

Abstract
Since the cloning of Aequorea victoria green fluorescent protein (GFP) in 1992, a family of known GFP-like proteins has been growing rapidly. Today, it includes more than a hundred proteins with different spectral characteristics cloned from Cnidaria species. For some of these proteins, crystal structures have been solved, showing diversity in chromophore modifications and conformational states. However, we are still far from a complete understanding of the origin, functions and evolution of the GFP family. Novel proteins of the family were recently cloned from evolutionarily distant marine Copepoda species, phylum Arthropoda, demonstrating an extremely rapid generation of fluorescent signal. Here, we have generated a non-aggregating mutant of Copepoda fluorescent protein and solved its high-resolution crystal structure. It was found that the protein β-barrel contains a pore, leading to the chromophore. Using site-directed mutagenesis, we showed that this feature is critical for the fast maturation of the chromophore.
Keywords: GFP, Arthropoda, TurboGFP, structure, chromophore maturation
Introduction
Protein-related fluorescence in hydromedusa Aequorea victoria was first studied more than 40 years ago (Johnson et al, 1962). Cloning (Prasher et al, 1992) and characterization of A. victoria green fluorescent protein (GFP) provided the scientific community with a protein capable of visible-spectrum fluorescence without the requirement of exogenous cofactors. Subsequently, GFP and its derivatives have been extensively used as non-invasive, in situ and in vivo markers (Lippincott-Schwartz & Patterson, 2003; Chudakov et al, 2005). In 1996, the crystal structure of GFP was solved (Ormo et al, 1996; Yang et al, 1996), which spurred the development of scientifically useful fluorescent variants.
Divergent evolution doctrine suggests that other fluorescent organisms are likely to contain GFP homologues. Indeed, more than 120 fluorescent and coloured GFP-like proteins were cloned from nearly 70 Cnidaria species (Labas et al, 2002; Matz et al, 2006). The similarity of these proteins to GFP ranges from 80–90% to less than 25% identity in amino-acid sequence, but all these proteins share the same β-barrel fold. One would expect GFP-like proteins to be found in organisms that are evolutionarily close to Cnidaria. However, surprisingly, we have recently reported several fluorescent proteins from evolutionarily distant Copepoda species (Arthropoda: Crustacea: Maxillopoda: Copepoda: Pontellidae; Shagin et al, 2004). The biological role for Pontellidae fluorescence is unclear. These organisms are not luminescent, which eliminates the need for the fluorescent protein as a chromatic shift agent. Presumably, visual mate recognition is important, as Pontellidae have elaborate eyes and show sexual dimorphism in eye design (Ohtsuka & Huys, 2001).
Some of the Copepoda GFPs, such as ppluGFP2 from Pontellina plumata (GenBank accession number AY268072), showed extremely rapid development of bright green fluorescent signal when expressed in Escherichia coli and mammalian cells (HeLa, mouse embryonic fibroblasts 3T3 and human embryonic kidney 293T). Solving the crystal structure of ppluGFP2 would increase understanding of this phenomenon. As Copepoda GFPs are prone to form aggregates, protein purification and crystallization experiments have been hindered. Overexpression of these proteins in mammalian cells also leads to formation of microcrystals that eventually rupture membranes, which limits their applicability as biological tags (Fig 1A).
Figure 1.
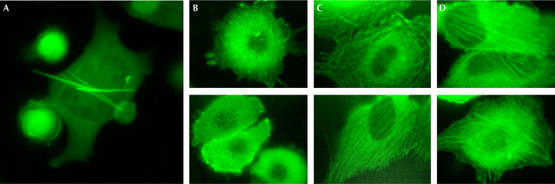
Confocal microscopy of fluorescent protein expression in HeLa cells. (A) ppluGFP2 (green fluorescent protein from Pontellina plumata) forms needles 3 days after transfection. (B) ppluGFP2–β-actin fusion protein. (C) TurboGFP–β-actin fusion protein. (D) Enhanced green fluorescent protein–β-actin fusion protein.
To overcome these problems, we performed solubility-enhancing optimization of ppluGFP2. We produced a highly soluble, rapidly maturing variant, named TurboGFP. Having solved its crystal structure, we were able to analyse the chromophore environment and explain the rapid development of the fluorescent signal in vivo.
Results and Discussion
Development of non-aggregating ppluGFP2
We have threaded the sequence of ppluGFP2 onto the A. victoria GFP structure (Protein Data Bank (PDB) ID: 1GFL) using Swiss-PdbViewer and HyperChem 5.01 (Hypercube Inc., Gainesville, FL, USA). The resulting model was used to generate a plausible ppluGFP2 molecular surface. Analysis of the computed electrostatic potential suggested that the protein surface is mostly negatively charged with the exception of a positively charged patch of residues near the amino and carboxyl termini. We supposed that aggregation might be caused by electrostatic interactions between the positively and the negatively charged surfaces (Himanen et al, 1997). With the aim of shielding the positively charged patch, we added polar, negatively charged amino acids to the protein termini. The resulting ppluGFP2 variant aggregated less in vitro. However, it still formed microcrystals 7 days after transient transfection of HeLa cells (see supplementary information 1 online for details). Importantly, it was shown to be tetrameric, according to the gel-filtration data. This suggests that wild-type ppluGFP2 is also tetrameric (ppluGFP2 was originally characterized tentatively as a monomer; Shagin et al, 2004).
We followed up with another cycle of mutagenesis, changing exposed hydrophobic residues to polar and charged ones. The initial ppluGFP2 model suggested oligomer interfaces, by comparison with structures of oligomeric fluorescent proteins. To loosen the ppluGFP2 tetramer and make the protein more suitable as a fusion tag, we proceeded to make non-conservative substitutions of several key residues in the hypothetical interfaces. Ultimately, we obtained a non-aggregating dimeric ppluGFP2 variant, designated TurboGFP (see supplementary Fig 1 online for sequence).
Characterization of TurboGFP
TurboGFP is a bright green fluorescent protein (see supplementary Fig 2 online for fluorescence spectrum) with a quantum yield of 0.53 and molar extinction coefficient of 70,000 M−1 cm−1. Its fluorescence has rather high pH stability (pKa 5.2). The protein is a dimer in solution, at least at concentrations up to 5 mg/ml (0.2 mM), according to gel-filtration (supplementary Fig 3 online) and light scattering (unimodal distribution of apparent molecular weight centred at 54 kDa) data. It is soluble under physiological conditions (pH 7.4, 20 mM phosphate-buffered 0.14 M NaCl), at least at concentrations up to 60 mg/ml.
We have succeeded in expressing high levels of TurboGFP in bacteria, yeast (see supplementary information 2 online) and mammalian cells. Unlike with the native ppluGFP2, stable cell lines expressing TurboGFP can be established, as was shown for HeLa, 3T3, mouse melanoma M3 and PC12 cell lines (Dr C. Petzelt, personal communication).
We have also generated a destabilized TurboGFP version with a rapid turnover, by fusing the protein with the degradation domain of mouse ornithine decarboxylase (Li et al, 1998). For evaluation of the construct, mammalian cells, transiently transfected with destabilized TurboGFP, were treated with 100 μg/ml cycloheximide (an inhibitor of protein synthesis). According to time-lapse measurements, the destabilized TurboGFP had a half-life of 2 h (supplementary Fig 4 online). Remarkably, the wild-type ppluGFP2 was insensitive to fusion of the degradation domain, presumably owing to aggregates being inaccessible to the proteolytic machinery. Rapidly maturing fluorescent proteins with a characteristically short half-life are particularly useful for studies of gene expression timelines.
To evaluate the suitability of TurboGFP as a fusion partner for in vivo fluorescent tagging, we compared the performance of ppluGFP2, TurboGFP and the commonly used enhanced green fluorescent protein (EGFP; Clontech, Mountain View, CA, USA) fusions with β-actin in HeLa cells. Fig 1C,D shows that the fluorescence patterns of mammalian cells expressing TurboGFP–actin are not visually distinguishable from those expressing EGFP–actin. Both show formation of typical elongated actin filaments and an apparently normal cytoskeleton. By contrast, the ppluGFP2–actin filaments are much shorter, leading to distortion of the cytoskeleton morphology (Fig 1B).
Similar to the wild-type ppluGFP2, TurboGFP is characterized with a rapid generation of fluorescent signal in living cells. In transient transfections of HeLa, 3T3 and Phoenix Eco cell lines, TurboGFP fluorescence was detectable 3–5 h earlier than that of EGFP. Furthermore, we compared TurboGFP and EGFP in developing Xenopus laevis embryos (Fig 2). TurboGFP fluorescence was about threefold brighter at the early (10 h after fertilization) and twofold brighter at the middle (12 h after fertilization) gastrula stages, demonstrating the speed advantage offered by this tool to visualize gene expression.
Figure 2.
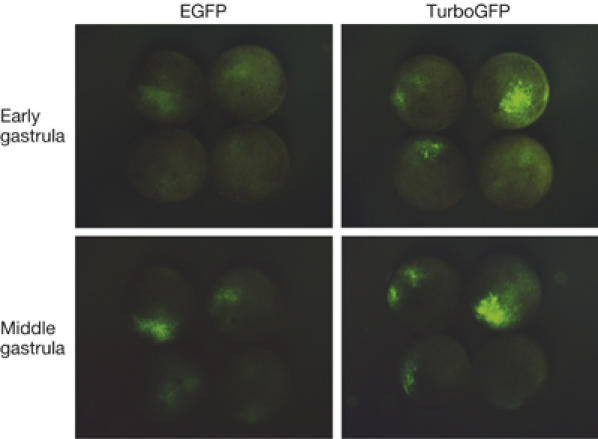
Comparison of TurboGFP and enhanced green fluorescent protein maturation speed in developing Xenopus laevis embryos. At the stage of two blastomeres, embryos were microinjected with TurboGFP-C1 and pEGFP-C1 vectors. Living embryos were photographed from the animal pole side at the early and mid-gastrula stages. EGFP, enhanced green fluorescent protein.
TurboGFP crystal structures
TurboGFP expression in E. coli easily yielded enough material to pursue structural studies. The details of cloning, protein production, crystallization and structure solution are provided in Methods and supplementary information 3 online. As expected, despite the modest degree of identity between TurboGFP and other members of the GFP family, the structure of this protein is a β-barrel fold (PDB IDs 2G6X and 2G6Y; Fig 3A). TurboGFP is a dimer in solution at concentrations at least up to 5 mg/ml, whereas it forms tetramers in the crystal form. It is likely that TurboGFP dimers undergo reversible association into tetramers at higher concentrations. There are virtually no differences in the structures of the two crystal forms (P21 and C2221; see supplementary Table 1 online for details)—they superimpose with inter-monomer and inter-tetramer Cα root-mean-square deviations (r.m.s.d.) of 0.35 and 0.5 Å, respectively. All four monomers in the TurboGFP asymmetric unit are also virtually identical (pairwise Cα r.m.s.d. of 0.3 Å), with the exception of residues 185–187 of chain B in the P21 crystal form, which adopt a slightly different conformation, probably because of a crystal contact. The quaternary structure of TurboGFP tetramer is similar to that of DsRed (Wall et al, 2000; Yarbrough et al, 2001). In fact, the two tetramers superimpose with a least-squares minimized Cα r.m.s.d. of 1.45 Å.
Figure 3.
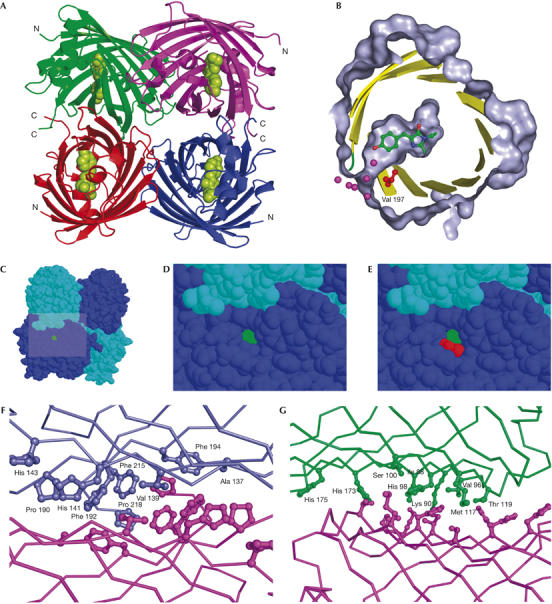
TurboGFP crystal structure. (A) The overall structure of TurboGFP. Cα traces are in green, violet, red and blue. The chromophore is shown as green van der Waals spheres. (B–E) A pore leading to the TurboGFP chromophore. The chromophore is highlighted in green and Val 197 in red. (B) Protein surface (grey) is cut away to show the pore and the chromophore cavity. Sections of secondary structure elements are shown as yellow cartoons. Relevant water molecules are depicted as magenta spheres. (C–E) The pore remains unobstructed on tetramerization. (F,G) Contacts between TurboGFP monomers. Two TurboGFP monomers are shown as Cα traces, with contact residues shown as balls and sticks of the same colour as the particular monomer. For clarity, only one set of residues is labelled. (F) The more extensive contact area having mainly hydrophobic character is shown. (G) The less extensive contact area, composed of a mixture of polar and hydrophobic interactions is shown.
The first dimer interface (monomers A∣B and C∣D) occludes about 1,093 Å2 of molecular surface per monomer. The interface is composed of residues A137, V139, H141, H143, M145, Y165, P190, F192, F194, F215 and P218 contributed by two-fold non-crystallographic symmetry-related monomers (Fig 3F). We believe this junction to be tight, as the interface residues and the interactions between them are similar to those found in protein hydrophobic cores. The atomic temperature factors of these residues are some of the lowest in the whole structure (18 Å2 on average, compared with the average B-factor of 38 Å2), which further underscores the stability of this interface. With the aim of producing monomeric TurboGFP, we mutated several clusters of residues within the first interface. Invariably, all mutants were completely insoluble and non-fluorescent on expression in E. coli, confirming the importance of the first dimer interface in the context of the overall TurboGFP structure. The second, less extensive dimer interface (monomers A∣D and B∣C) occludes 703 Å2 of molecular surface per monomer and is composed of a mixture of polar and hydrophobic interactions involving residues I88, K90, V96, H98, S100, M117, T119, G146, H173 and H175 (Fig 3G).
In general, the crystal structure of TurboGFP has validated the assumptions made during the design of non-aggregating ppluGFP2 derivatives. The addition of charged termini shields the positively charged patch on the TurboGFP surface and probably contributes to mutual electrostatic repulsion of the C termini. Most of the other surface mutations loosen the weaker dimer–dimer interface, with the exception of L200D, which probably contributes to the overall reduction in protein hydrophobicity.
Least-squares superposition of atomic coordinates of TurboGFP monomer with those of GFP, DsRed, pocilloporin, eqFP611, KFP1 and zFP538 results in Cα r.m.s.d. of 1.25, 1.07, 1.04, 1.00, 1.08 and 1.11 Å, respectively. As expected from the sequence alignment, the geometry of the loops (residues 29–32, 66–72, 78–82, 92–95, 105–110, 143–151, 160–165, 175–179, 199–207) that join the conserved structural elements of TurboGFP differ in other members of the GFP family, whereas the core secondary structure elements themselves superimpose well—the exceptions are the extra helix present only in TurboGFP (residues 180–188) and the N-terminal capping helix of GFP (residues 2–9).
Electron density maps indicate complete maturation of all four chromophores in the tetramer. The geometry of the mature TurboGFP chromophore is identical to that of GFP. It is noteworthy that the coloration of the frozen protein crystal completely disappeared after a brief exposure to X-ray radiation. To test this further, a new crystal was tested with the intensity of the beam attenuated to the lowest transmittance (∼1%), and still the coloration was eliminated in the course of a single 1-s exposure. As atomic motion is highly restricted in a crystal at 100 K, it is likely that only an electronic configuration rearrangement is taking place, possibly initiated by excitation of outer shell electrons of the S atom of M34, located in the vicinity of the chromophore. Alternatively, a process similar to that described earlier (Matsui et al, 2002) might be responsible for this effect.
Overall, the milieu of the TurboGFP chromophore is similar to that of other GFPs, which show an abundance of buried charged side chains typical of internalized catalytic centres. The residues that contribute functional groups to this environment vary between the members of the family, but the chromophore-relative positions of important functional groups are conserved—for example, R87 is the equivalent of GFP R96 and E210 is the equivalent of GFP E222.
The putative basis for fast TurboGFP maturation
Our data show that TurboGFP fluorescence appears significantly earlier than that of the commonly used EGFP under similar in vivo conditions. To understand the basis of this phenomenon, we compared protein-folding and chromophore maturation kinetics for EGFP and TurboGFP in renaturation and renaturation/maturation assays.
First, the recovery of fluorescence during refolding of preheated urea-denatured fluorescent proteins was monitored (see Methods). Denaturation does not affect the chemical structure of the chromophore; therefore, such experiments only show protein-refolding kinetics. In these experiments, TurboGFP demonstrated extremely fast protein-folding kinetics, with a half-life of 11 s, whereas for EGFP the half-life was about 1.5 min (Table 1; Fig 4A). In general, chromophore formation, but not protein folding, is known to be the slowest and time-limiting step in fluorescent protein maturation. However, it is presumed that the fast and effective folding of TurboGFP contributes to the development of early fluorescent signal in vivo, by allowing a higher percentage of the protein to escape the protein degradation machinery of the cell. Notably, in vivo, faster folding of TurboGFP and ppluGFP2 is due to the absence of cis-prolines, which are reported to slow down the folding of A. victoria GFP (Enoki et al, 2004).
Table 1.
Spectral characteristics and parameters of refolding and refolding/maturation kinetics
| Fluorescent protein | ɛ (λmax)* | QY (λmax)‡ | Refolding half-time (s) | Maturation half-time (s) | kox (10−4 s−1) |
|---|---|---|---|---|---|
| EGFP |
55,000 (489) |
0.60 (509) |
90.6 |
3,915 |
1.77 |
| Venus§ |
110,000 (515) |
0.63 (527) |
46.2 |
4,076 |
1.70 |
| SYFP2§ |
101,000 (515) |
0.68 (527) |
69.3 |
3,300 |
2.10 |
| TurboGFP |
70,000 (482) |
0.53 (502) |
11.0 |
1,468 |
4.72 |
| TurboGFP-V197L | 73,000 (482) | 0.47 (502) | 9.5 | 2,493 | 2.78 |
Protein refolding and maturation were followed by measuring the recovery of fluorescence at 25°C. Maturation rate constants (kox) were determined by computer-fitting the kinetic data to the first-order exponential decay (Origin 6.0).
EGFP, enhanced green fluorescent protein; GFP, green fluorescent protein.
*Extinction coefficient (M−1 cm−1) with excitation maximum (nm) in parentheses.
‡Quantum yield with emission maximum (nm) in parentheses.
§Data from Kremers et al (2006).
Figure 4.
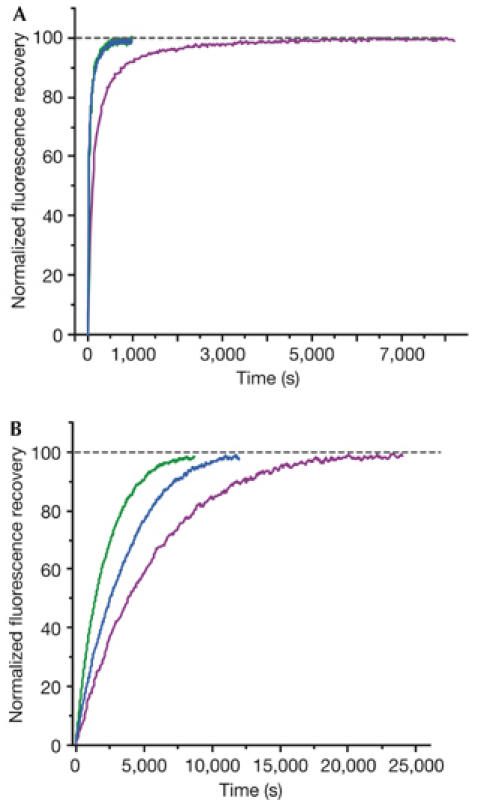
Comparison of refolding and maturation speed in vitro of EGFP, TurboGFP and TurboGFP-V197L. Normalized fluorescence recovery plots are shown. EGFP, violet lines; TurboGFP, green lines; TurboGFP-V197L, blue lines. See Methods and Table 1 for details. (A) Refolding kinetics. (B) Chromophore maturation kinetics.
Next, we compared chromophore maturation rates for EGFP and TurboGFP. We used dithionite reduction of a chromophore of urea-denatured protein, commonly used to follow-up fluorescent protein maturation starting from the native polypeptide (Reid & Flynn, 1997; Nagai et al, 2002). Maturation curves fitted first-order exponential decay (Origin 6.0), enabling an estimation of maturation rate constants (kox). For EGFP, kox was found to be 1.77 × 10−4 s−1, close to the previously reported value (Reid & Flynn, 1997). At the same time, the maturation rate was significantly higher for TurboGFP, with kox of 4.72 × 10−4 s−1 (Table 1; Fig 4B).
Ultimately, we believe that both rapid protein folding and chromophore maturation contribute to early fluorescent signal development by Copepoda fluorescent proteins. Although protein folding depends on many parameters, faster chromophore maturation could be explained by the chromophore environment.
It is considered that chromophore maturation in GFP-like proteins proceeds in two stages: cyclization of amino acids 65–67 (using GFP residue numbers), followed by the slowest and thus time-limiting step, dehydrogenation of the Y66 Cα–Cβ bond (Reid & Flynn, 1997). In the presence of molecular oxygen, this reaction proceeds spontaneously, although protein environment strongly influences the reaction kinetics (Kojima et al, 1998). The recently proposed chromophore maturation mechanism (Rosenow et al, 2004) implies a dehydrogenation step initiated by Y66 Cα deprotonation by base. Interestingly, TurboGFP contains a unique E89 residue, which could aid in proton abstraction (see supplementary information 4 online).
Another unique feature of TurboGFP compared with other fluorescent proteins is the water-filled pore, leading from the outside of the protein barrel to Y58 of the chromophore (Fig 3B–E). This pore remains unobstructed on tetramerization. Importantly, molecular oxygen is required for the rate-limiting step of chromophore dehydrogenation (Cody et al, 1993; Heim et al, 1994; Inouye & Tsuji, 1994). Therefore, it can be presumed that the pore facilitates oxygen conveyance to the premature chromophore, thus speeding up maturation.
To verify this hypothesis, we performed site-directed mutagenesis of TurboGFP, introducing leucine instead of valine in position 197 (205 in GFP), shown in red in Fig 3B,E. This mutation should narrow the pore and therefore impair the proposed oxygen-conveyance mechanism. The resulting protein TurboGFP-V197L was brightly fluorescent, although its fluorescence signal was generated much more slowly when expressed in E. coli. Its renaturation kinetics was shown to be as fast as that of TurboGFP, indicating that this mutation does not impair protein folding (Table 1; Fig 4A). Also, the fluorescence characteristics of mutants, such as excitation/emission spectra, fluorescence quantum yield and extinction coefficient, were almost identical to those of the parent TurboGFP (Table 1), indicating that the chromophore environment was not significantly altered. The chromophore maturation kinetics of TurboGFP-V197L was markedly slower (Table 1; Fig 4B). This suggests that the pore found in TurboGFP/ppluGFP2 is essential for fast maturation of the chromophore, most probably as an access route for oxygen conveyance to the premature chromophore.
As an alternative hypothesis, it is proposed that water molecules coordinated in the pore can participate in proton abstraction from the Y66 Cα atom. This facilitates the dehydrogenation step of chromophore maturation, in common with the model of Rosenow et al (2004), implying that the water molecule is the base responsible for proton abstraction.
Rapidly maturing fluorescent proteins are highly desirable tools for: monitoring the activity of promoters; studying the protein degradation machinery of the cell; and a wide range of applications in which a rapid generation of fluorescent signal is crucial. TurboGFP can be used in lieu of EGFP in applications that are not strictly dependent on the monomeric nature of the tag, but demand rapid signal detection. We believe that our findings could contribute to the rational design of rapidly maturing monomeric fluorescent proteins, suitable for fluorescence resonance energy transfer-based protein interaction studies and for fastidious fusions.
In general, the TurboGFP structure is intriguing, as it is the first reported non-Cnidarian fluorescent protein structure solved. We believe that it will have a significant impact on the further understanding of the evolution, functions and chromophore biophysics of GFP-like proteins.
Methods
Construction of protein mutants. For bacterial expression, the DNA encoding the codon-optimized version of ppluGFP2 from P. plumata (Shagin et al, 2004) was amplified using specific primers and cloned into Qiagen pQE30 vector using BamHI–HindIII restriction sites. Site-directed mutagenesis was performed by overlap-extension PCR, with primers containing the appropriate target substitutions (Ho et al, 1989).
Expression in mammalian cell lines and imaging. For expression in eukaryotic cells, a PCR-amplified AgeI–BglII fragment encoding TurboGFP or ppluGFP2 was swapped with EGFP in pEGFP-C1 vector (Clontech), resulting in TurboGFP-C1 or ppluGFP2-C1 plasmid. To generate TurboGFP–β-actin or ppluGFP2–β-actin fusion proteins, the corresponding gene was swapped with EGFP in a pEGFP–actin vector (Clontech). Fluorescence images of transfected HeLa cells were obtained using the Olympus CK40 inverted microscope equipped with the Olympus DP50 camera.
Protein purification. Proteins fused to the N-terminal polyhistidine tag were expressed in E. coli XL1-blue strain (Stratagene, La Jolla, CA, USA) and purified using TALON metal-affinity resin (Clontech). The polyhistidine tag was not removed.
Spectral measurements. Absorption spectra were recorded with a Beckman DU520 UV/VIS Spectrophotometer. The Varian Cary Eclipse Fluorescence Spectrophotometer was used for measuring excitation–emission spectra. For molar extinction coefficient determination, we relied on estimating the mature chromophore concentration. An alkali-denatured GFP chromophore absorbs at 446 nm with a molar extinction coefficient 44 000 M−1 cm−1. Molar extinction coefficients for the native state were estimated from the absorption of denatured proteins. For quantum yield determination, the fluorescence of the proteins was compared with equally absorbing EGFP (quantum yield 0.60; Patterson et al, 2001).
Refolding and maturation kinetics. Samples of fluorescent proteins were heated to 95°C in denaturation solution (8 M urea, 1 mM dithiothreitol) for 4 min. Refolding reactions were initiated following 100-fold dilution in the renaturation buffer (35 mM KCl, 2 mM MgCl2, 50 mM Tris pH 7.5, 1 mM dithiothreitol). In the maturation assay, 5 mM freshly dissolved dithionite was added to the denaturation solution (Reid & Flynn, 1997). Owing to the instability of dithionite at high temperatures and to provide for complete chromophore reduction, the sample was cooled to 25°C and the addition of 5 mM dithionite followed by heating to 95°C were repeated. Protein refolding and maturation were followed by measuring the recovery of fluorescence using the Varian Cary Eclipse Fluorescence Spectrophotometer, with the chamber temperature maintained at 25°C.
Supplementary information is available at EMBO reports online (http://www.emboreports.org).
Supplementary Material
Supplementary Information
supplementary Table and Figures
Acknowledgments
A.G.E. thanks K. Greis and T. Burt for the mass spectrometry measurements. This work was supported by grants from Molecular and Cell Biology Program (MCBR) RAS and EC FP-6 Integrated Project LSHG-CT-2003-503259 (to K.A.L. and D.M.C.). D.M.C. is supported by grants from the President of Russian Federation MK-8236.2006.4 and Russian Science Support Foundation. X-ray data were collected at SER-CAT 22-ID beamline at the Advanced Photon Source (APS), Argonne National Laboratory. The use of the APS was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences, Contract No. W-31-109-Eng-38. This work was partially supported by Howard Hughes grant 55000344 and MCBR RAS and Russian Fund of Basic Research grant 01-04-49258 (to A.G.Z.).
References
- Chudakov DM, Lukyanov S, Lukyanov KA (2005) Fluorescent proteins as a toolkit for in vivo imaging. Trends Biotechnol 23: 605–613 [DOI] [PubMed] [Google Scholar]
- Cody CW, Prasher DC, Westler WM, Prendergast FG, Ward WW (1993) Chemical structure of the hexapeptide chromophore of the Aequorea green-fluorescent protein. Biochemistry 32: 1212–1218 [DOI] [PubMed] [Google Scholar]
- Enoki S, Saeki K, Maki K, Kuwajima K (2004) Acid denaturation and refolding of green fluorescent protein. Biochemistry 43: 14238–14248 [DOI] [PubMed] [Google Scholar]
- Heim R, Prasher DC, Tsien RY (1994) Wavelength mutations and posttranslational autoxidation of green fluorescent protein. Proc Natl Acad Sci USA 91: 12501–12504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himanen JP, Popowicz AM, Manning JM (1997) Recombinant sickle hemoglobin containing a lysine substitution at Asp-85(α): expression in yeast, functional properties, and participation in gel formation. Blood 89: 4196–4203 [PubMed] [Google Scholar]
- Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR (1989) Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77: 51–59 [DOI] [PubMed] [Google Scholar]
- Inouye S, Tsuji FI (1994) Evidence for redox forms of the Aequorea green fluorescent protein. FEBS Lett 351: 211–214 [DOI] [PubMed] [Google Scholar]
- Johnson FH, Shimomura O, Saiga Y, Gershman LC, Reynolds GT, Waters JR (1962) Quantum efficiency of Cypridina luminescence, with a note on that of Aequorea. J Cell Comp Physiol 60: 85–103 [Google Scholar]
- Kojima S, Ohkawa H, Hirano T, Maki S, Niwa H, Ohashi M, Inouye S, Tsuji FI (1998) Fluorescent properties of model chromophores of tyrosine-66 substituted mutants of Aequorea green fluorescent protein (GFP). Tetrahedron Lett 39: 5239–5242 [Google Scholar]
- Kremers GJ, Goedhart J, van Munster EB, Gadella TW Jr (2006) Cyan and yellow super fluorescent proteins with improved brightness, protein folding, and FRET Forster radius. Biochemistry 45: 6570–6580 [DOI] [PubMed] [Google Scholar]
- Labas YA, Gurskaya NG, Yanushevich YG, Fradkov AF, Lukyanov KA, Lukyanov SA, Matz MV (2002) Diversity and evolution of the green fluorescent protein family. Proc Natl Acad Sci USA 99: 4256–4261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zhao X, Fang Y, Jiang X, Duong T, Fan C, Huang CC, Kain SR (1998) Generation of destabilized green fluorescent protein as a transcription reporter. J Biol Chem 273: 34970–34975 [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Patterson GH (2003) Development and use of fluorescent protein markers in living cells. Science 300: 87–91 [DOI] [PubMed] [Google Scholar]
- Matsui Y, Sakai K, Murakami M, Shiro Y, Adachi S, Okumura H, Kouyama T (2002) Specific damage induced by X-ray radiation and structural changes in the primary photoreaction of bacteriorhodopsin. J Mol Biol 324: 469–481 [DOI] [PubMed] [Google Scholar]
- Matz MV, Labas YA, Ugalde J (2006) Evolution of function and color in GFP-like proteins. Methods Biochem Anal 47: 139–161 [DOI] [PubMed] [Google Scholar]
- Nagai T, Ibata K, Park ES, Kubota M, Mikoshiba K, Miyawaki A (2002) A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat Biotechnol 20: 87–90 [DOI] [PubMed] [Google Scholar]
- Ohtsuka S, Huys R (2001) Sexual dimorphism in calanoid copepods: morphology and function. Hydrobiologia 453–454: 441–466 [Google Scholar]
- Ormo M, Cubitt AB, Kallio K, Gross LA, Tsien RY, Remington SJ (1996) Crystal structure of the Aequorea victoria green fluorescent protein. Science 273: 1392–1395 [DOI] [PubMed] [Google Scholar]
- Patterson G, Day RN, Piston D (2001) Fluorescent protein spectra. J Cell Sci 114: 837–838 [DOI] [PubMed] [Google Scholar]
- Prasher DC, Eckenrode VK, Ward WW, Prendergast FG, Cormier MJ (1992) Primary structure of the Aequorea victoria green-fluorescent protein. Gene 111: 229–233 [DOI] [PubMed] [Google Scholar]
- Reid BG, Flynn GC (1997) Chromophore formation in green fluorescent protein. Biochemistry 36: 6786–6791 [DOI] [PubMed] [Google Scholar]
- Rosenow MA, Huffman HA, Phail ME, Wachter RM (2004) The crystal structure of the Y66L variant of green fluorescent protein supports a cyclization–oxidation–dehydration mechanism for chromophore maturation. Biochemistry 43: 4464–4472 [DOI] [PubMed] [Google Scholar]
- Shagin DA et al. (2004) GFP-like proteins as ubiquitous metazoan superfamily: evolution of functional features and structural complexity. Mol Biol Evol 21: 841–850 [DOI] [PubMed] [Google Scholar]
- Wall MA, Socolich M, Ranganathan R (2000) The structural basis for red fluorescence in the tetrameric GFP homolog DsRed. Nat Struct Biol 7: 1133–1138 [DOI] [PubMed] [Google Scholar]
- Yang F, Moss LG, Phillips GN Jr (1996) The molecular structure of green fluorescent protein. Nat Biotechnol 14: 1246–1251 [DOI] [PubMed] [Google Scholar]
- Yarbrough D, Wachter RM, Kallio K, Matz MV, Remington SJ (2001) Refined crystal structure of DsRed, a red fluorescent protein from coral, at 2.0-Å resolution. Proc Natl Acad Sci USA 98: 462–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information
supplementary Table and Figures