478 [DOI] [PubMed] [Google Scholar]
- Kurata M, Hirata M, Watabe S, Miyake M, Takahashi SY, Yamamoto Y (2003) Expression, purification, and inhibitory activities of mouse cytotoxic T-lymphocyte antigen-2a. Protein Expr Purif 32: 119–125 [DOI] [PubMed] [Google Scholar]
- Lisi S, Mazzon I, White K (2000) Diverse domains of THREAD/DIAP1 are required to inhibit apoptosis induced by REAPER and HID in Drosophila. Genetics 154: 669–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier JK et al. (2002) The neuronal apoptosis inhibitory protein is a direct inhibitor of caspases 3 and 7. J Neurosci 22: 2035–2043 [DOI] [PMC free article] [PubMed]
- Marusawa H, Matsuzawa S, Welsh K, Zou H, Armstrong R, Tamm I, Reed JC (2003) HBXIP functions as a cofactor of survivin in apoptosis suppression. EMBO J 22: 2729–2740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morizane Y, Honda R, Fukami K, Yasuda H (2005) X-linked inhibitor of apoptosis functions as ubiquitin ligase toward mature caspase-9 and cytosolic Smac/DIABLO. J Biochem (Tokyo) 137: 125–132 [DOI] [PubMed] [Google Scholar]
- Olayioye MA, Kaufmann H, Pakusch M, Vaux DL, Lindeman GJ, Visvader JE (2005) XIAP-deficiency leads to delayed lobuloalveolar development in the mammary gland. Cell Death Differ 12: 87–90 [DOI] [PubMed] [Google Scholar]
- Qiu XB, Goldberg AL (2005) The membrane-associated inhibitor of apoptosis protein, BRUCE/Apollon, antagonizes both the precursor and mature forms of Smac and caspase-9. J Biol Chem 280: 174–182 [DOI] [PubMed] [Google Scholar]
- Riedl SJ, Shi Y (2004) Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol Cell Biol 5: 897–907 [DOI] [PubMed] [Google Scholar]
- Riedl SJ, Renatus M, Schwarzenbacher R, Zhou Q, Sun S, Fesik SW, Liddington RC, Salvesen GS (2001) Structural basis for the inhibition of caspase-3 by XIAP. Cell 104: 791–800 [DOI] [PubMed] [Google Scholar]
- Roy N, Deveraux QL, Takahashi R, Salvesen GS, Reed JC (1997) The c-IAP-1 and c-IAP-2 proteins are direct inhibitors of specific caspases. EMBO J 16: 6914–6925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan CA, Stennicke HR, Nava VE, Lewis J, Hardwick JM, Salvesen GS (2002) Inhibitor specificity of recombinant and endogenous caspase 9. Biochem J 366: 595–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rydel TJ, Ravichandran KG, Tulinsky A, Bode W, Huber R, Roitsch C, Fenton JW (1990) The structure of a complex of recombinant hirudin and human α-thrombin. Science 249: 277–280 [DOI] [PubMed] [Google Scholar]
- Salvesen GS, Duckett CS (2002) IAP proteins: blocking the road to death's door. Nat Rev Mol Cell Biol 3: 401–410 [DOI] [PubMed] [Google Scholar]
- Schimmer AD, Dalili S (2005) Targeting the IAP family of caspase inhibitors as an emerging therapeutic strategy. Hematology (Am Soc Hematol Educ Program) 1: 215–219 [DOI] [PubMed] [Google Scholar]
- Scott FL, Denault JB, Riedl SJ, Shin H, Renatus M, Salvesen GS (2005) XIAP inhibits caspase-3 and -7 using two binding sites: evolutionarily conserved mechanism of IAPs. EMBO J 24: 645–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin H, Renatus M, Eckelman BP, Nunes VA, Sampaio CA, Salvesen GS (2005) The BIR domain of IAP-like protein 2 is conformationally unstable: implications for caspase inhibition. Biochem J 385: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiozaki EN, Chai J, Rigotti DJ, Riedl SJ, Li P, Srinivasula SM, Alnemri ES, Fairman R, Shi Y (2003) Mechanism of XIAP-mediated inhibition of caspase-9. Mol Cell 11: 519–527 [DOI] [PubMed] [Google Scholar]
- Silke J, Ekert PG, Day CL, Hawkins CJ, Baca M, Chew J, Pakusch M, Verhagen AM, Vaux DL (2001) Direct inhibition of caspase 3 is dispensable for the anti-apoptotic activity of XIAP. EMBO J 20: 3114–3123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silke J, Kratina T, Chu D, Ekert PG, Day CL, Pakusch M, Huang DC, Vaux DL (2005) Determination of cell survival by RING-mediated regulation of inhibitor of apoptosis (IAP) protein abundance. Proc Natl Acad Sci USA 102: 16182–16187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasula SM et al. (2001) A conserved XIAP-interaction motif in caspase-9 and Smac/DIABLO regulates caspase activity and apoptosis. Nature 410: 112–116 [DOI] [PubMed] [Google Scholar]
- Stennicke HR, Ryan CA, Salvesen GS (2002) Reprieval from execution: the molecular basis of caspase inhibition. Trends Biochem Sci 27: 94–101 [DOI] [PubMed] [Google Scholar]
- Sun C et al. (1999) NMR structure and mutagenesis of the inhibitor-of-apoptosis protein XIAP. Nature 401: 818–822 [DOI] [PubMed] [Google Scholar]
- Sun C, Cai M, Meadows RP, Xu N, Gunasekera AH, Herrmann J, Wu JC, Fesik SW (2000) NMR structure and mutagenesis of the third BIR domain of the inhibitor of apoptosis protein XIAP. J Biol Chem 275: 33777–33781 [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Nakabayashi Y, Takahashi R (2001) Ubiquitin-protein ligase activity of X-linked inhibitor of apoptosis protein promotes proteasomal degradation of caspase-3 and enhances its anti-apoptotic effect in Fas-induced cell death. Proc Natl Acad Sci USA 98: 8662–8667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenev T, Zachariou A, Wilson R, Ditzel M, Meier P (2005) IAPs are functionally non-equivalent and regulate effector caspases through distinct mechanisms. Nat Cell Biol 7: 70–77 [DOI] [PubMed] [Google Scholar]
- Vaux DL, Silke J (2003) Mammalian mitochondrial IAP binding proteins. Biochem Biophys Res Commun 304: 499–504 [DOI] [PubMed] [Google Scholar]
- Vaux DL, Silke J (2005) IAPs, RINGs and ubiquitylation. Nat Rev Mol Cell Biol 6: 287–297 [DOI] [PubMed] [Google Scholar]
- Vaux DL, Silke J (2005) IAPs—the ubiquitin connection. Cell Death Differ 12: 1205–1207 [DOI] [PubMed] [Google Scholar]
- Vucic D, Stennicke HR, Pisabarro MT, Salvesen GS, Dixit VM (2000) ML-IAP, a novel inhibitor of apoptosis that is preferentially expressed in human melanomas. Curr Biol 10: 1359–1366 [DOI] [PubMed] [Google Scholar]
- Vucic D et al. (2005) Engineering ML-IAP to produce an extraordinarily potent caspase 9 inhibitor: implications for Smac-dependent anti-apoptotic activity of ML-IAP. Biochem J 385: 11–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan N, Wu JW, Chai J, Li W, Shi Y (2004) Molecular mechanisms of DrICE inhibition by DIAP1 and removal of inhibition by Reaper, Hid and Grim. Nat Struct Mol Biol 11: 420–428 [DOI] [PubMed] [Google Scholar]
- Zhou Q, Snipas S, Orth K, Dixit VM, Salvesen GS (1997) Target protease specificity of the viral serpin CrmA: analysis of five caspases. J Biol Chem 273: 7797–7800 [DOI] [PubMed] [Google Scholar]
- Zhou Q, Krebs JF, Snipas SJ, Price A, Alnemri ES, Tomaselli KJ, Salvesen GS (1998) Interaction of the baculovirus anti-apoptotic protein p35 with caspases: specificity, kinetics, and characterization of the caspase/p35 complex. Biochemistry 37: 10757–10765 [DOI] [PubMed] [Google Scholar]