

Abstract
Cdc20 and cdh1 are coactivators of the anaphase-promoting complex (APC). APCcdc20 is necessary for the metaphase–anaphase transition and, at the end of mitosis, vertebrate cdc20 itself becomes a target for degradation through KEN-box-dependent APCcdh1 activity. By studying the degradation of fluorescent protein chimaeras in mammalian oocytes and early embryos, we found that cdc20 was degraded through two independent degradation signals (degrons), the KEN box and a newly described CRY box. In both oocytes and G1-stage embryos, the rate of degradation through the CRY box was greater than through the KEN box, although both were mediated by APCcdh1. Thus, mammalian oocytes and embryos have the capacity to recognize two degrons in cdc20.
Keywords: cell-cycle proteins, metaphase, meiosis, oocytes, ubiquitin-ligase protein complexes
Introduction
Cdc20, an essential activator of the anaphase-promoting complex/cyclosome (APC), is an important cell-cycle protein involved universally in the metaphase–anaphase transition. It is unstable during the cell cycle, and its instability seems to be due to more than one discrete degradation signal (referred to as a ‘degron') located in its primary sequence (Fang et al, 1998; Prinz et al, 1998; Pfleger & Kirschner, 2000; Huang et al, 2001).
In the budding yeast Saccharomyces cerevisiae, APCcdh1-mediated cdc20 degradation during the G1 phase of the cell cycle is mediated by one of its two amino-terminal destruction (D)-box degrons (Prinz et al, 1998; Huang et al, 2001). Mammalian cdc20, like budding yeast, is unstable in G1 but lacks a D box (Fang et al, 1998). Instead, its stability seems to be regulated by a single degron, the KEN box (Pfleger & Kirschner, 2000). The KEN box is exclusively recognized by APCcdh1 (Zur & Brandeis, 2002) and has been found on other proteins, such as securin, in which it confers degradation during late M and early G1 (Hagting et al, 2002).
Interestingly, APCcdh1 activity is not confined to late mitosis. Cdh1 has been found to be present in postmitotic neurons (Gieffers et al, 1999), in which it could be co-purified with the APC and was capable of ubiquitin-ligase activity. In fact, APCcdh1 functions in axonal morphogenesis in brain neurons (Konishi et al, 2004). Similarly, we have recently observed that APCcdh1 also functions in maintaining germinal-vesicle (GV)-stage oocytes at prophase I (Reis et al, 2006). These oocytes are from antral follicles and, within the follicle, remain GV arrested until a mid-cycle luteinizing hormone surge. In that study, it was observed that cdc20-green fluorescent protein (GFP) is degraded in oocytes, which is consistent with APCcdh1 activity. Here, we show that such degradation is indeed mediated by APCcdh1, but rather surprisingly it can occur through either the KEN box or a second portable degron, which is called the CRY box (CRYxPS).
Results and Discussion
KEN-box-independent cdc20 degradation in GV oocytes
GV-stage mouse oocytes were maintained in arrest at prophase I and microinjected with complementary RNA to either GFP or full-length cdc20, coupled to GFP at the carboxyl terminus (cdc20–GFP). GFP and cdc20–GFP were allowed to express for a few hours; then, further protein expression was blocked by either the addition of cycloheximide (Fig 1A) or microinjection of an antisense oligonucleotide to GFP (supplementary Figure S1 online), which we have used previously to block expression of GFP chimaeras (Nixon et al, 2002). Similar observations were made with both procedures. After blocking expression of cdc20–GFP, we observed its steady degradation; by contrast, GFP alone was unstable, with little to no degradation. Such observations are consistent with our recent finding that APCcdh1 is active in GV oocytes and contributes to GV arrest (Reis et al, 2006). The rate of degradation is slow compared with the degradation of various cell-cycle proteins in mitosis (Hagting et al, 2002; Lindon & Pines, 2004), but the rate of degradation is similar to that previously observed for other APCcdh1 substrates (Reis et al, 2006). It is possible that, in a large oocyte, slow degradation is accounted for by spatially restricted degradation or by the presence of APCcdh1 inhibitors such as Emi1 (Margottin-Goguet et al, 2003; Ohsumi et al, 2004).
Figure 1.
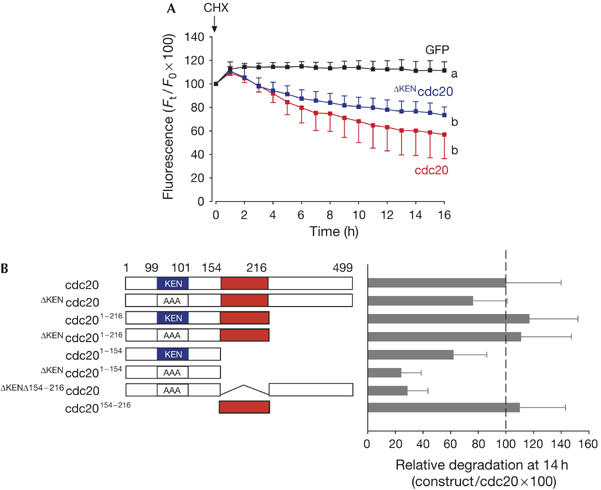
Identification of a second degradation signal in cdc20. (A) Germinal-vesicle-arrested oocytes were microinjected with complementary RNA to green fluorescent protein (GFP; black, n=16), cdc20–GFP (red, n=18) or ΔKENcdc20–GFP (blue, n=14). Allowing 2–3 h for expression, oocytes were then incubated with cycloheximide (CHX). a and b denote significantly different fluorescence at the 16 h time point (P<0.05 χ2 test). (B) Various cdc20 constructs, coupled at their carboxyl terminus to GFP, were assayed for their degradation in oocytes on addition of CHX (n=15–25 per construct). Mean GFP fluorescence loss in oocytes after 14 h was expressed as a quotient with respect to cdc20–GFP, such that full-length cdc20 was assigned 100% degradation. From these deletion and mutation experiments, we identified two regions that were determinants of cdc20 stability: the KEN box (in blue) and residues 154–216 in cdc20 (red).
We assumed that cdc20–GFP was degraded by APCcdh1 in GV oocytes; so we mutated the KEN degron to AAA anticipating that this would block degradation. However, the KEN mutant construct (ΔKENcdc20) was also degraded on addition of cycloheximide or after antisense injection (Fig 1A; supplementary Figure S1 online). In fact, the removal of the KEN degron had only a small impact on the rate of cdc20 degradation, as degradation rates of ΔKENcdc20–GFP were less than those of full-length cdc20–GFP; however, this difference was not statistically significant. There are two possible explanations for the lack of effect of mutating the KEN box on cdc20 degradation: (i) the recognition sequence for the KEN degron is relaxed in oocytes when compared with somatic cells, such that APCcdh1 can effectively ubiquitinate cdc20 using residues neighbouring the KEN sequence; or (ii) degradation is conferred by a novel degron that may or may not be recognized by APCcdh1.
Identification of a second degradation signal in cdc20
To further characterize the putative new degron, various cdc20 deletion constructs coupled to GFP were expressed in GV-arrested oocytes; cycloheximide was then added to block further synthesis. To perform a large number of experiments, cycloheximide was used in preference to the GFP-oligonucleotide antisense construct, which would involve further microinjection. Removal of the C-terminal half (residues 217–499) of either cdc20 or ΔKENcdc20 removed six of the seven predicted WD repeats (residues 182–211 form the first most N-terminal WD repeat identified by http://bmerc-www.bu.edu/projects/wdrepeat/) and the C-terminal IR (Ile/Arg) tail involved in APC binding (Vodermaier et al, 2003), but the resulting constructs (cdc201−216 and ΔKENcdc201−216) were still degraded with rates similar to those of full-length cdc20 (Fig 1B). However, removal of a further 62 residues from cdc201−216 generated a construct (cdc201−154) that showed greater stability than the other constructs. cdc201−154 was further stabilized by mutating the KEN box (ΔKENcdc201−154). These data suggest the existence of a second, KEN-independent degron between residues 154 and 216. This was confirmed when this region alone was tagged to GFP (cdc20154−216) and was degraded with the same kinetics as full-length cdc20 (Fig 1B). There were no other observable degradation signals outside these two regions (the KEN box and residues 154–216), as the double degron mutant, ΔKENΔ154−216cdc20, was stable. Therefore, with respect to its degradation in GV oocytes, cdc20 seems to contain two distinct degrons: the KEN box and a second degron between residues 154 and 216.
The cdc20 CRY box is recognized by APCcdh1
To narrow down the degron further, we made various N-terminal deletions of this construct. The Venus fluorochrome was used in preference to GFP for these experiments because we obtained a significantly brighter signal. The cdc20154−216–Venus and cdc20164−216–Venus constructs were degraded in oocytes (Fig 2A,B, red traces) but cdc20174−216 and cdc20184−216 were not (Fig 2A,B, black traces), suggesting that residues 164–174 may contain the degron (Fig 2A). Sequential point mutation of all ten amino-acid residues 164–173 of cdc20 demonstrated that residues 165-CRYxPS-170 were required for degradation (Fig 2C). This second degradation signal in cdc20 (CRYxPS) was therefore named the CRY box.
Figure 2.
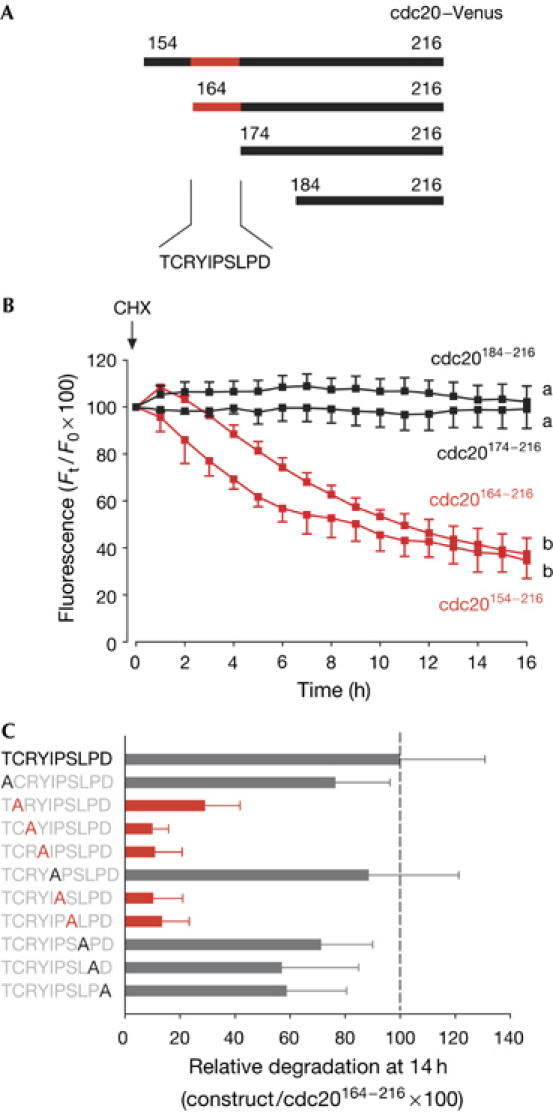
Characterization of the CRY degron in cdc20. (A,B) Cdc20–Venus complementary RNA constructs (A) were expressed in oocytes; cycloheximide (CHX) was then added to block further protein translation. (B) Cdc20174−216 (n=13) and cdc20184−216 (n=17; black traces) were stable, whereas cdc20154−216 (n=20) and cdc20164−216 (n=15; red traces) were degraded. The cdc20 sequence became stable when residues 164–174 were removed. a and b denote significantly different fluorescence at the 16 h time point (P<0.05 χ2 test). (C) To examine which residues were crucial for degradation in cdc20 residues 164–174, point mutations of each residue were made to alanine, as indicated. Mean loss of fluorescence of green fluorescent protein (GFP) in oocytes after 14 h was expressed as a quotient with respect to cdc20164−216–GFP, such that cdc20164−216 was assigned 100% degradation (n=15–20 per construct). Mutations that are defined as very stable are marked in red.
The CRY box in cdc20 conferred instability on cRNA constructs of fluorescent protein chimaeras in oocytes when further protein expression was blocked by either antisense or cycloheximide. To determine the mechanism of CRY-mediated cdc20 degradation, oocytes expressing cdc20154−216–GFP were either microinjected with methylubiquitin (Hershko et al, 1991) or incubated with the proteasomal inhibitor MG132 (Lee & Goldberg, 1996), and then challenged with cycloheximide. Both microinjection of methylubiquitin and addition of the proteasomal inhibitor MG132 acted to prevent cdc20154−216–GFP degradation (Fig 3A). Polyubiquitination of in vitro-translated (IVT) full-length cdc20–GFP, cdc20154−216–GFP and ΔKENcdc20–GFP could also be detected in rabbit reticulocyte lysates. The IVT product was incubated with unlabelled cdh1, E1, E2, rabbit reticulocyte lysate and an ATP-regenerating system. The reticulocyte lysate contains weak endogenous APCcdh1 activity (Shteinberg et al, 1999; Yudkovsky et al, 2000); therefore, we added excess unlabelled cdh1 to improve ubiquitination. In the presence of added ubiquitin, but not in its absence, high-molecular-weight cdc20–GFP, cdc20154−216–GFP and ΔKENcdc20–GFP could be observed on blots (Fig 3B).
Figure 3.
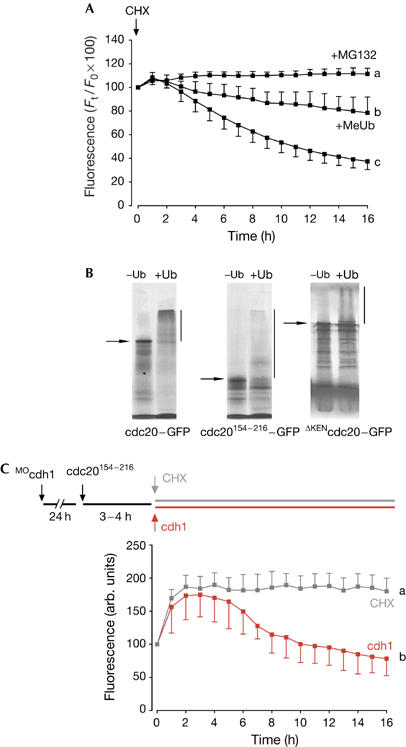
Degradation of CRY-box-containing constructs requires APCcdh1 activity. (A) Fluorescence levels of 154−216cdc20–green fluorescent protein (GFP) on cycloheximide (CHX) addition. In some oocytes, methylubiquitin (MeUb; 1 μg/ml) was microinjected or MG132 (50 μg/ml) was added to the culture medium before imaging. a–c denote significantly different fluorescence at the 16 h time point (P<0.05 χ2 test). (B) In vitro-translated cdc20–GFP, cdc20154−216–GFP or ΔKENcdc20–GFP was incubated in a ubiquitination mix with or without ubiquitin (Methods; predicted molecular weights are indicated by arrows); in the presence of ubiquitin, proteins show loss of full-length band intensity and gain of high-molecular-weight polyubiquitination adducts (vertical line). (C) Germinal-veside-stage V oocytes were microinjected with cdh1 morpholino (MOcdh1) and cultured for 24 h. A further microinjection of complementary RNA to cdc20154−216–GFP was made and allowing a few hours for expression, at time t=0 h, either CHX was added (n=14; grey trace) or cdh1 cRNA was microinjected (n=21; red trace). a and b denote significantly different fluorescence at the 16 h time point (P<0.05 χ2 test).
As degradation requires polyubiquitination and the 26S proteasome, one possibility is that CRY-box-mediated degradation could also involve the APC, especially given that we measured APCcdh1 activity in GV oocytes (Reis et al, 2006). To determine whether APCcdh1 is the E3 ligase responsible for CRY-box ubiquitination, oocytes were incubated with a morpholino antisense oligonucleotide against cdh1 (MOcdh1), which we found to be effective in reducing cdh1 protein by about 90% in 24 h (Reis et al, 2006). In these oocytes, which were allowed to express cdc20154−216–GFP, there was no CRY-box-mediated degradation on addition of cycloheximide (Fig 3C, grey trace), because the construct seemed to be stable. However, CRY-box-mediated degradation in cdh1-knockdown oocytes could be rescued by microinjection of exogenous cdh1 cRNA (Fig 3C, red trace). To prove that this approach of knocking down cdh1 and performing a cdh1 rescue experiment would work on an already-established APCcdh1 degron, we repeated these experiments for a cdc20 construct that carried only the KEN degron. Such a construct, cdc201−154, was stable in oocytes after morpholino knockdown of cdh1, but degradation could be rescued by cdh1 cRNA (supplementary Figure S2 online). We conclude that, in oocytes, both KEN- and CRY-box degradation rely on APCcdh1 activity. These experiments, along with those in supplementary Figure S1 online, also show that both KEN- and CRY-box-mediated degradation are not dependent on the presence of cycloheximide.
CRY-box degradation in G1 embryos
So far, we have described APCcdh1-mediated CRY-box degradation only in GV-stage, prophase I oocytes. APCcdh1 activity at this time in the cell cycle, equivalent to G2/M, is unusual, as it is associated with late M and early G1 of the mitotic cell cycle. It was therefore useful to determine whether the CRY box was a degron unique to APCcdh1 recognition in GV oocytes. Mouse embryos, unlike those of frog, contain distinct gaps in their cell cycle between S and M phases (Bolton et al, 1984; Moore et al, 1996). Two-cell embryos in early G1 were used, as early G1 is the cell-cycle point at which cells have maximal APCcdh1 activity (Huang et al, 2001; Rape & Kirschner, 2004). One-cell embryos were recovered from superovulated and mated females, and cultured in vitro after microinjection of various cdc20 constructs that contained no degron (ΔKENcdc201−154), the KEN box only (cdc201−154), the CRY box only (ΔKENcdc201−216) or both degrons (cdc201−216; Fig 4A). By monitoring for nuclear envelope breakdown every 10 min, we could record GFP levels when the first mitotic division was complete, as judged by a visible nuclear envelope. As predicted, the presence of either of the two degrons was necessary to observe degradation (Fig 4B). In fact, the constructs containing the CRY degron were degraded better in G1 embryos than GV oocytes (Figs 1A, 4B), which is consistent with high APCcdh1 activity at this time. Interestingly, the CRY box conferred much greater levels of degradation on GFP than did the KEN box. The presence of both boxes had no additive effect compared with the CRY box only (Fig 4).
Figure 4.
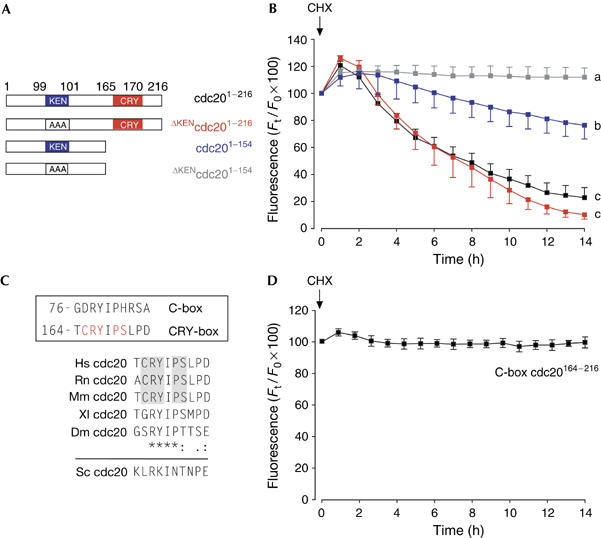
CRY-box degradation in G1 embryos. (A,B) Four green fluorescent protein (GFP)-containing constructs were used: construct containing both KEN box and CRY box (cdc201−216; n=20; black); construct containing CRY box only (ΔKENcdc201−216; n=22; red); construct containing KEN box only (cdc201−154; n=23; blue); or construct without degron (ΔKENcdc201−154; n=22; grey). Two-cell G1 embryos expressing one of these four constructs were imaged on addition of cycloheximide (CHX). a–c denote significantly different fluorescence at the 16 h time point (P<0.05 χ2 test). (C) Boxed: comparison of the C-box and the CRY-box degron in human cdc20. Residues needed for CRY-box degradation are marked in red. Comparison of the CRY box in human, rat, mouse, Xenopus and Drosophila (asterisk denotes identical residues, colon conserved residues and dot semiconserved residues). The equivalent sequence in budding yeast is also shown. (D) Lack of degradation for the construct C-box cdc20164−216 on addition of CHX at the time indicated (n=19). This construct is similar to cdc20164−216–GFP used previously (Fig 2B), except that 165-CRYIPSL-171 is changed to 165-DRYIPHR-171 to mimic the cdc20 C box.
C box and CRY box
In conclusion, here we describe a second degron in mammalian cdc20 that contributes to its degradation in oocytes and embryos; we have named this the CRY box. Identification of this sequence as a degron is interesting given that a near-match sequence is found in the N terminus of cdc20 (Fig 4C) and is known to be involved in APC binding. The N-terminal repeat forms part of the so-called conserved box (C box), which takes its name from the fact that it is present in both cdh1 and cdc20, and is involved in APC binding. Cdc20 binds to the APC through two distinct domains—its C-terminal IR tail (Vodermaier et al, 2003) and the C box, 77-DRYIPHR-83 (Schwab et al, 2001).
The C box is sufficiently distinct from the CRY box to account for the fact that it is not itself a degron, as N-terminal cdc20 constructs containing the C box, and not the CRY or KEN box, are stable (Fig 1B). Moreover, mutation of CRY-box residues to those present in the C box (CRYIPSL to DRYIPHR; C-box cdc20164−216) gives a stable construct (Fig 4D). In addition, CRY-box-mediated degradation of cdc20 can take place in the absence of a C box (Fig 2B). Therefore, the two boxes behave independently. However, it is interesting as to how two similar primary sequences in cdc20 have distinct functions: the C box in APC binding and the CRY box in cdc20 degradation. Further studies are needed to determine what, if any, functional consequence exists in terms of APCcdc20 activity and cdc20 degradation for removing both or either one of the two cdc20 degrons in oocytes and embryos. In this case, it will be necessary to knock out endogenous cdc20 expression and add back cdc20 constructs carrying one or both degrons to determine whether these constructs still retain cdc20 activity and are degraded as endogenous cdc20 in the embryonic mitotic divisions.
Methods
Oocyte and embryo culture The collection and culture of GV oocytes has been described previously (Reis et al, 2006). For one-cell embryos, superovulated females were mated and embryos recovered from the oviduct 20–22 h later. To induce GV arrest, media were supplemented with either 0.1 mM dibutyryl cyclic AMP or 1 μM milrinone. Cycloheximide was used at 50 μg/ml.
Complementary RNA Cdc20 and ΔKENcdc20 were made as described previously (Chang et al, 2004). C-terminal truncations of cdc20 and ΔKENcdc20 were made using standard PCR-based techniques for cloning into a modified pRN3 vector (Levasseur & McDougall, 2000), designed to produce either C-terminally coupled GFP or Venus. Unlabelled cdh1 was cloned from mouse testis complementary DNA using pRN3 that lacked any C-terminal tag. Δ154−216cdc20 and ΔKENΔ154−216cdc20 were made by linearizing cdc20–GFP or ΔKENcdc20–GFP, respectively, with the restriction enzyme BbsI (New England BioLabs, Hitchin, UK). This cuts cdc20 once at 486-GAAGACCTvGCCGv-497, corresponding to amino-acid residues 163-KTCR-166. The BbsI-linearized construct was used as template in a PCR reaction with primers 5′-GATCGAATTCCTGCAGCTTTTGCAAATGGAG- 3′ (forward) and 5′-GATCGAATTCGCTGTAGAGTACTTTCAGTCT- 3′ (reverse). The PCR product was cut with EcoR1 restriction enzyme, ligated and cloned using standard techniques to generate Δ154−216cdc20 and ΔKENΔ154−216cdc20. cRNA was synthesized using T3 mMESSAGE mMACHINE (Ambion, Huntingdon, UK).
Antisense constructs An antisense morpholino (5′-CCTTCGCTCATAGTCCTGGTCCATG-3′) designed to recognize the start region of the cdh1 gene was manufactured by Genetools (Philomath, OR, USA). It was used at a micropipette concentration of 1.5 mM. The extent of the knockdown of cdh1 was assessed in 50 oocytes, as described previously (Reis et al, 2006). An antisense oligonucleotide to GFP was used, as described previously (Nixon et al, 2002).
Microinjection and imaging All microinjections were made on the stage of a Nikon TE300 inverted microscope, as described previously (Nixon et al, 2002), using the negative capacitance overcompensation facility on an electrophysiological amplifier. Bright-field and epifluorescence images were recorded using an Interline MicroMax CCD camera (Sony). MetaMorph and MetaFluor software (UIC, PA) were used for image capture and data analysis. In all figures showing real-time GFP/Venus measurements, the mean GFP/Venus fluorescence levels are plotted, with standard errors, as a fluorescence reading at time t (in hours), expressed as quotient with respect to time 0 h.
In vitro ubiquitination [35S]methionine-labelled full-length cdc20–GFP and cdc20154−216–GFP were IVT in a rabbit reticulocyte lysate (Promega, Southampton, UK) by addition of cRNA, according to the manufacturer's instructions. The endogenous APC activity of the rabbit reticulocyte lysate (Shteinberg et al, 1999; Yudkovsky et al, 2000) was harnessed in an in vitro ubiquitination assay comprising 50 μM ubiquitin, 1 μM ubiquitin aldehyde, 1 pmol/10 μl E1, 5 pmol/10 μl E2, an ATP-regenerating system (1 mM ATP, 30 U/ml rabbit creatine phosphokinase type 1, 7.5 mM creatine phosphate), unlabelled cdh1, methionine-labelled GFP construct and 50 μg/ml cycloheximide. This ubiquitination mix had 3.5 μl of IVT radiolabelled APC substrate and 1.15 μl of IVT cdh1 in a total volume of 15 μl, in buffer QA (Hagting et al, 2002). Cycloheximide was added in the mix to prevent any further protein translation during the reaction period of 2 h at 30°C, after which the reaction was stopped by adding 15 μl of 2 × SDS sample loading buffer. A 10 μl portion of the reaction mixes was analysed by SDS–polyacrylamide gel electrophoresis and autoradiography.
Supplementary information is available at EMBO reports online (http://www.emboreports.org).
Supplementary Material
supplementary Figure S1
Acknowledgments
We thank J. Gannon and T. Hunt for the cdh1 antibody and J. Pines for the Venus construct. This work was supported by a Wellcome Trust Project Grant (075744) to K.T.J.
References
- Bolton VN, Oades PJ, Johnson MH (1984) The relationship between cleavage DNA replication and gene expression in the mouse 2-cell embryo. J Embryol Exp Morphol 79: 139–163 [PubMed] [Google Scholar]
- Chang HY, Levasseur M, Jones KT (2004) Degradation of APCcdc20 and APCcdh1 substrates during the second meiotic division in mouse eggs. J Cell Sci 117: 6289–6296 [DOI] [PubMed] [Google Scholar]
- Fang G, Yu H, Kirschner MW (1998) Direct binding of CDC20 protein family members activates the anaphase-promoting complex in mitosis and G1. Mol Cell 2: 163–171 [DOI] [PubMed] [Google Scholar]
- Gieffers C, Peters BH, Kramer ER, Dotti CG, Peters JM (1999) Expression of the CDH1-associated form of the anaphase-promoting complex in postmitotic neurons. Proc Natl Acad Sci USA 96: 11317–11322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagting A, Den Elzen N, Vodermaier HC, Waizenegger IC, Peters JM, Pines J (2002) Human securin proteolysis is controlled by the spindle checkpoint and reveals when the APC/C switches from activation by Cdc20 to Cdh1. J Cell Biol 157: 1125–1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko A, Ganoth D, Pehrson J, Palazzo RE, Cohen LH (1991) Methylated ubiquitin inhibits cyclin degradation in clam embryo extracts. J Biol Chem 266: 16376–16379 [PubMed] [Google Scholar]
- Huang JN, Park I, Ellingson E, Littlepage LE, Pellman D (2001) Activity of the APCCdh1 form of the anaphase-promoting complex persists until S phase and prevents the premature expression of Cdc20p. J Cell Biol 154: 85–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi Y, Stegmuller J, Matsuda T, Bonni S, Bonni A (2004) Cdh1-APC controls axonal growth and patterning in the mammalian brain. Science 303: 1026–1030 [DOI] [PubMed] [Google Scholar]
- Lee DH, Goldberg AL (1996) Selective inhibitors of the proteasome-dependent and vacuolar pathways of protein degradation in Saccharomyces cerevisiae. J Biol Chem 271: 27280–27284 [DOI] [PubMed] [Google Scholar]
- Levasseur M, McDougall A (2000) Sperm-induced calcium oscillations at fertilisation in ascidians are controlled by cyclin B1-dependent kinase activity. Development 127: 631–641 [DOI] [PubMed] [Google Scholar]
- Lindon C, Pines J (2004) Ordered proteolysis in anaphase inactivates Plk1 to contribute to proper mitotic exit in human cells. J Cell Biol 164: 233–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margottin-Goguet F, Hsu JY, Loktev A, Hsieh HM, Reimann JDR, Jackson PK (2003) Prophase destruction of Emi1 by the SCFβTrCP/Slimb ubiquitin ligase activates the anaphase promoting complex to allow progression beyond prometaphase. Dev Cell 4: 813–826 [DOI] [PubMed] [Google Scholar]
- Moore GD, Ayabe T, Kopf GS, Schultz RM (1996) Temporal patterns of gene expression of G1–S cyclins and cdks during the first and second mitotic cell cycles in mouse embryos. Mol Reprod Dev 45: 264–275 [DOI] [PubMed] [Google Scholar]
- Nixon VL, Levasseur M, McDougall A, Jones KT (2002) Ca2+ oscillations promote APC/C-dependent cyclin B1 degradation during metaphase arrest and completion of meiosis in fertilizing mouse eggs. Curr Biol 12: 746–750 [DOI] [PubMed] [Google Scholar]
- Ohsumi K, Koyanagi A, Yamamoto TM, Gotoh T, Kishimoto T (2004) Emi1-mediated M-phase arrest in Xenopus eggs is distinct from cytostatic factor arrest. Proc Natl Acad Sci USA 101: 12531–12536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfleger CM, Kirschner MW (2000) The KEN box: an APC recognition signal distinct from the D box targeted by Cdh1. Genes Dev 14: 655–665 [PMC free article] [PubMed] [Google Scholar]
- Prinz S, Hwang ES, Visintin R, Amon A (1998) The regulation of Cdc20 proteolysis reveals a role for APC components Cdc23 and Cdc27 during S phase and early mitosis. Curr Biol 8: 750–760 [DOI] [PubMed] [Google Scholar]
- Rape M, Kirschner MW (2004) Autonomous regulation of the anaphase-promoting complex couples mitosis to S-phase entry. Nature 432: 588–595 [DOI] [PubMed] [Google Scholar]
- Reis A, Chang HY, Levasseur M, Jones KT (2006) APCcdh1 activity in mouse oocytes prevents entry into the first meiotic division. Nat Cell Biol 8: 539–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab M, Neutzner M, Mocker D, Seufert W (2001) Yeast Hct1 recognizes the mitotic cyclin Clb2 and other substrates of the ubiquitin ligase APC. EMBO J 20: 5165–5175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shteinberg M, Protopopov Y, Listovsky T, Brandeis M, Hershko A (1999) Phosphorylation of the cyclosome is required for its stimulation by Fizzy/cdc20. Biochem Biophys Res Commun 260: 193–198 [DOI] [PubMed] [Google Scholar]
- Vodermaier HC, Gieffers C, Maurer-Stroh S, Eisenhaber F, Peters JM (2003) TPR subunits of the anaphase-promoting complex mediate binding to the activator protein CDH1. Curr Biol 13: 1459–1468 [DOI] [PubMed] [Google Scholar]
- Yudkovsky Y, Shteinberg M, Listovsky T, Brandeis M, Hershko A (2000) Phosphorylation of Cdc20/fizzy negatively regulates the mammalian cyclosome/APC in the mitotic checkpoint. Biochem Biophys Res Commun 271: 299–304 [DOI] [PubMed] [Google Scholar]
- Zur A, Brandeis M (2002) Timing of APC/C substrate degradation is determined by fzy/fzr specificity of destruction boxes. EMBO J 21: 4500–4510 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supplementary Figure S1