

Abstract
Incorporation of uracil during DNA synthesis is among the most common types of endogenously generated DNA damage. Depletion of Caenorhabditis elegans dUTPase by RNA interference allowed us to study the role of DNA damage response (DDR) pathways when responding to high levels of uracil in DNA. dUTPase depletion compromised development, caused embryonic lethality and led to activation of cell-cycle arrest and apoptosis. These phenotypes manifested as a result of processing misincorporated uracil by the uracil-DNA glycosylase UNG-1. Strikingly, abrogation of the clk-2 checkpoint gene rescued lethality and developmental defects, and eliminated cell-cycle arrest and apoptosis after dUTPase depletion. These data show a genetic interaction between UNG-1 and activation of the CLK-2 DDR pathway after uracil incorporation into DNA. Our results indicate that persistent repair intermediates and/or single-stranded DNA formed during repair of misincorporated uracil are tolerated in the absence of the CLK-2 checkpoint in C. elegans.
Keywords: base-excision repair, checkpoints, CLK-2, UNG, uracil
Introduction
Cellular responses to DNA damage include the activation of DNA repair and a highly conserved signal-transduction cascade, called the DNA damage response (DDR), which leads to cell-cycle arrest or apoptosis. Activation of the DDR by endogenously generated DNA base damage has not been studied extensively, partly because methods to introduce such lesions above steady-state levels have not been readily available. Incorporation of uracil into DNA due to the use of dUMP instead of dTMP by DNA polymerases is the most common type of endogenous DNA damage (Guillet & Boiteux, 2003). Uracil misincorporation is counteracted by dUTP nucleotidohydrolase (dUTPase), which maintains a low intracellular pool of dUTP (Fig 1; Tye & Lehman, 1977). Hence, uracil incorporation into DNA can be promoted by depleting dUTPase (dut-1). Using RNA interference (RNAi) in Caenorhabditis elegans to deplete dUTPase allowed us to circumvent the lethality observed in dut-1 loss-of-function mutants and to expand on previous studies in single-celled dut-1 mutants. Thus, we could assess the activation of DDR pathways in response to misincorporation of uracil in DNA in an animal model.
Figure 1.
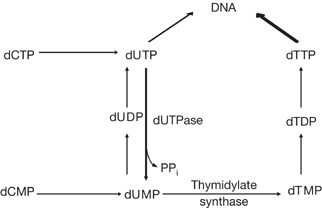
The de novo pathway for thymidine biosynthesis. A simplified scheme is shown. Use of dUTP for DNA synthesis in place of dTTP can be promoted by depletion of dUTPase.
C. elegans is the simplest animal that allows the study of the DDR (Stergiou & Hengartner, 2004). Genotoxic insult or replicative stress leads to two spatially distinct checkpoint responses in the C. elegans germ line. Cell-cycle arrest can be assessed by the presence of fewer but enlarged mitotic nuclei, whereas DNA-damage-induced apoptosis is restricted to the pachytene region of the meiotic compartment, in which dying cells can be monitored by their distinct morphological appearance. Both responses require DNA-damage-induced signalling through conserved pathways (Stergiou & Hengartner, 2004). In response to ionizing radiation (IR)- and hydroxyurea (HU)-induced replication stress, recruitment of replication factor A (RPA-1) and ATL-1 (C. elegans ATR) to chromatin is responsible for activation of the DDR checkpoint (Garcia-Muse & Boulton, 2005). The proliferating cell nuclear antigen-like complex (9-1-1 complex), consisting of a heterotrimer of the HPR-9/HUS-1/MRT-2 proteins (Hofmann et al, 2002), is required for IR-induced checkpoint responses but is dispensable for the checkpoint that follows replication stress. Checkpoint responses to IR- and HU-induced replication stress also require CLK-2 (Ahmed et al, 2001), but the precise molecular function of this protein is unknown.
Here, we identify a novel function for the CLK-2 checkpoint protein in the activation of cell-cycle arrest and apoptosis in response to dut-1(RNAi). Furthermore, we show a genetic interaction between the base-excision repair (BER) enzyme UNG-1 and activation of the DDR signalling pathway through CLK-2 after uracil excision from DNA.
Results and Discussion
dut-1(RNAi) is lethal in Caenorhabditis elegans
To increase uracil incorporation into DNA, dUTPase was depleted by RNAi feeding (dut-1(RNAi)). Larval stage 4 (L4) N2 wild-type worms subjected to dut-1(RNAi) resulted in embryonic lethality in the offspring (72% dead embryos of 1,147 inspected), indicating that dUTPase is essential for viability (Table 1). The progeny that hatched (28%) remained thin and elongated, consistent with developmental arrest, and only 0.8% grew to an L4-like size. A total of 26% of the offspring showed a severe protruding vulva (Pvl) phenotype (supplementary Fig 1 online). Reduction of the dut-1(RNAi) dose by lowering the IPTG (isopropyl-beta-D-thiogalactopyranoside) concentration gave fewer animals with a Pvl phenotype. However, the vulval structures still remained dysfunctional, resulting in egg-laying-defective (Egl) animals (supplementary Fig 1 online). Partial rescue of lethality and emergence of Egl rather than Pvl phenotypes with thymidine-supplemented growth medium (data not shown) strongly suggests that the observed defects are related directly to dUTPase depletion. Thus, dut-1(RNAi) interfered with germline and vulva development, the two proliferating organs of the adult worm.
Table 1.
dut-1(RNAi) is embryonic lethal in N2 wild-type Caenorhabditis elegans
| RNAi | Animals inspected | Embryonic lethal (%) | Hatching (%) | Adult (%) | t-Test (P)* |
|---|---|---|---|---|---|
|
L4440 |
1,260 |
6 (0.5) |
1,254 (99.5) |
1,254 (99.5) |
NA |
| dut-1 | 1,147 | 824 (72) | 324 (28) | 9 (0.8)‡ | <0.001 |
NA, not applicable; RNAi, RNA interference.
*P-values calculated from two-tailed Student's t-test after dut-1(RNAi) compared with empty vector control.
‡L4-like phenotype characterized by adult-sized animals but no formation of oocytes or embryos.
Cell-cycle arrest and apoptosis after dut-1(RNAi)
The incomplete penetrance of dut-1(RNAi) allows the observation of post-embryonic phenotypes resulting from dUTPase depletion. Detailed analysis of 4,6-diamidino-2-phenylindole (DAPI)-stained, dissected germ lines of dut-1(RNAi)-fed animals showed the presence of enlarged mitotic nuclei (Fig 2A). Also, the number of mitotic nuclei was significantly reduced from about 65 nuclei in worms fed the empty vector control RNAi to fewer than 40 fed on dut-1(RNAi) (Fig 2A). This reduction, combined with the increased nuclear size, is consistent with checkpoint activation and induction of cell-cycle arrest (Stergiou & Hengartner, 2004). Scoring of apoptotic corpses showed 1–2 apoptotic corpses per gonad arm in the control RNAi-fed animals, whereas 6–12 corpses were seen in dut-1(RNAi)-fed animals, consistent with activation of apoptosis. We conclude that the DDR checkpoint is activated in the C. elegans germ line in response to dUTPase depletion.
Figure 2.
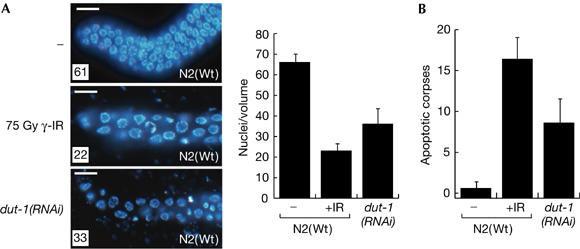
dut-1(RNAi) induces cell-cycle arrest and apoptosis in wild-type animals. (A) The presence of fewer but enlarged 4,6-diamidino-2-phenylindole-stained nuclei in the mitotic region of dissected gonads shows induction of cell-cycle arrest in dut-1(RNAi)-fed N2 animals. Germ lines extruded from untreated (−) and γ-irradiated (75 Gy γ-IR) animals are shown for comparison. The number of nuclei was counted in a volume of 54,000 μm3 16 h after the exposure of L4 larvae to the indicated treatments in 25 germ lines. Scale bars, 5 μm. (B) Induction of apoptosis was scored by counting apoptotic corpses in the pachytene region of the germ line under differential interference contrast before (−) and after (+) ionizing radiation (IR), and after dut-1(RNAi). Error bars indicate the standard error of the mean for at least 20 adult worms from three independent experiments. RNAi, RNA interference; Wt, wild type.
dut-1(RNAi) lethality is suppressed by ung-1(RNAi)
The BER pathway is the principal repair pathway for endogenous DNA damage (Barnes & Lindahl, 2004). The lethality resulting from dUTPase depletion might therefore be caused by excision of uracil by UNG-1, the only uracil-DNA glycosylase encoded in the C. elegans genome. Indeed, co-depletion of dut-1 and ung-1 in the RNAi-sensitive strain rrf-3 partly rescued germline development, allowing formation of oocytes and sperm in 3% of the animals inspected (Table 2). Rescue was never observed when dut-1(RNAi) was fed together with the empty vector control, although a fraction of the offspring (0.8%) grew to an adult size, later arresting with a Pvl phenotype (Table 2). The rescue of dut-1(RNAi) lethality by ung-1(RNAi) strongly suggests that the phenotype observed is a direct consequence of BER-dependent processing of misincorporated uracil.
Table 2.
Suppressors of (dut-1)RNAi lethality in rrf-3
| RNAi | Animals inspected | Adults (% of total brood) | t-Test (P)* |
|---|---|---|---|
|
L4440 |
1,399 |
1,385 (99) |
NA |
| dut-1 | 1,965 | 7 (0.4)‡ | NA |
|
L4440;dut-1 |
1,593 |
13 (0.8)‡ |
NA |
| ung-1 | 1,227 | 1206 (98) | NA |
|
ung-1;dut-1 |
1,993 |
52 (3) |
0.08 |
| clk-2 | 571 | 535 (95) | NA |
|
clk-2;dut-1 |
489 |
34 (7) |
0.013 |
| chk-1 | 1,156 | 481 (42)§ | NA |
| chk-1;dut-1 | 1,016 | 46 (5)§ | 0.011 |
NA, not applicable; RNAi, RNA interference.
*P-values calculated from one-tailed Student's t-test between either dut-1(RNAi) in combination with empty vector or dut-1(RNAi) in combination with test constructs as indicated.
‡L4-like phenotype characterized by adult-sized animals but no formation of oocytes or embryos.
§Sterile offspring.
Screen for suppressors of dut-1(RNAi)
To identify genes required for cell-cycle arrest and apoptosis in response to dut-1(RNAi), we co-depleted genes known to be involved in DDR pathways in C. elegans. Surprisingly, co-depletion of dUTPase with either clk-2 or chk-1 checkpoint genes resulted in 7% and 5% of the offspring, respectively, developing into adults (Table 2). Although the clk-2(RNAi);dut-1(RNAi) offspring could be studied further, the chk-1(RNAi);dut-1(RNAi) offspring were all sterile, preventing further analyses of germline phenotypes as with the phenotype observed for chk-1(RNAi) alone (Kalogeropoulos et al, 2004). Co-depletion of hpr-17, the C. elegans homologue of the human RAD17 (RFC1-like alternative clamp loader), or hus-1, failed to rescue the lethality. This indicates that signalling through the 9-1-1 complex is dispensable for the dut-1(RNAi) phenotypes (supplementary Table 1 online). This was supported by the lack of rescue in the mrt-2(e2663) mutant (data not shown). Moreover, a loss-of-function mutation in CEP-1 (cep-1 (gk138)), the C. elegans homologue of p53, did not rescue dut-1(RNAi) lethality (data not shown), consistent with the fact that CEP-1 is dispensable for checkpoint-dependent cell-cycle arrest (Schumacher et al, 2001).
Although ATL-1 and CLK-2 are both required for the S-phase checkpoint (Garcia-Muse & Boulton, 2005), co-depletion of atl-1 with dUTPase failed to rescue the dut-1(RNAi) phenotype. These different phenotypic outcomes might point to a function not shared by CLK-2 and ATL-1.
Genetic mutants rescue dut-1(RNAi) lethality
The double-RNAi screen identified genes that provided an obvious, but not highly penetrant, rescue of the dut-1(RNAi) phenotype. Therefore, clk-2 and ung-1 mutants (supplementary Fig 2 online) were used to confirm our previous findings. Depletion of dUTPase in either clk-2 or ung-1 mutants robustly rescued the dut-1(RNAi) phenotype compared with the N2 control, with about 30% of the progeny developing into adults (Table 3). Germline development after dut-1(RNAi) was also rescued in both clk-2 (Fig 3A,e) and ung-1 (Fig 3A,f) mutants. Although the clk-2 mutant completely rescued vulva development and functionality (Fig 3Ae), some ung-1 mutants developed a Pvl phenotype (Fig 3Af, inset), suggesting incomplete rescue. Importantly, both mutants abrogated activation of cell-cycle arrest, as no significant reduction in the number of mitotic nuclei, or enlarged mitotic nuclei (data not shown), were seen in either mutant (Fig 3B). Similarly, no elevation in the number of apoptotic corpses was observed in response to dut-1(RNAi) in either mutant (Fig 3C). We conclude that both excision of uracil by UNG-1 and a functional CLK-2 checkpoint are required to activate cell-cycle arrest and apoptosis in response to uracil misincorporation into DNA in C. elegans.
Table 3.
Rescue of dut-1(RNAi) lethality in genetic mutants
| Strain | RNAi | Adults (% of total (n))* | t-Test§ |
|---|---|---|---|
| N2 |
L4440 |
99.5±0.4 (1260) |
|
| |
dut-1 |
0.8±0.5 (1147)‡ |
NA |
|
clk-2 |
L4440 |
98.4±2.4 (502) |
|
| |
dut-1 |
24.5±14 (1230) |
P<0.001 |
|
ung-1 |
L4440 |
99.8±0.4 (535) |
|
| dut-1 | 30±8 (686) | P=0.001 |
NA, not applicable; RNAi, RNA interference.
*Survival was scored as the number of offspring that developed into adults and is given (±s.e.m.) for at least five independent experiments. n is the total number of animals inspected.
‡L4-like phenotype characterized by adult-sized animals but no formation of oocytes or embryos.
§P-values calculated from two-tailed Student's t-test after dut-1(RNAi) in N2 compared with clk-2 or ung-1.
Figure 3.
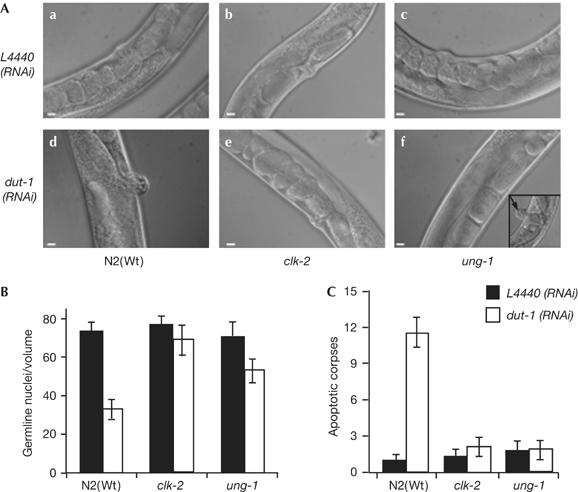
Rescue of dut-1(RNAi) in ung-1 and clk-2 mutants. (A) Differential interference contrast images of representative phenotypes observed in the progeny in empty vector L4440(RNAi) control-fed (a–c) compared with dut-1(RNAi)-fed (d–f) animals. N2 worms fed dut-1(RNAi) develop a strong protruding vulva (Pvl) phenotype (d), which was fully rescued in the clk-2 mutant (e) but only partly rescued in ung-1, as some animals showed a Pvl phenotype (f, arrow pointing to Pvl in the inset). Scale bars, 10 μm. (B) Cell-cycle arrest and (C) apoptotic corpses per germ line were counted 16 h after exposing L4 to dut-1(RNAi) (open bars) compared with animals fed empty vector control, L4440(RNAi) (filled bars). Error bars indicate the standard error of the mean for at least 20 adult worms from three independent experiments. RNAi, RNA interference; Wt, wild type.
Repair intermediates are tolerated in clk-2
Consistent with the activation of cell-cycle arrest and apoptosis after dut-1(RNAi), dUTPase depletion in the N2 strain resulted in the accumulation of RPA-1 (data not shown) and ATL-1 (Fig 4A,B) foci on chromatin, which indicates S-phase checkpoint activation (Garcia-Muse & Boulton, 2005). Uracil-containing DNA is itself not sufficient to trigger the DNA damage checkpoint, as co-depletion of ung-1 and dut-1 abolished RPA-1 and ATL-1 focus formation. This result confirms that processing of misincorporated uracil by UNG-1 is required to trigger checkpoint activation. Furthermore, clk-2 mutants under dut-1(RNAi) conditions also accumulate RPA-1 (data not shown) and ATL-1 foci, similar to that observed after dut-1 depletion in the wild type. Immunostaining with anti-RAD-51 antibodies, the most accepted marker for double-strand break (DSB) formation in C. elegans (Alpi et al, 2003), showed a significant increase in RAD-51 foci after dut-1(RNAi) in both wild type and clk-2 mutants. RAD-51 focus formation depended on the UNG-1 enzyme (Fig 4C,D). This result indicates that the different outcomes of dut-1(RNAi) in the wild-type and clk-2 mutant strains are not due to a reduction in DSB formation in the mutant. Collectively, these results indicate that the rescue of dut-1(RNAi) in the clk-2 mutants is not due to a defect in incorporation or excision of uracil, but that the DNA repair intermediates arising from the UNG-1-mediated excision of uracil are tolerated in the absence of CLK-2. These data further suggest a role for CLK-2 downstream from RPA-1 and ATL-1 in the S-phase checkpoint pathway (Garcia-Muse & Boulton, 2005).
Figure 4.

Recruitment of ATL-1 and RAD-51 to chromatin after dut-1(RNAi) depends on UNG-1. Representative images of anti-ATL-1 (top left, A) and anti-RAD-51 (top right, C) staining of fixed mitotic nuclei from N2 wild-type and clk-2 mutant animals 16 h after exposure of L4 larvae to the indicated RNAi. (B,D) Quantification of the data is shown for N2 (filled) and clk-2 (open) bars. Error bars indicate the standard error of the mean for at least 20 adult worms from two independent experiments. RNAi, RNA interference; Wt, wild type.
In conclusion, depletion of dUTPase activity by RNAi enhances uracil misincorporation into DNA. This leads to checkpoint activation only after processing of uracil by UNG-1. Uracil excision leads to recruitment of RPA-1 and ATL-1 to chromatin and subsequent activation of the CLK-2 checkpoint, which manifests as cell-cycle arrest and activation of apoptosis in the C. elegans germ line, independent of the 9-1-1 complex. Thus, the genetic requirements for activation of the DDR in response to dut-1(RNAi) differ from those primarily responding to ionizing radiation. Previous studies of molecular events leading to lethality in dUTPase mutants concentrated on the network of enzymes directly processing uracil in DNA (Guillet et al, 2006). However, we report a novel genetic interaction between UNG-1 and CLK-2 demonstrating that activation of the DDR in response to misincorporated uracil depends on DNA repair. Indeed, abrogation of the CLK-2 checkpoint leads to tolerance to DNA repair intermediates. Consequently, misincorporated uracil, a presumed innocuous lesion, can be processed by UNG-1 to yield repair intermediates that might be carried into the next generation as a mutagenic and cytotoxic lesion in the absence of CLK-2.
Recent reports demonstrated that DDR pathways are constitutively activated in pre-neoplastic human cells (Bartkova et al, 2005; Gorgoulis et al, 2005). It was suggested that inappropriate stimulation of replication could lead to replication stress and activation of the DNA damage checkpoint, and that this would provide selection pressure for the subsequent inactivation of tumour suppressor genes. The results presented here confirm that endogenous DNA base damage might contribute to replication stress and activation of DDR pathways. Therefore, we speculate that elevated steady-state levels of uracil could contribute to the development of cancer, not only by increasing the mutation rate (An et al, 2005) but also by contributing to replication stress. Our data indicate that CLK-2 could function to prevent mutagenesis caused by propagation of repair intermediates arising from the processing of endogenous DNA damage.
Methods
Strains and culture conditions. C. elegans strains were cultured using standard procedures. All experiments were performed at 15°C unless stated otherwise. The Caenorhabditis Genetics Centre (University of Minnesota, St Paul, MN, USA) provided the following strains: the reference strain Bristol N2, clk-2(mn159), NL2099 rrf-3(pk1426), VC172 cep-1(gk138) and CB5348 mrt-2(e2663). A frozen trimethylpsoralen-UV (TMP-UV) deletion library was prepared essentially as described (Liu et al, 1999). The ung-1(qa7600) allele was identified by poison primer PCR screening (Edgley et al, 2002). The ung-1 mutant was recovered by sibling selection and was backcrossed six times into the N2 wild-type strain. RNAi was performed by feeding, as described previously (Boulton et al, 2002). Double-feeding experiments were performed in the RNAi-sensitive strain rrf-3(pk1426) (Simmer et al, 2002) by mixing fresh overnight cultures of Escherichia coli strains harbouring dut-1(RNAi) feeding construct at 1:5 ratio with strains expressing either test plasmids or the empty vector control, pL4440, to establish baseline survival. Survival in RNAi feeding experiments was scored as the fraction of the progeny that developed past L4.
Gateway recombination cloning. Open-reading-frame-specific primers compatible with the Gateway system (Invitrogen, UK) were designed for dut-1 (K07A1.2), ung-1 (Y56A3A.29a), clk-2 (C07H6.6), chk-1 (Y39H10A.7), rad-51 (Y43C5A.6), chk-2 (Y60A3A.12), atl-1 (T06E4.3), hpr-17 (F32A11.2), hus-1 (H26D21.1) and cep-1 (F52B5.5), PCR amplified and cloned into pL4440-dest, as described previously (Boulton et al, 2002).
Cytological preparation and immunostaining. After L4 animals were subjected to RNAi for 16 h, the germ lines of gravid hermaphrodites were extruded and processed for immunostaining, as described previously (Garcia-Muse & Boulton, 2005). Primary antibodies were diluted in TBS-T (TBS+0.5% BSA+0.1% Triton X-100; 1:200 for anti-RAD-51, 1:500 for anti-RPA-1 and 1:500 for anti-ATL-1) and incubated overnight at 4°C in a humid chamber. The secondary antibody, anti-rabbit Cy3 (Sigma, UK), was used at a 1:10,000 dilution. Apoptotic corpses were scored using differential interference contrast (DIC) microscopy, and cell-cycle arrest was scored in extruded germ lines stained with DAPI, as described previously (Gartner et al, 2004).
Microscopy. DIC microscopy was performed on a Zeiss Axiovert 200M inverted microscope with × 20 Plan-Apochromat 0.8 NA or × 63Plan-Apochromat 1.4 NA objectives. Deltavision microscopy was used to examine germ lines after immunostaining with × 40 or × 63 1.4 NA Planachromat objectives, as described previously (Garcia-Muse & Boulton, 2005).
Supplementary information is available at EMBO reports online (http://www.emboreports.org).
Supplementary Material
Supplementary Information
Acknowledgments
We thank the C. elegans Genetics Centre for providing C. elegans and bacterial strains, and Dr E. Boye and S.S. Salus for a critical reading of the manuscript. This work was supported by the University of Oslo (H.N., M.D.) and by Breast Cancer Campaign (GA3221) and Cancer Research UK (S.J.B. and T.G.-M.).
References
- Ahmed S, Alpi A, Hengartner MO, Gartner A (2001) C. elegans RAD-5/CLK-2 defines a new DNA damage checkpoint protein. Curr Biol 11: 1934–1944 [DOI] [PubMed] [Google Scholar]
- Alpi A, Pasierbek P, Gartner A, Loidl J (2003) Genetic and cytological characterization of the recombination protein RAD-51 in Caenorhabditis elegans. Chromosoma 112: 6–16 [DOI] [PubMed] [Google Scholar]
- An Q, Robins P, Lindahl T, Barnes DE (2005) C → T mutagenesis and λ-radiation sensitivity due to deficiency in the Smug1 and Ung DNA glycosylases. EMBO J 24: 2205–2213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes DE, Lindahl T (2004) Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Annu Rev Genet 38: 445–476 [DOI] [PubMed] [Google Scholar]
- Bartkova J et al. (2005) DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 434: 864–870 [DOI] [PubMed] [Google Scholar]
- Boulton SJ, Gartner A, Reboul J, Vaglio P, Dyson N, Hill DE, Vidal M (2002) Combined functional genomic maps of the C. elegans DNA damage response. Science 295: 127–131 [DOI] [PubMed] [Google Scholar]
- Edgley M, D'Souza A, Moulder G, McKay S, Shen B, Gilchrist E, Moerman D, Barstead R (2002) Improved detection of small deletions in complex pools of DNA. Nucleic Acids Res 30: e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Muse T, Boulton SJ (2005) Distinct modes of ATR activation after replication stress and DNA double-strand breaks in Caenorhabditis elegans. EMBO J 24: 4345–4355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gartner A, MacQueen AJ, Villeneuve AM (2004) Methods for analyzing checkpoint responses in Caenorhabditis elegans. Methods Mol Biol 280: 257–274 [DOI] [PubMed] [Google Scholar]
- Gorgoulis VG et al. (2005) Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 434: 907–913 [DOI] [PubMed] [Google Scholar]
- Guillet M, Boiteux S (2003) Origin of endogenous DNA abasic sites in Saccharomyces cerevisiae. Mol Cell Biol 23: 8386–8394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillet M, Van Der Kemp PA, Boiteux S (2006) dUTPase activity is critical to maintain genetic stability in Saccharomyces cerevisiae. Nucleic Acids Res 34: 2056–2066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann ER, Milstein S, Boulton SJ, Ye M, Hofmann JJ, Stergiou L, Gartner A, Vidal M, Hengartner MO (2002) Caenorhabditis elegans HUS-1 is a DNA damage checkpoint protein required for genome stability and EGL-1-mediated apoptosis. Curr Biol 12: 1908–1918 [DOI] [PubMed] [Google Scholar]
- Kalogeropoulos N, Christoforou C, Green AJ, Gill S, Ashcroft NR (2004) chk-1 is an essential gene and is required for an S–M checkpoint during early embryogenesis. Cell Cycle 3: 1196–1200 [PubMed] [Google Scholar]
- Liu LX et al. (1999) High-throughput isolation of Caenorhabditis elegans deletion mutants. Genome Res 9: 859–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher B, Hofmann K, Boulton S, Gartner A (2001) The C. elegans homolog of the p53 tumor suppressor is required for DNA damage-induced apoptosis. Curr Biol 11: 1722–1727 [DOI] [PubMed] [Google Scholar]
- Simmer F, Tijsterman M, Parrish S, Koushika SP, Nonet ML, Fire A, Ahringer J, Plasterk RH (2002) Loss of the putative RNA-directed RNA polymerase RRF-3 makes C. elegans hypersensitive to RNAi. Curr Biol 12: 1317–1319 [DOI] [PubMed] [Google Scholar]
- Stergiou L, Hengartner MO (2004) Death and more: DNA damage response pathways in the nematode C. elegans. Cell Death Differ 11: 21–28 [DOI] [PubMed] [Google Scholar]
- Tye BK, Lehman IR (1977) Excision repair of uracil incorporated in DNA as a result of a defect in dUTPase. J Mol Biol 117: 293–306 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information