

Abstract
The recent finding of intrinsically unstructured proteins defies the classical structure–function paradigm. However, owing to their flexibility, intrinsically unstructured proteins generally escape detailed structural investigations. Consequently little is known about the extent of conformational disorder and its role in biological functions. Here, we present the X-ray structure of the unbound ribosomal protein L20, the long basic amino-terminal extension of which has been previously interpreted as fully disordered in the absence of RNA. This study provides the first detailed picture of two protein folding states trapped together in a crystal and indicates that unfolding occurs in discrete regions of the whole protein, corresponding mainly to RNA-binding residues. The electrostatic destabilization of the long α-helix and a structural communication between the two L20 domains are reminiscent of those observed in calmodulin. The detailed comparison of the two conformations observed in the crystal provides new insights into the role of unfolded extensions in ribosomal assembly.
Keywords: intrinsically unstructured protein, ribosome assembly, RNA binding
Introduction
Eubacterial L20, a small ribosomal protein (∼118 aa), is essential for the first steps of 50S ribosomal subunit assembly (Nowotny & Nierhaus, 1980; Guillier et al, 2005a) and regulates the translation of its own messenger RNA (Guillier et al, 2005b) in binding to two RNA partners: the 23S RNA and its mRNA operator. L20 shows a highly charged α-helical amino-terminal extension (aa 1–59) that contains clusters of conserved basic residues involved in RNA contacts in the ribosome (supplementary Fig S1 online). Recent X-ray structures of ribosomal particles have shown that many ribosomal proteins have long extensions that penetrate deep into the RNA core (Harms et al, 2001; Schuwirth et al, 2005). As in many other intrinsically unstructured proteins involved in RNA folding and nucleic acid binding (Wright & Dyson, 1999; Tompa & Csermely, 2004), these extensions are unfolded in the absence of nucleic acids. Indeed, nuclear magnetic resonance studies have indicated that L20 extension is fully unstructured in the absence of its RNA target (Raibaud et al, 2002). As unstructured proteins generally escape detailed structural analysis, little is known about their degree of order and their conformations in the unfolded state. Such detailed information is, however, crucial for explaining the role of disorder in biological function. In this paper, we present the X-ray analysis of two folded states of L20 trapped together in the same crystal unit cell. This unique situation allows a detailed comparison of the two forms in identical physico-chemical conditions and provides a detailed picture of a partially unfolded protein.
Results and Discussion
The structure of a selenomethionyl derivative of Aquifex aeolicus L20 was determined by single-wavelength anomalous dispersion at 2.8 Å resolution (Table 1). The triclinic cell contains four L20 molecules organized into a heterotetramer in such way that a dimer of partially unfolded monomers (form 2) tightly embraces a dimer of folded monomers (form 1; Fig 1; supplementary Fig S2a,b online); the two forms are stabilized by their mutual fit. Interestingly, in each dimer, the protein interfaces correspond to those of L20/L21 in the ribosome (supplementary Fig S2c online). In form 1, the long N-terminal helix α2 (aa 30–70) is fully folded despite the absence of RNA. However, in contrast to L20 bound to 23S RNA of Deinococcus radiodurans (Harms et al, 2001) and Escherichia coli (Schuwirth et al, 2005) in the large ribosomal subunit, helix α2 is perfectly straight in the present structure (Fig 2A). The carboxy-terminal half of form 1 forms a compact globular domain. Helix α3 (aa 74–83), loop 3–4 (aa 84–89), helix α4 (aa 90–100) and helix α5 (aa 101–118) stack around a hydrophobic core maintained by evolutionarily conserved residues (supplementary Fig S1 online).
Table 1.
Data collection, single-wavelength anomalous dispersion phasing and refinement statistics
|
Data collection | ||||
| Space group |
P1 |
|
|
|
| Unit cell |
a=44.91 Å |
b=45.22 Å |
c=67.06 Å |
|
| |
α=104.06° |
β=106.20° |
γ=97.76° |
|
| Wavelength (Å) |
Peak |
Edge |
Remote |
Peak (three crystals) |
| |
0.97961 |
0.97971 |
0.97697 |
0.97961 |
| Resolution (Å) |
2.8 |
3.0 |
3.0 |
2.7 |
| Observations |
21,490 |
17,185 |
14,284 |
44,569 |
| Unique |
11,349 |
8,745 |
8,119 |
13,043 |
| Completeness |
96.2 (95) |
96.2 (96.2) |
88.0 (85.2) |
98.9 (93.5) |
| Multiplicity |
1.9 (1.5) |
2.0 (2.0) |
1.8 (1.7) |
3.5 (3.2) |
| Rsym |
0.064 (0.218) |
0.056 (0.097) |
0.112 (0.214) |
0.144 (0.223) |
| I/σ |
10.6 (3.3) |
11.9 (6.8) |
5.5 (3.0) |
3.6 (2.5) |
| SAD phasing power |
0.58 |
|
|
0.60 |
| Figure of merit (before/after solvent flattening) |
0.12/0.90 |
|
|
0.14/0.91 |
| Rcullis |
0.93 |
|
|
0.93 |
|
Refinement statistics | ||||
| Mol/asymmetric unit |
4 |
|
|
|
| Atom |
3,238 |
|
|
|
| Solvent |
80 |
|
|
|
| Number of reflections |
10,246 |
|
|
|
| R/Rfree |
0.22/0.29 |
|
|
|
| R.m.s.d. bond lengths (Å) |
0.0076 |
|
|
|
| R.m.s.d. angles (deg) |
1.10946 |
|
|
|
| Average B (Å2) | 69 | |||
Rsym=∑∣I−〈I〉∣/∑∣〈I〉∣, where I is the measured intensity of each reflection and 〈I〉 the intensity averaged from multiple observation of symmetry-related reflections.
Phasing power=〈FH/LOC〉, where LOC is lack of closure.
Rcullis=∣∣FPH±FP∣−FH∣/∣FPH±FP∣.
SAD, single-wavelength anomalous dispersion.
Figure 1.
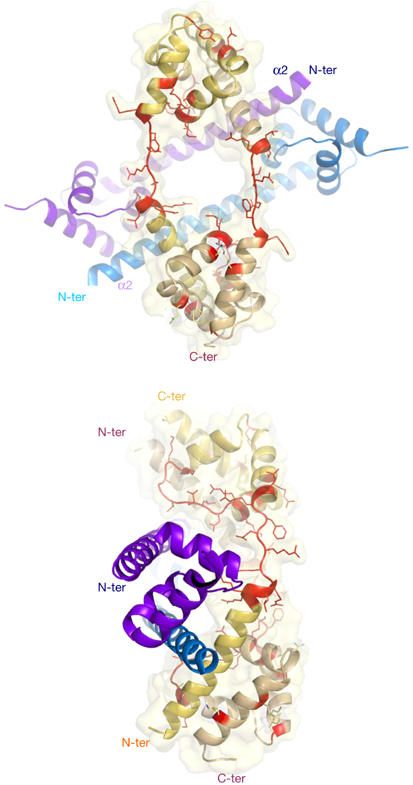
Two views of the heterotetramer in the asymmetric unit of P1 cell. The two folded monomers (form 1) are represented by deep-blue and light-blue ribbons. The two partly unfolded monomers (form 2) are represented by yellow and orange ribbons. The unfolded regions in the carboxy- and amino-terminal domains of form 2 are represented by red sticks. The tetramer association buries 8,017 Å2. The two dimers share a common two-fold non-crystallographic axis.
Figure 2.
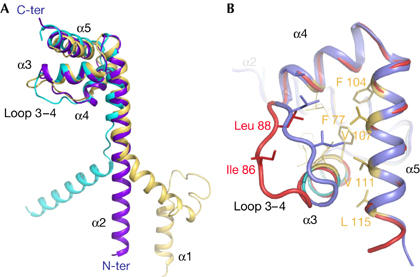
L20 is partly unfolded in both carboxy- and amino-terminal domains. (A) Comparison of three forms of L20 protein. In Escherichia coli, the Cα atoms of the carboxy-terminal domains of the ribosome-bound form (yellow), the unbound form 1 (violet) and form 2 (light blue) are superimposed, showing the different paths of the long amino-terminal α2-helix. Least-square superimposition was performed with the program O (33) to compare the Cα coordinates of residues 59–115 of Aquifex aeolicus form 1 with the corresponding forms of the other L20 crystal structures. This provided r.m.s.d. values of 1.5 Å (form 2), 2.0 Å (E. coli 1, Protein Data Bank (PDB) code 2AW4), 1.9 Å (E. coli 2, PDB code 2AWB) and 1.5 Å (Deinococcus radiodurans, PDB code 1KC9). In the ribosome, both the RNA backbone and the extension of its neighbour L21 deviate the path of α2 and impose a sharp kink around residue Leu 50 of A. aeolicus. (B) Weakened hydrophobic core in folding intermediate. Comparison of the C-terminal domains of form 1 (blue) and form 2 (red). The opening out of loop 3–4 moves hydrophobic residues away and disrupts the hydrophobic core. Hydrophobic residues are represented by yellow sticks. Cα atom Ile 86 of loop 3–4 is displaced by up to 7.9 Å in form 2 relative to its corresponding positions in form 1.
A detailed picture of a partially unfolded protein
The folded form (form 1) coexists with a well-defined partially unfolded form of L20 (form 2) in the crystal unit cell. Oligomerization has frozen unstructured regions of form 2 and has made possible their crystallographic analysis. Notably, both the N- and C-terminal domains of form 2 are partially unfolded, as if they had not yet reached the corresponding secondary and tertiary structures found in form 1. The N-terminal helix α2 is unwound and stretched, from Arg 48 to Arg 57 (Figs 1,3A). The comparison of the two forms indicates that helical destabilization in this region is due to an electrostatic repulsion between the positive charges of the cluster of conserved basic residues, Arg 48, Lys 49, Lys 52, Arg 53 and Arg 56. Indeed, form 1 shows that these residues are disposed on the same face of helix α2 and create a highly positively charged surface spanning three helical turns (Fig 3B). In the E. coli ribosome, these positive charges are neutralized by neighbouring phosphate groups of two distinct RNA helices—H25 and the H40–H41 junction (supplementary Fig S4 online). The unwinding of this region in form 2 separates the charged side chains and minimizes their electrostatic repulsion. In form 2, the unfolding of loop 3–4 also disrupts the hydrophobic core by moving hydrophobic residues away from helices α4 and α5. The side chain of Ile 86 is displaced by almost 8 Å relative to its corresponding positions in form 1 and can no longer pack onto the hydrophobic core (Fig 2B). Consequently, the C-terminal end of helix α3 has lost half a turn. The two forms also differ by a set of tertiary contacts. Tertiary interactions anchoring α-helices in the C-terminal domain that involve direct hydrogen bonds in form 1 are solvent mediated (Arg 62–Ala 94) or are simply broken (Tyr 70–Glu 105, Lys 110–Ala 84) in form 2 (Figs 1, 4A,B). Fig 3 shows that unfolding has markedly remodelled the pattern of electrostatic potential in both the N- and C-terminal domains and indicates that the two forms have distinct RNA-binding properties. The C-terminal domain is more positively charged in form 2.
Figure 3.
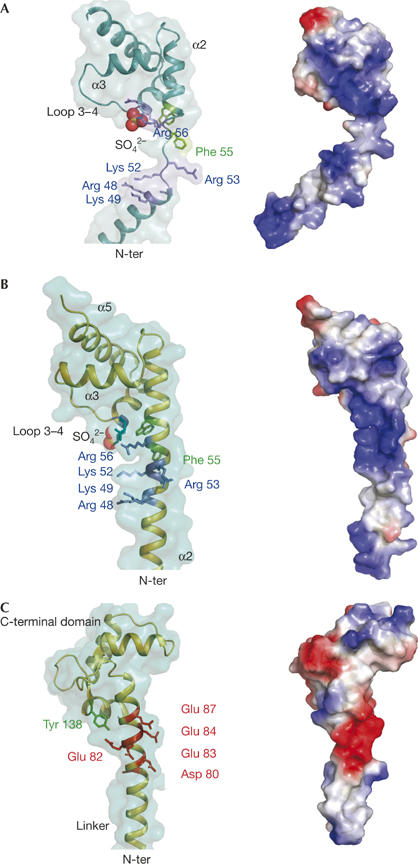
α-Helix stability and electrostatic potential in clusters of charged amino acids. (A) Unwinding of the region containing the cluster of basic residues 49–58 in form 2. Helix–coil transition separates charged side chains (left) and fully modifies the distribution of charge on the surface of L20 (right). (B) Fully folded α2-helix in form 1. The cluster of basic residues Arg 48, Lys 49, Lys 52, Arg 53 and Arg 56 of α2 and the two amino-terminal residues Arg 90 and Lys 91 (depicted in blue) of α4 generate a highly charged positive surface in the vicinity of the carboxy-terminal domain (right). Electrostatic potential surfaces were generated with Pymol. The sulphate ion bound by Arg 90 and Lys 91 is represented by CPK spheres. (C) The structure (residues 65–148) and electrostatic potential map of calmodulin (CaM; Protein Data Bank code 1CLL) are shown to underline the analogy with L20. A highly charged surface is also generated by a similar cluster of charged amino acids in the vicinity of the globular C-terminal region. Arg 49, Lys 52, Arg 53 and Arg 56 in L20 correspond to Asp 80, Glu 83, Glu 84 and Glu 87 in CaM and produce a similar helix instability.
Figure 4.
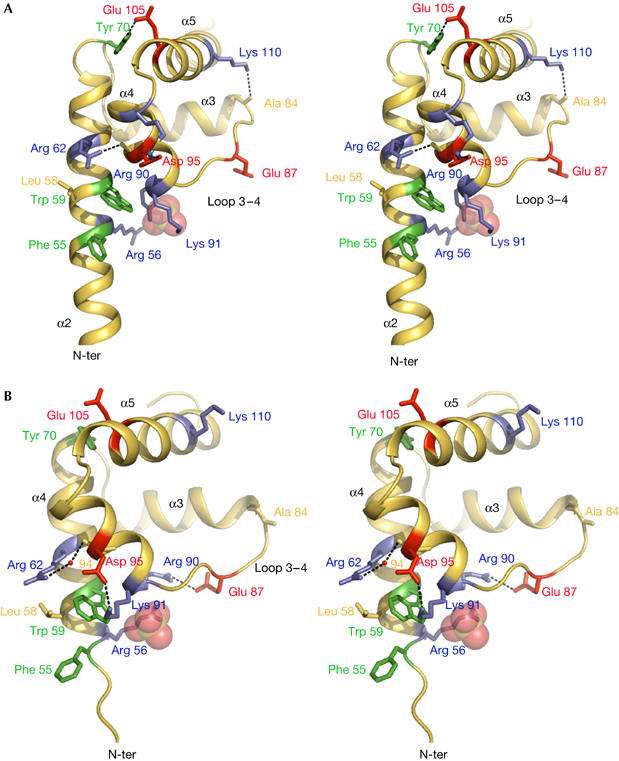
A conformational switch mediates a structural communication between the amino- and carboxy-terminal domains. Side chains of basic, acid and aromatic residues are represented by blue, red and green sticks, respectively. (A) Folded C-terminal domain. Arg 90 and Lys 91 bridge a sulphate ion (represented by transparent spheres) located at the N-terminal end of α4. The guanidinium group of Arg 62 and the hydroxyl group of Tyr 70 are involved in direct hydrogen bonds with the backbone carbonyl group of Ala 94 and the carboxyl group of Glu 105, respectively (dashed lines). Arg 56 is in close proximity to Arg 90. The amine group of Lys 110 bridges the carbonyl group of Ala 84. Asp 95 forms a salt bridge with Arg 99. (B) Partially unfolded C terminus of form 2. Arg 90 and Lys 91 form salt bridges with Glu 87 and Asp 95, respectively. The sulphate ion is bridged by Arg 56. Tyr 70 and Lys 110 are too far (4.5 and 15.0 Å) to interact with Glu 105 and Ala 84, respectively. Arg 62–Ala 94 contact mediated by a water molecule is represented in red.
Conformational switch and domain communication
In addition, two conserved basic residues, Arg 90 and Lys 91, located at the N terminus of helix α4 contribute to a marked reorganization of salt bridges. In form 1, their side chains are oriented along the helix axis and point towards the solvent, bridging a sulphate ion clearly defined in the electron density map. This anion represents a strong RNA-binding site, as it occupies a position similar to that of a phosphate group in E. coli ribosomal RNA (supplementary Fig S4 online; Schuwirth et al, 2005). Form 1 mimics the folded conformation of L20 in the ribosome, as, in the E. coli 50S particle, both Arg 90 and Lys 91 residues adopt a similar geometry (supplementary Fig S4 online). In form 2, although their Cα atoms occupy nearly identical positions, the side chains of Arg 90 and Lys 91 have a different geometry. Arg 90 forms a salt bridge with Glu 87 brought into close proximity by the opening out of loop 3–4 (Fig 4B). Conversely, Lys 91 has pivoted by 180° and establishes a salt bridge with Asp 95, located in the middle of helix α4. This salt bridge generates a steric hindrance that modifies the side-chain geometry of residues located in the vicinity of helix α2. It pushes the indole ring of Trp 59 towards Leu 58 and Arg 62, and tilts the aromatic ring of Phe 55 by about 60° relative to its orientation in form 1. Despite the shift in the position of Arg 90 and Lys 91 side chains, the sulphate ion occupies the same site in form 2, at the N-terminal end of helix α4. However, in this case, the guanidinium group of Arg 56 takes the position of that of Arg 90 and replaces it for bridging the sulphate ion. Thus, the conformational switch involving Arg 90 and Lys 91, which occupy a crucial position between loop 3–4 and helix α2, establishes a structural communication between the C- and N-terminal domains.
Implications for protein flexibility
L20 shows a noticeable analogy with calmodulin (CaM), a ubiquitous Ca2+-binding protein that has a key role in Ca2+-dependent signalling pathways in eukaryotic cells. Crystal structures of Ca2+-CaM have shown a dumbbell-shaped structure consisting of the N- and C-terminal Ca2+-binding domains separated by a long α-helical linker (27 residues; Babu et al, 1988). L20 and CaM have a common spatial arrangement of a long solvent-exposed α-helix connected to a C-terminal globular domain (Fig 3B,C). In both proteins, the helices show a highly unstable region located in the vicinity of the C-terminal globular domains. Helix–coil transition in this region confers on CaM the required plasticity for binding to an exceptional variety of distinct partners (Hoeflich & Ikura, 2002). Our study shows that the flexible regions of L20 and CaM share a similar sequence motif that confers helical instability, that is a cluster of charged amino acids distributed on the same helical face and spanning three helical turns (CxxCCxxC, where C is a charged amino acid of the same polarity; Fig 3B,C). In CaM, however, the polarity of the charged amino acids is reversed. In both cases, side-chain electrostatic repulsion seems to be responsible for helical instability. These sequence patterns might therefore constitute a hot spot for α-helix unwinding. However, in the present structure, this sequence exists in both folded and unfolded conformations, indicating that its stability is fine-tuned by the tertiary interactions with the C-terminal domain. Recent experimental data have shown that the flexibility of the CaM linker is also modulated by a structural communication between domains. Structural changes induced by either calcium or target binding in the CaM C-terminal domain are transmitted to the central helix through Tyr 138 located on the C-terminal domain and affect its stability (Sun et al, 2001; Slaughter et al, 2005; Fig 3C). Thus, our data show that both local sequence and tertiary interactions influence the conformational order/disorder equilibrium in proteins.
An unusual observation is that transient salt bridges stabilize form 2. Although the role of salt bridges in protein stability is still a matter of debate (Kumar & Nussinov, 2002), the formation of salt bridges during protein folding has been previously proposed to stabilize transition states (Tissot et al, 1996). Here, we show that the salt bridge has to be disrupted and reorganized in passing from one form to another. This unusual feature might be explained by the fact that the L20 sequence is poised to be unstructured in the absence of its binding partner. In conclusion, our data provide structural insights into how the sequences of intrinsically unstructured proteins (Wright & Dyson, 1999; Fink, 2005) are designed by evolution for maintaining partly or fully unfolded conformations under physiological conditions.
Speculation
Our study shows that the helix–coil transition in L20 extension occurs in a segment involved in 23S RNA recognition. The knowledge that L20 extension is strictly required for ribosomal assembly (Guillier et al, 2005a) suggests that the flexibility of this region is important during ribosome assembly. We propose a model involving the gradual co-folding of L20 and 23S RNA. Consistent with the current views (Klein et al, 2004), the first step would involve the binding of the partially folded C-terminal domain on its recognition site on the H40–H41 junction. This form has an enhanced positive electrostatic charge on its C-terminal domain surface and a decreased charge on helix α2 (Fig 3A,B). This reinforces the affinity of the globular domain for its RNA target and decreases that of α2-helix. The less ‘sticky' N-terminal helix is therefore free to rotate, owing to the flexibility of segment spanning aa 49–57. Consequently, once anchored to the H40–H41 junction through the C-terminal domain, α2 might scan around for binding to the distant H25 RNA helix, similar to the ‘fishing' model used in coated pit formation (Dafforn & Smith, 2004). In the next step, binding to the distant H25 RNA helix causes the full folding of the α2-helix. Coil–helix transition reduces the size of the stretched segment and brings the two RNA helices into close proximity. Finally, the fully folded extension locks the compact ternary complex.
Methods
L20 of A. aeolicus was overexpressed and purified, as described previously (Raibaud et al, 2002). Selenium was incorporated into the protein by growth on minimal media containing selenomethionine. The protein was purified by ion-exchange chromatography on an SP-Sepharose FF column, solubilized in a solution containing 1.5 M NaCl and 100 mM sodium acetate (pH 5.2) and then concentrated up to 15 mg/ml. Crystals were grown by the vapour diffusion method at 21°C. Hanging drops were made by mixing 2 μl of protein solution (15 mg/ml) with an equivalent volume of well solution (50 mM Tris (pH 7), 200 mM ammonium sulphate and 25% of PEG monoethyl ether 5000). Fine platelets appeared in drops equilibrated against well solution. It took several months for crystals to grow to a size suitable for data collection (30 × 300 × 300 μm). Data sets were collected at B30A beamline (ESRF). One crystal was used for collecting data at three wavelengths for multiple-wavelength anomalous dispersion experiments. Triclinic crystals diffracted up to 2.7 Å resolution (Table 1). Data were processed with MOSFLM and scaled with SCALA of the CCP4 package (Collaborative Computing Project Number 4, 1994). Self-rotation function indicated the presence of a two-fold non-crystallographic axis. The heavy atom search was processed with crystallography and nuclear magnetic resonance system (CNS; Brunger et al, 1998). Eight peaks related by the two-fold non-crystallographic axis were found by anomalous Patterson map analysis. Selenium peaks were attributed to the expected selenomethionines 38 and 96 of each of L20 monomers. Two dimers related by the two-fold axis occupied the asymmetric unit. Single-wavelength anomalous dispersion phasing, by using the peak data set and solvent flattening, was performed with CNS. The two long α2-helices related by the non-crystallographic axis clearly appeared in first experimentally phased maps and their quality allowed one to build the main chain of most of the four molecules. Subsequent cycles of building and refinement with the program O (Jones et al, 1991) and CNS allowed one to build the main chain and to include well-defined solvent molecules. Disordered regions 1–18 of form 1 and 1–26 of form 2 were not included in the final model. The small N-terminal helix α1 observed in 50S ribosome particles is unstructured in both unbound forms. Final refinement statistics are given in Table 1. A 2Fo−Fc electron density map centred on the two-fold non-crystallographic axis is shown in supplementary Fig S3 online. Two-fold non-crystallographic symmetry restraint, whether applied or not during refinement, produced results that were similar to refining the four molecules independently. Our model was subsequently checked by an independent refinement by using a peak data set merged from three crystals (Table 1). Buried area was computed with Areaimol CCP4. Figures were made with the program Pymol (DeLano, 2002). The coordinates and the structure factors are deposited in the Protein Data Bank (code 2GHJ).
Supplementary information is available at EMBO reports online (http://www.emboreports.org).
Supplementary Material
supplementary Fig S1
Acknowledgments
We thank P. Dumas and D. Picot for helpful discussions. We are grateful to the people of the beamline BM30A team (The European Synchrotron Radiation Facility, France). We thank A. Rak and M. Garber for the gift of A. aeolicus L20 complementary DNA and L. Dubois for participating in the crystallographic refinement during her Diplome d'Etudes Approfondies stage. We thank C. Condon, J.L. Sikorav and E. Westhof for a critical reading of the manuscript. This work was supported by Centre National de la Recherche Scientifique, Agence Nationale de la Recherche and Dynamique et Réactivité des Assemblages Biologiques.
References
- Babu YS, Bugg CE, Cook WJ (1988) Structure of calmodulin refined at 2.2 Å resolution. J Mol Biol 204: 191–204 [DOI] [PubMed] [Google Scholar]
- Brunger AT et al. (1998) Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D 54: 905–921 [DOI] [PubMed] [Google Scholar]
- Collaborative Computing Project. Number 4 (1994) Acta Crystallogr D 50: 760–76315299374 [Google Scholar]
- Dafforn TR, Smith CJI (2004) Natively unfolded domains in endocytosis: hook, lines and linkers. EMBO Rep 5: 1046–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano WL (2002), The PyMOL molecular graphics system on World Wide Web; http://www.pymol.org
- Fink A (2005) Natively unfolded proteins. Curr Opin Struct Biol 15: 35–41 [DOI] [PubMed] [Google Scholar]
- Guillier M, Allemand F, Graffe M, Raibaud S, Dardel F, Springer M, Chiaruttini C (2005a) The N-terminal extension of Escherichia coli ribosomal protein L20 is important for ribosome assembly, but dispensable for translational feedback control. RNA 11: 728–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillier M, Allemand F, Dardel F, Royer CA, Springer M, Chiaruttini C (2005b) Double molecular mimicry in Escherichia coli: binding of ribosomal protein L20 to its two sites in mRNA is similar to its binding to 23S rRNA. Mol Microbiol 56: 1441–1456 [DOI] [PubMed] [Google Scholar]
- Harms J, Schluenzen F, Zarivach R, Bashan A, Gat S, Agmon I, Bartels H, Franceschi F, Yonath A (2001) High resolution structure of the large ribosomal subunit from a mesophilic eubacterium. Cell 107: 679–688 [DOI] [PubMed] [Google Scholar]
- Hoeflich KP, Ikura M (2002) Calmodulin in action: diversity in target recognition and activation mechanisms. Cell 108: 739–742 [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M (1991) Improved methods for building protein models in electron density maps and location of errors in these models. Acta Crystallogr A 47: 110–119 [DOI] [PubMed] [Google Scholar]
- Klein DJ, Moore PB, Steitz TA (2004) The roles of ribosomal proteins in the structure assembly and evolution of the large ribosomal subunit. J Mol Biol 340: 141–177 [DOI] [PubMed] [Google Scholar]
- Kumar S, Nussinov R (2002) Close range electrostatic interactions in proteins. Chembiochem 3: 604–617 [DOI] [PubMed] [Google Scholar]
- Nowotny V, Nierhaus KH (1980) Protein L20 from the large subunit of Escherichia coli ribosomes is an assembly protein. J Mol Biol 137: 391–399 [DOI] [PubMed] [Google Scholar]
- Raibaud S, Lebars I, Guillier M, Chiaruttini C, Bontems F, Rak A, Garber M, Allemand F, Springer M, Dardel F (2002) NMR structure of bacterial ribosomal protein L20: implications for ribosome assembly and translational control. J Mol Biol 323: 143–151 [DOI] [PubMed] [Google Scholar]
- Schuwirth BS, Borovinskaya MA, Hau CA, Zhang W, Vila-Sanjurjo A, Holton JM, Doudna Cate JH (2005) Structures of the bacterial ribosome at 3.5 Å resolution. Science 310: 827–834 [DOI] [PubMed] [Google Scholar]
- Slaughter BD et al. (2005) Conformational substates of calmodulin revealed by single-pair fluorescence resonance energy transfer: influence of solution conditions and oxidative modification. Biochemistry 44: 3694–3707 [DOI] [PubMed] [Google Scholar]
- Sun H, Yin D, Coffeen LA, Shea MA, Squier TC (2001) Mutation of Tyr 138 disrupts the structural coupling between the opposing domains in vertebrate calmodulin. Biochemistry 40: 9605–9617 [DOI] [PubMed] [Google Scholar]
- Tissot AC, Vuilleumier S, Fersht AR (1996) Importance of two buried salt bridges in the stability and folding pathway of barnase. Biochemistry 35: 6786–6794 [DOI] [PubMed] [Google Scholar]
- Tompa P, Csermely P (2004) The role of structural disorder in the function of RNA and protein chaperones. FASEB J 18: 1169–1175 [DOI] [PubMed] [Google Scholar]
- Wright PE, Dyson HJ (1999) Intrinsically unstructured proteins: re-assessing the protein structure–function paradigm. J Mol Biol 293: 321–331 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supplementary Fig S1