

Abstract
Dysregulation of cyclin-dependent kinases (cdks) and cytoskeletal protein hyperphosphorylation characterizes a subset of human neurodegenerative diseases, including Alzheimer’s disease, amyotrophic lateral sclerosis, and Niemann-Pick Type C (NPC). It is thought that these cytoskeletal changes lead eventually to development of hallmark cytoskeletal lesions such as neurofibrillary tangles and axonal spheroids. Although many studies support an involvement of cdks in these neurodegenerative cascades, it is not known whether cdk activity is essential. The naturally occurring npc-1 mutant mouse mimics human NPC, in displaying activation of cdk5, mitotic cdc2, and cdk4, with concomitant cytoskeletal pathology and neurodegeneration. We availed of this model and specific pharmacological inhibitors of cdk activity, to determine whether cdks are necessary for NPC neuropathology. The inhibitors were infused intracerebroventricularly for a 2-week period, initiated at a pathologically incipient stage. While an inactive stereoisomer, iso-olomoucine, was ineffective, two potent inhibitors, roscovitine and olomoucine, attenuated significantly the hyperphosphorylation of neurofilament, tau, and mitotic proteins, reduced the number of spheroids, modulated Purkinje neuron death, and ameliorated motor defects in npc mice. These results suggest that cdk activity is required for neuropathology and subsequent motor impairment in NPC. Studies aimed at knocking down individual cdks in these mice will help identify the specific cdk(s) that are essential, and delineate their precise roles in the neurodegenerative process.
Cytoskeletal lesions like neurofibrillary tangles (NFT) and axonal spheroids are hallmarks of many human neurodegenerative diseases.1,2 NFT characterize Alzheimer’s disease (AD), axonal spheroids typify amyotrophic lateral sclerosis (ALS), and NFT and spheroids mark affected brain regions in Niemann-Pick disease Type C (NPC). NFT are composed primarily of hyperphosphorylated tau,3 whereas spheroids are rich in hyperphosphorylated neurofilament proteins.4 In general, hyperphosphorylation precedes formation of cytoskeletal lesions, which has led to the well-accepted notion that aberrant post-translational modification is a prerequisite for cytoskeletal pathology and neurodegeneration.5,6 Multiple members of the cyclin-dependent kinase (cdks) family have been implicated in this pathogenic cascade. Cdc2 and cdk4 are cdks that function in cell division, but are not normally expressed in post-mitotic neurons.7 Their unscheduled activation in degenerating neurons taken together with the presence of duplicated DNA in similar cells, has established the hypothesis that inappropriate re-entry into the cell cycle results in neurodegenerative pathology.8,9 Cdk5 is a neuronal-specific cdk that does not play a role in cell division but is normally active in differentiated neurons influencing neuronal migration and development. Hyperactivation of cdk5 in degenerating neurons may be a result of accumulation of the pathological activator, p25, a calpain-generated proteolytic fragment of the normal neuronal cdk5 activator, p35.10 P25 deregulates cdk5 activity by prolonging its activation and changing its cellular location.11
Concurrent activation of cdc2, cdk4, and cdk5 in degenerating neurons has emerged as a unifying theme in AD,7,8 ALS,12 and NPC.13 A connection between these cdks and neuronal death has been substantiated by several in vitro and in vivo studies. Aberrant activation of cdc2 and cdk4 has been implicated in neuronal apoptosis triggered by a range of neurotoxic insults.14–16 Cdk4 is activated in neurons damaged by ischemic stroke in vivo, and pharmacological inhibitors of cdk activity reverse this damage.17,18 Cdc2 activation marks dying dopaminergic neurons after intrastriatal injection of the neurotoxin 6-hydroxydopamine or medial forebrain bundle axotomy.19 In addition, co-activation of cdc2, cdk4, and cdk5 has been observed after axotomy-induced retinal ganglion cell death.20 The cdk5 kinase is activated in cultured neurons following p25 overexpression, and leads to tau hyperphosphorylation and apoptosis.21 In vitro, certain neurotoxic insults, including exposure to β-amyloid (Aβ), trigger p25 accumulation, cdk5 activation, and neuronal death, which is averted by pharmacological cdk inhibitors.21,22 Moreover, extracellular Aβ deposits induce cdk5 activation and tau hyperphosphorylation in mutant APP transgenic mice.23 It has been shown that double transgenic mice expressing mutant tau and p25 show even more extensive NFT formation than those expressing mutant tau alone.24 Thus, while the bulk of these studies are consistent with an involvement of cdks in neuropathology, identifying the precise kinase(s) and mechanism leading to cytoskeletal pathology in specific neurodegenerative disorders has been hampered by the paucity of comprehensive animal models for these disorders, particularly for NFT production from normal, rather than mutant, tau. Fortunately, excellent mouse models for human NPC and ALS are available. These models not only recapitulate the neuropathologic features and symptoms of their cognate human diseases, but also the activation of cdc2, cdk4, and cdk5 in affected neurons.13,25 They are therefore excellent models for exploring the roles of these cdks in neurodegenerative pathology.
The npcm1N mouse (henceforth referred to as “npc mouse”) arose from a spontaneous mutation in the npc-1 gene,26 the orthologous gene mutated in human NPC and encoding for a late endosomal/lysosomal lipid transporter. NPC is essentially a ubiquitous lipid storage disorder culminating in a profound neurological syndrome, which results in premature death.27 The neurological syndrome is characterized by progressive demyelination, neuroaxonal dystrophy with formation of spheroids, NFT formation, and extensive neurodegeneration.27 Although neurodegeneration in NPC is quite widespread, Purkinje neurons are particularly vulnerable, and their loss accounts for NPC-associated ataxic gait.28 The NFT of NPC are ultrastructurally and antigenically similar to those of AD, and have a similar regional distribution.29 Although npc mice replicate most of the human NPC phenotype, they fail to develop frank NFT.13,30 Nevertheless, they exhibit the characteristic tau hyperphosphorylation profile that precedes and signifies development of NFT in human NPC and AD,13,30 suggesting that they enlist similar neuropathological signaling mechanisms. Accordingly, p25 levels are increased and cdk5 activity is elevated in affected brain regions.31 Additionally, cdc2/cyclin B and cdk4/cyclin D are up-regulated, and many of their cell cycle regulatory substrates are hyperphosphorylated13 [Klunemann H, Bitao B, Zhang M, Vincent I, unpublished observations].
Pathological cdk activation in npc mice is detected before development of cytoskeletal lesions or any motor defects, and cdks co-localize with hyperphosphorylated cytoskeletal proteins in axon spheroids and degenerating neurons suggestive of a cause and effect relationship.31 These observations support an involvement of cdks in cytoskeletal pathology in NPC. As a first step in determining whether cdk activity is necessary for NPC neuropathogenesis, we have analyzed the effects of pan-cdk inhibitors on the progression of cytoskeletal pathology in npc mice. Roscovitine and olomoucine are two selective active site cdk inhibitors32 that have been used extensively for inhibiting cdks in dividing tissues33 and for inhibition of brain cdk5 activity.34 We infused these inhibitors directly to the brains of npc mice, to minimize toxicity that could result from cdk inhibition in dividing tissues. Because NPC is a progressive disease, we opted for continuous infusion into the lateral ventricle. Our study shows that each of these inhibitors, but not an inactive stereoisomer, iso-olomoucine, markedly attenuates protein phosphorylation and cytoskeletal pathology in npc mice, providing a mechanistic view of the involvement of cdk activity in neurodegenerative pathology in this disease model.
Materials and Methods
Npc Mice
A breeding pair of heterozygous npc1m1N mice of the BALB/cnih strain was obtained from the Jackson Laboratory (Bar Harbor, ME) and bred to generate homozygous mice for this study. Genotyping was done by PCR.26 All animal procedures for this study were approved by the Animal Use and Care Committee of the University of Washington.
Cdk Inhibitors
Roscovitine, olomoucine, and iso-olomoucine, obtained from LC laboratories (Woburn, MA), were dissolved in 100% dimethyl sulfoxide (DMSO, Sigma, St. Louis, MI) and diluted with saline to the desired concentrations. Two-week infusions were performed with four different concentrations of roscovitine, 72, 144, 300, and 600 nmoles/day, and 4-week roscovitine infusion involved delivery of 200 nmoles/day. In initial studies, we found that roscovitine above 600 nmoles/day in wild-type mice caused tremor, malaise, and weight loss and had to be euthanized within 1 week. Therefore, doses higher than 600 nmoles/day were discontinued. Olomoucine and iso-olomoucine were infused at 2.5 μmoles/day. All controls received the vehicle solution containing the appropriate percentage of DMSO.
Intracerebroventricular Infusion
ALZET microosmotic pumps (Model 1002, DURECT, Cupertino, CA) were used to infuse cdk inhibitors as described previously.35 Two-week pumps filled with 100 μl inhibitor solution except for higher doses (600 nmoles/day roscovitine, and 2.5 μmoles/day olomoucine and iso-olomoucine each), which were placed in a coiled tube connected with the pump, because the higher percentage of DMSO (75%) required to maintain solubility at these concentrations is not compatible with the osmotic pumps. Because the weight of npc mice does not meet the criteria for implantation of the larger 4-week pumps, all 4-week treatments were performed using 2-week pumps, with replacement of the empty pump with a fresh one at the 2-week midpoint. Following the infusion, treatment mice were euthanized and the brains were removed rapidly and divided mid-sagittally. One half was fixed and paraffin-embedded for immunohistochemistry, and the other half was frozen at −80°C for immunoblotting analysis.31
Antibodies and Immunological Analysis
SMI31 and SMI34 (recognizing distinct phospho-NF-H epitopes) and SMI32 (recognizing a dephosphoyrlated NF-H phosphoepitope) were from Sternberger Monoclonals (Lutherville, MD); TG-3 (Phospho-Thr 231 of tau), TG-5, PHF-1, MC-15, and CP-13 were a kind gift from Dr. Peter Davies (Albert Einstein Medical College, New York, NY); AP-18 was from Dr. Lester Binder (Northwestern University, Evanston, IL); AP20 and MPM-2 (mitotic phosphoepitopes) were obtained from Sigma (St. Louis, MI) and Upstate, New York, respectively; RMd09 (dephospho-NF-H), RMO24 (P-NF-H), RMO32 (P-NF-M), and RMO255 (P-independent-NF-M) were kindly provided by Dr. Virginia Lee (University of Pennsylvania, Philadelphia, PA). The antibodies recognizing total ERK1/2; P-ERK1/2, ERK substrates (total ELK1, P-ELK1, P-BAD, and P-RSK), total GSK-3β and P-GSK-3β were obtained from Cell Signaling Technology (Beverly, MA); NeuN from Chemicon (Temecula, CA); cdk5 from Santa Cruz Biotechnology (Santa Cruz, CA). Immunohistochemical analyses of paraffin-embedded mouse brain sections and immunoblot analyses of brain lysates were done as described before.31 Enhanced chemiluminescence films were scanned and the appropriate band densities were measured using ImageQuant (Molecular Dynamics, Sunnyvale, CA). The band intensities for a particular phospho- or non-phospho-epitope were pooled from triplicate gel runs of samples from 6 to 10 mice, and normalized against band intensities for loading control antigens, NeuN or cdk5, on the same blots. While significant differences were observed with many of the phospho-epitopes as indicated, none of the non-phospho-epitopes was significantly altered by any concentration of inhibitor. Hence, for simplicity, only one loading control is shown in most figures.
Body Weight and Motor Activity
Before infusion and at weekly intervals till the end of the treatment, mice were weighed and evaluated for limb motor activity using the coat hanger test.36 Before and every 4 days during the drug treatment, these mice were tested for motor activity and overall muscle strength. The animals were allowed to grab a metal coat hanger suspended 20 cm above a flat surface, observed for 2 minutes, and the length of time the mouse remained on the hanger measured (hanging time). The ratio of the hanging time at the end of the treatment and before treatment served as a measure of the decline (control) or sustenance (cdk inhibitor). These analyses were performed blinded.
Spheroid and Purkinje Cell Counting
Axon spheroids visualized by SMI32 as well as hematoxylin and eosin (H&E) staining were counted and images collected digitally (Olympus DP10 camera attached to an Olympus BX40F4 microscope, Tokyo, Japan). Four different subregions of the brain (see Figure 4A; region 1 in the white matter of cerebellum, regions 2 and 3 in the dorsal and ventral regions of the pons, and region 4 in the midbrain inferior colliculus) from sections 30 μm lateral from the midline were analyzed. Spheroid counting was performed by two separate individuals, both blinded to the experimental details. The two sets of numbers were averaged, and then statistical analyses were performed using the Student’s t-test.
Figure 4.

Roscovitine reduces axon spheroid number in npc mice. A: Results of 2-week roscovitine infusion. a, Schematic showing 4 regions (regions 1 to 4) used for spheroid counting; b, axon spheroids (arrows) stained by SMI32 (brown, bar, 25 μm); c–h, SMI32 staining showing spheroid distribution in regions 1, 2, and 4 of control (c, e, and g) or roscovitine-treated (d, f, and h) mice (bar, 50 μm); i, summary of spheroid numbers from regions 1 to 4, normalized against the average spheroid number in the same sex controls: control (0 nmoles/day, blue), 72 nmoles/day (red), 144 nmoles/day (yellow, n = 10), 300 nmoles/day (green), and 600 nmoles/day (purple, n = 7). All of the photomicrographs were taken at ×20 magnification except for b which was taken at ×40. B: Results of 4-week roscovitine infusion. SMI32 staining shows spheroid distribution in control, a and c, and mice treated with 200 nmole/day roscovitine, b and d. Quantitative data for the 4-week roscovitine treatment is shown in e. Roscovitine caused a significant decrease in spheroids in all subregions.
Purkinje cells were counted in H&E-stained sections from control and inhibitor-treated mice. To standardize the counting, the same lobe was selected in each section, and all visible Purkinje neurons were counted in that lobe. The data were analyzed statistically as described below.
Statistics
At least 6 to 10 mice in each category were analyzed for all of the experiments described here, and usually contained equivalent numbers of each gender. For each assay, the data from each gender were analyzed separately and also in a combined fashion. Statistical differences were observed in both cases so only the combined results are presented here. Throughout, a P value of 0.05 was used for statistical significance but in some instances lower values were obtained.
Results
Roscovitine Causes a Decrease in Phosphoepitopes Generated by cdk5
The IC50 of roscovitine for inhibition of cdk5-p25 is 0.16 μmol/L, and for cdc2-cyclin B is 0.45 μmol/L. The extracellular signal-regulated kinases, ERK1 and ERK2, are inhibited at 5- to 100-fold, and cdk4 and GSK3β at 1000-fold higher concentrations.37 Thus, roscovitine is more likely to inhibit cdk5 and cdc2 than any other kinase in brain. In our previous study, we suggested that deregulation of cdk5 contributes to NPC neuropathology by extensive hyperphosphorylation of cytoskeletal proteins such as tau, NFs, and MAP2a, b.31 We therefore examined the phosphorylation status of cdk5 consensus sites in cytoskeletal proteins in npc mice after administering a similar dose of roscovitine, ie, 144 nmoles/day that was used previously to prove that cdk5 plays an essential role during opiate withdrawal.34 We found that a 2-week intraventricular infusion of roscovitine, initiated in 5-week-old npc mice, alleviates hyperphosphorylation of cytoskeletal proteins, whereas a control DMSO vehicle treatment of npc mice was ineffective. Cdk5-phosphorylated motifs in the C-terminal region of NF were probed by using the SMI31 and SMI34 monoclonal antibodies. We observed a 150 to 200% decrease in SMI31 immunoreactivity with NF-H and NF-M, and a decrease in SMI34 immunoreactivity with NF-H, following 144 nmoles/day and higher doses of roscovitine (Figure 1A, SMI31 and SMI34, shown for 144 nmole/day only). Dephospho-NF-H antibodies SMI32 and RMd09 as well as phospho-independent antibody to NF-M showed no significant changes, indicative of equivalent NF-H and NF-M levels in the roscovitine-treated and untreated groups. Similarly, cdk5-generated phosphoepitopes on tau were examined using the PHF-1, CP13, and MC15 antibodies.31 A 50 to 100% decrease in PHF-1, CP-13, and MC-15 immunoreactivity was observed starting with 72 nmoles/day and higher doses of roscovitine (Figure 1A, Tau). The tau primary sequence antibody, TG-5, showed a predominance of hypophosphorylated tau, consistent with tau dephosphorylation with increasing doses of roscovitine. We also found that a dose of 300 nmoles/day roscovitine treatment reverted the hyperphosphorylated status of NF and tau in npc mice (−/−) to their normal levels as seen in wild-type mice (+/+) (Figure 1B, shown for SMI31 and PHF-1 only). The TG-5 antibody showed that the relatively hypophosphorylated 43-kDa tau band was the major tau band in mice treated with 300 nmoles/day roscovitine. In contrast to these roscovitine-mediated decreases in NF and tau phosphorylation, phosphorylation of MAP2b at the AP-18 epitope increased even beyond its typical increase in NPC, while the levels of MAP2a and MAP2b as visualized with AP-20 were unchanged (Figure 1A, MAP-2). In summary, these results suggest that cdk activity promotes hyperphosphorylation of NF and tau, but may have a negative effect on phosphorylation of MAP2 in npc mice.
Figure 1.

Roscovitine inhibits phosphorylation of cytoskeletal proteins in npc mice. Mice either treated with different doses of roscovitine or the vehicle only for 2 weeks were analyzed for the presence of various epitopes. The antibodies used are indicated on the right of the corresponding blots. A: Immunoblots of control or roscovitine-treated mice (duplicate animals from a total of 6 to 10 treated with 72 or 144 nmoles/day shown only). Cdk5 is the representative loading control for all antigens. B: Samples from the untreated (−/−), control mice (control), or 300 nmoles/day roscovitine-treated mice (300) are compared with samples from wild-type (+/+) to illustrate normalization of protein phosphorylation in npc mice after roscovitine treatment (NeuN, loading control). The bands corresponding to NF-H and NF-M are labeled as H and M, respectively.
Roscovitine Treatment Causes a Decrease in Phosphoepitopes Generated by cdc2
Neurofibrillary degeneration in NPC has been characterized by accumulation of cdc2-generated mitotic phospoepitopes.38 The MPM-2 monoclonal antibody reacts with a phosphoepitope present in a discrete set of proteins that function in mitosis, and is present in AD and NPC.38 Additionally, the TG-3 antibody reacts with cdc2-phosphorylated nucleolin.39 Compared to control npc mice infused with DMSO, npc mice receiving a 2-week regimen of roscovitine at doses ranging from 72 to 300 nmoles/day showed dramatic reductions of TG-3 and MPM-2 immunoreactivity (Figure 2).
Figure 2.
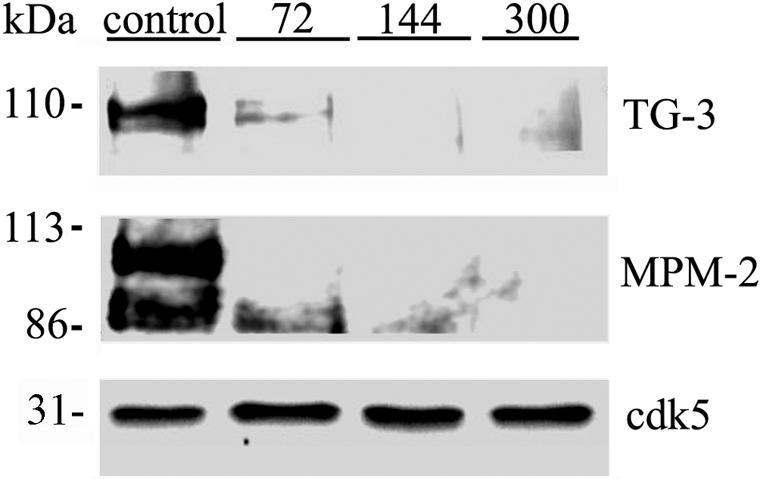
Roscovitine treatment can rescue abnormal mitotic phosphoepitopes in npc mice. Mice treated either with the indicated doses of roscovitine (top) or the vehicle (control) for 2 weeks were analyzed for the presence of various cdc2 generated epitopes. Cdk5 has been used as loading control for both the blots. The antibodies used are indicated on the right of the corresponding blots.
Ameliorating Effects of Roscovitine on NPC Pathology Are Mediated by cdk Inhibition
Because roscovitine inhibits cdks reversibly, it is not feasible to verify the extents of in vivo inhibition by subsequent immunoprecipitation and kinase activity assays in vitro. As an attempt to rule out possible effects of roscovitine on non-cdk kinases that might also modulate cytoskeletal function,6,40 we compared in vivo indices of ERK or GSK3β activity in roscovitine-treated and control mice. We analyzed the immunoreactivities of phospho site-specific antibodies recognizing proven phosphoepitopes generated by these kinases in brain. Activated ERKs phosphorylate cytosolic proteins eg, the p90RSK41 or the pro-apoptotic Bad protein,42 and also nuclear transcription factors, eg, ELK-1.41 A modest increase in tyrosine phosphorylation, indicative of ERK activation, was observed after roscovitine treatment, but none of the ERK-generated phosphoepitopes in p90RSK (not shown), BAD, or ELK-1, increased appreciably (Figure 3A).
Figure 3.

Immunoblotting analyses of ERK (A), and GSK-3β (B), and their phospho-substates in control and inhibitor-treated mice. Mice treated with roscovitine for 2 weeks were analyzed for the presence or absence of either ERK or GSK3β-generated epitopes (cdk5, loading control). The epitopes being detected are indicated on the right of the corresponding blot. The figures on top indicate the inhibitor dose (nmoles/day). The bands corresponding to NF-H and NF-M are labeled as H and M, respectively.
GSK3β is a PDK that phosphorylates cytoskeletal proteins on sites that are also potentially recognized by cdks.43 GSK-3β produces the RMO24 phosphoepitope in NF, and phosphorylates β-catenin leading to its degradation by proteasomes. The kinase is inactivated by phosphorylation of its Ser-9 residue by the Akt kinase, leading to neuronal apoptosis. Following treatment of npc mice with 72 and 144 nmoles/day of roscovitine, GSK3β kinase was hyperactivated, as evidenced by reduced immunoreactivity with the P-Ser9 antibody, and a simultaneous decrease of β-catenin levels. However, RMO24 immunoreactivity remained unaltered under these conditions (Figure 3B). Thus, roscovitine treatment does not lead to inhibition of ERK or GSK3β. Rather, transient activation, occurring simultaneously with the inhibition of NF or tau phosphorylation, makes it unlikely that ERK or GSK3β contribute to the effects of roscovitine in npc mice. Instead, the inhibition of NPC pathology by roscovitine occurs most probably via cdk inhibition, a conclusion supported by the effects with another inhibitor, olomoucine (see Figure 8 below)
Figure 8.

Olomoucine can ameliorate the neuropathological phenotype in npc mice. Two-week olomoucine (olo), but not iso-olomoucine (iso), treatment improves motor activity (a). Olomoucine treatment (c) (bar, 50 μm), decreased SMI32-stained spheroid count compared to iso-olomoucine-treated mice (b). The results from regions (reg) 1 to 4 are summarized (d); spheroid numbers are expressed as percentage control; control mice, blue bars; iso-treated mice, red bars; olo-treated mice, yellow bars. Olomoucine treatment resulted in a decrease of SMI31 and PHF-1 immunoreactivity compared to iso-olomoucine treated mice (e). NeuN shows the loading control in this panel.
Roscovitine Ameliorates Cytoskeletal Lesion Formation in npc Mice
To test the hypothesis that hyperphosphorylation of cytoskeletal proteins is a precursor for cytoskeletal lesion formation, we examined the distribution of axon spheroids in the brains of roscovitine-treated mice having decreased phosphorylation and control mice having hyperphosphorylation. Mouse brain sections were immunostained with SMI32. Total SMI32 immunoreactivity was not affected at any dose of roscovitine (Figure 1A) making this antibody a reliable reagent for comparing spheroid numbers in control and roscovitine-treated mice (Figure 4). H&E staining of the spheroids essentially yielded similar results (data not shown). Spheroids were counted in four subregions of the hindbrain that are affected prominently in NPC (Figure 4A, schematically represented in a). Using anatomically matched sagittal brain sections from 2-week roscovitine-treated mice (Figure 4A, d, f, and h) and control mice (Figure 4A, c, e, and g), we found a dramatic decrease in spheroid number following roscovitine treatment. This decrease was significant at roscovitine doses of 144 nmoles/day or higher in regions 1, 3, and 4, and at 300 nmoles/day or higher in region 2 (Figure 4A, i). Following a 4-week 200 nmoles/day roscovitine infusion, spheroid numbers were also significantly reduced in the four regions analyzed (Figure 4B, a to d). Roscovitine treatment also led to a reduction in number of spheroids recognized by the MPM-2 antibody (Figure 5b) relative to controls (Figure 5a, arrows). Overall, these data show that a decrease in the number of spheroids at 144 nmoles/day (Figure 4A, i) coincides with a decrease in phosphorylation of NF, tau, and mitotic proteins (Figure 1A). However, no significant decrease in spheroid number (Figure 4A, i) is apparent at a dose 72 nmoles/day, when tau phosphorylation is reduced significantly (Figure 1A). Thus, spheroid numbers correlate better with the extent of cdk5- and cdc2-mediated phosphorylation of NF and mitotic proteins than with tau or MAP2. This is consistent with previous reports of hyperphosphorylated NF being the major protein constituent of spheroids.4
Figure 5.
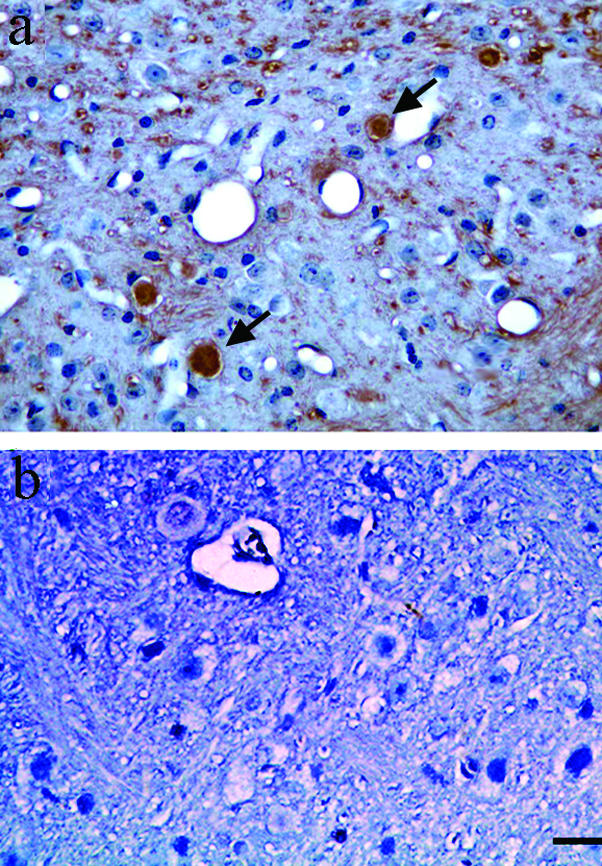
Immunohistochemical analyses of mitotic phosphoepitopes in roscovitine-treated and control mice. A 2-week roscovitine treatment ameliorates MPM-2 phosphoepitopes (b), compared to the control animals treated with vehicle alone (a) (bar, 50 μm). Arrows indicate MPM-2-labeled spheroids. The photomicrographs were taken at ×40 magnification.
Roscovitine Ameliorates But Does Not Spare Purkinje Cell Loss in npc Mice
Progressive loss of Purkinje cells in npc mice starts at 6.5 weeks, leading to gradual ataxia and paralysis. The number of Purkinje neurons in a selected lobe of the cerebellum was scored in DMSO- and roscovitine-treated mice. These cells, stained with H&E, were barely detected in DMSO-treated 7-week-old npc mice where empty spaces were apparent in locations where Purkinje cells once lived (Figure 6A, a, arrows). In contrast, significant numbers of Purkinje neurons were apparent in 7-week-old mice that received roscovitine for 2 weeks initiated at 5 weeks of age (Figure 6A, b, arrows). This effect was evident from doses of 144 nmoles/day and above (Figure 6A, c). However, in mice receiving roscovitine for 4 weeks, there was no sparing of Purkinje neuron loss (Figure 6B, a and b). These results suggest that introducing roscovitine between 4 to 5 weeks of age in npc mice modulates the rate of, but does not prevent, death of Purkinje neurons. They also indicate that the entire Purkinje neuronal layer is lost rapidly in npc mice, during a 3-week interval starting at about 5 weeks of age.
Figure 6.

Roscovitine infusion effectively reduces Purkinje neuron loss in npc mice. Mice treated with either roscovitine or DMSO only (control) for 2 weeks (144 nmoles/day) (A) or 4 weeks (200 nmoles/day) (B) were analyzed for the presence or absence of Purkinje neurons Aa: H&E staining showing Purkinje neurons missing in control (arrow). Ab: H&E staining showing Purkinje neurons surviving in roscovitine-treated mice (arrows). Ac: Quantitative summary of Purkinje cell numbers in control and 2-week roscovitine-treated mice. The x-axis shows the dose of roscovitine treatment (nmoles/day). Note, mean values at 144, 300, and 600 nmoles/day roscovitine, are not statistically different from each other, but are statistically increased relative to control and 72 nmoles/day roscovitine. B: H&E staining showing missing Purkinje neurons (arrows) in 4-week control (a), and in roscovitine-treated mice (b). All of the photomicrographs were taken at ×40 magnification.
Roscovitine Alleviates Motor Defects and Decreases Weight Loss in npc Mice
The sparing of Purkinje cells following 2-week treatment of npc mice with roscovitine, corresponded with visible improvement in ataxia. To assess whether axon spheroids underlie the characteristic weakness in the limbs of npc mice, we performed quantitative analyses of motor activity using the coat hanger test.36 Npc mice treated with DMSO vehicle alone displayed the expected decline in motor activity at 7 weeks of age compared to 5 weeks, but this symptom was improved in a dose-dependent fashion on roscovitine treatment during this interval (Figure 7A). Similar results were obtained when roscovitine was administered at 200 nmoles/day for 4 weeks, starting at 5 weeks and ending at 9 weeks of age. Here, roscovitine improved the performance of the mice by almost 300% relative to control mice (Figure 7B). With respect to body weight, no significant difference was noted in either control or roscovitine-treated groups after a 2-week roscovitine treatment. However, following a 4-week treatment, the control mice lost 21% of their original weight, and administration of roscovitine abolished this decrease (Figure 7C). These combined results suggest that roscovitine inhibits the progression of motor dysfunction, and also the weight loss associated with NPC.
Figure 7.

Roscovitine (ros) improves motor activity and inhibits weight loss in npc mice. The hanging time on a coat hanger and body weight were compared before (white bars) and after treatment (black bars) with either control or roscovitine-treated mice. A: Motor activity before and after 2-week infusion with roscovitine at the indicated concentrations (144 and 600 nmoles/day, n = 10 and 7 respectively, for the other doses tested, n = 6). B: Motor activity before and after 4-week roscovitine treatment at 200 nmoles/day, n = 8, P = 0.03. C: Body weight after 2- or 4-week roscovitine treatment at 200 nmoles/day, n = 8, P = 0.007.
Roscovitine-Induced Changes Are Replicated by Another Specific cdk Inhibitor, Olomoucine
To further verify that the effects of roscovitine treatment are specifically mediated by cdk inhibition, we infused npc1 mice with another cdk inhibitor, olomoucine, or its inactive stereoisomer, iso-olomoucine.44 Olomoucine is a potent and very selective, competitive inhibitor of cdc2 (IC50 = 7 μmol/L) and cdk2/cdk5 (IC50 = 3 μmol/L) at the ATP binding site. It inhibits ERK1 with a IC50 of 30 μmol/L and is inactive against cdk4 and cdk6.44 The effects of olomoucine treatment were essentially similar to those observed with roscovitine. A 2-week intraventricular treatment regime of 2.5 μmoles/day of olomoucine resulted in a tremendous improvement in motor activity (Fig. 8, a). There was a concomitant decrease in spheroid number (Figure 8, b to d), amelioration of Purkinje neuron loss (data not shown), and a significant decrease of cdk5-generated NF and tau phosphoepitopes (Figure 8e). A similar treatment group that received equivalent doses of the inactive stereoisomer, iso-olomoucine, did not show any significant change in these neuropathological states (Figure 8, d and e). As before, a corresponding group of mice receiving only DMSO did not show any improvement in either motor activity or the pathological parameters tested (Figure 8a). These effects of olomoucine lend further credence to the essential role of cdk activity in NPC neuropathology.
Discussion
In previous studies, we determined that elevation of cdk5 and cdc2 activity coincides spatially and temporally with the appearance of hyperphosphorylated cytoskeletal proteins in NPC. These cdks and their hyperphosphorylated cytoskeletal protein products precede development of, but are eventually incorporated into, cytoskeletal lesions called spheroids. Here we show that administration of cdk inhibitors to npc mice during the period of cdk activation but before spheroid formation results in attenuation of protein hyperphosphorylation and reduced formation of spheroids, reduced neurodegeneration, and reduced motor defects. These findings support the hypothesis that cdk activity is essential for progression of cytoskeletal pathology in NPC.
Concurrent activation of cdk5 and cdc2 appears to trigger hyperphosphorylation of a variety of neuronal proteins including NF, tau MAP2, and mitotic substrates. Our studies suggest that of these, only NF and mitotic protein hyperphosphorylations contribute directly to formation of axonal spheroids. Decreases in NF and mitotic protein hyperphosphorylation, and spheroid number, following cdk inhibition, coincide with a marked improvement in performance on the coat-hanger test, indicating that the prevalence of axonal spheroids underlies limb weakness in npc mice. On the other hand, cdk inhibition only modulates the rate of, but does not prevent, the death of Purkinje neurons, suggested that the signaling pathways leading to loss of these cells may be somewhat different from those involved in spheroid formation. It has been found that in npc mice Purkinje neurons die in waves patterned as “stripes” throughout the region.45 Zebrin II-negative “stripes” die earlier, the alternate zebrin II-positive stripes being more resistant. It is possible that cdk activity only plays a role in death of some of these cells but does not affect the others. More detailed studies of the effects of cdk inhibition on the pattern of Purkinje death would be important. Evidence has linked cdk5 activation to NF phosphorylation and axonal transport and trafficking,46 and it has been proposed that disruption of this normal scheme gives rise to cytoskeletal pathology.47 Defects in axonal transport have been implicated in cytoskeletal pathology in AD48 and ALS49 as well. Thus it seems reasonable that aberrant cdk5 activation in NPC interferes with cytoskeletal protein function and axonal transport and trafficking, culminating in spheroid formation and neurodegeneration, both of which contribute to motor defects. On the other hand, the activation of cdc2 in post-mitotic neurons is thought to signal an abortive attempt at cell cycle re-entry.7 Mitotic changes are also likely to impact cytoskeletal protein function and axonal transport, thereby contributing to spheroid formation as well. At present it is not clear why cdk inhibition prevents weight loss in npc mice, and it is unknown whether roscovitine or olomoucine have any effects on lipid storage in NPC. Nevertheless, to our knowledge, this is the first in vivo study demonstrating an ability of cdk inhibitors to retard development of multiple neuropathological hallmarks and symptoms of a neurodegenerative disease. The confirmation of an essential role for cdk activity in NPC opens the way to identify which cdk(s) are crucial, and in what aspects of the disease. RNA-mediated interference (RNAi)-based approaches aimed at specific knockdown of cdk5 or cdc2 might be useful in determining which phosphoepitopes are produced by each of these cdks, and how phosphorylation of specific substrates participates in NPC neuropathology. Similar approaches would facilitate evaluation of a role for cdk4 in NPC. Furthermore, studies with cdk inhibitors in other neurodegenerative disease models such as the SOD1 mutant ALS mice and P301L tauopathy mice would help determine whether hyperactivation of cdks is paramount in these neuropathological states as well.
In agreement with the pathological scheme suggested above, p25 overexpression driven by the PDGF promoter in transgenic mice leads to cdk5 activation and axonopathy with formation of axon spheroids and motor defects.50 Cytoskeletal changes were observed in the absence of spheroid formation in another line overexpressing p25, under control of the neuron-specific enolase promoter, but this pathology was not accompanied by a net increase in cytoskeletal protein phosphorylation.51 Conditional, neuronal-specific p25 expression resulted in tau hyperphosphorylation, neuronal death, and NFT formation but no evidence of spheroids.52 Because similar increases of cdk5 activity were reported in all these lines, it appears that the outcome of cdk5 activation depends more heavily on which neurons are targeted, and on the timing and subcellular localization of cdk5 activation during the period of gestation through adulthood. Other studies also support a connection between cdc2 activation and neurodegeneration in vivo.19,20,53 However, a more detailed analysis of cell cycle and apoptotic markers in NPC will be necessary for understanding the mechanism by which cdc2 operates in this disease.
Presently, it is unclear why a mutation in NPC-1, a protein involved in intracellular transport of cholesterol to postendosomal/lysosomal destinations, leads to activation of multiple cdks, and perhaps even other types of protein kinases.54 A possible means of hyperactivation of cdk5 is chronic, accelerated calpain-mediated conversion of p35 to p25.21 NPC-1, calpain, and p35 all appear to be membrane-associated proteins colocalizing in “signal transduction centers”, which are distinct entities located in lipid rafts.31 Thus, it is possible that NPC-1 interacts with and influences the functions of calpain and p35 in localized vesicle or membrane structures. NPC-1 mutations precipitate defects in transport and trafficking of cholesterol in neurons.55 It would be worth investigating possible direct effects of NPC-1 mutations on calpain activation and p25 production, which may occur independently of their effects on lipid transport. Curiously, APP and SOD1 are also localized in vesicle and membrane fractions,56,57 and may trigger neurodegenerative changes similarly by impacting p25 production in these subcellular locations. How NPC-1 mutations cause activation of cdc2 in degenerating neurons is more difficult to explain. In dividing cells, cdc2 activation is orchestrated with successful and fidelous completion of DNA replication.58 Duplicated chromosomes, along with accumulation of mitotic phosphoepitopes, have been detected in degenerating neurons in AD,59 but there have been no reports to date regarding the status of DNA in other neurodegenerative disorders. Cdc2 activation in post-mitotic neurons has also been implicated in certain types of neuronal apoptosis, but without involvement of prior DNA synthesis.60 Thus cdc2/cyclin B activation may not necessarily signify cell cycle dysregulation, but may constitute a more general response to adverse conditions and insults. In earlier studies we found that normal differentiated neurons express all of the known upstream cdc2 regulators, the Wee1 tyrosine kinase, and the Cdc25A and B phosphatases, in a constitutive fashion even though they lack cdc2 and cyclin B.7 In the brain, these enzymes may support full activation of cdc2, even in the absence of prior DNA synthesis or cell cycle entry. Others have shown that the cdc2 activating cdk7 kinase and the CIP-1-associated regulator of cyclin B have elevated expression in affected neurons in AD.61,62 Examining similar cell cycle and apoptotic markers in NPC will be necessary for understanding how cdc2 becomes activated and induces its downstream neuropathological effects. Such studies should also provide new insights into potential therapeutic targets for the treatment of NPC.
In light of the efficacious management of lipid abnormalities in NPC using lipid-lowering drugs,63–65 an exciting possibility for comprehensive treatment of NPC might be a lipid-lowering drug for normalizing lipid levels used in conjunction with a specific cdk inhibitor such as roscovitine for normalizing neuronal cytoskeletal protein function. Current phase II clinical trials with roscovitine for human cancer therapy33 promise that the application of cdk inhibitors as a general mode of intervention for neurodegeneration may materialize in the near future.
Acknowledgments
We thank Drs. Peter Davies, Harish Pant, Virginia Lee, Jean Campbell, and Lester Binder for kindly supplying us with antibodies. We also thank Chong In Pae for technical assistance, Jan Hallows for an excellent review of the manuscript, Jan Hallows and John Ho for help with genotyping, and Michael Parker and Rebecca Biasell for their excellent care of the npc mice.
Footnotes
Address reprint requests to Inez Vincent, Ph.D, Research Associate Professor, Department of Pathology, University of Washington, Box 357705, K-078A, 1959 N.E. Pacific Street, Seattle, WA 98195. E-mail: ivincent@u.washington.edu.
Supported by National Institutes of Health grant AG12721, the Seattle Jim Lambright Niemann-Pick Foundation, the Ara Parseghian Medical Research Foundation, and an anonymous foundation.
References
- Goldman JE, Yen SH. Cytoskeletal protein abnormalities in neurodegenerative diseases. Ann Neurol. 1986;19:209–223. doi: 10.1002/ana.410190302. [DOI] [PubMed] [Google Scholar]
- Trojanowski JQ, Lee VM. Phosphorylation of neuronal cytoskeletal proteins in Alzheimer’s disease and Lewy body dementias. Ann NY Acad Sci. 1994;747:92–109. doi: 10.1111/j.1749-6632.1994.tb44403.x. [DOI] [PubMed] [Google Scholar]
- Goedert M. Neurofibrillary pathology of Alzheimer’s disease and other tauopathies. Prog Brain Res. 1998;117:287–306. doi: 10.1016/s0079-6123(08)64022-4. [DOI] [PubMed] [Google Scholar]
- Elleder M, Jirasek A. Neuropathology of various types of Niemann-Pick disease. Acta Neuropathol Suppl (Berl) 1981;7:201–203. doi: 10.1007/978-3-642-81553-9_60. [DOI] [PubMed] [Google Scholar]
- Iqbal K, Grundke-Iqbal I. Molecular mechanism of Alzheimer’s neurofibrillary degeneration and therapeutic intervention. Ann NY Acad Sci. 1996;777:132–138. doi: 10.1111/j.1749-6632.1996.tb34411.x. [DOI] [PubMed] [Google Scholar]
- Grant P, Sharma P, Pant HC. Cyclin-dependent protein kinase 5 (Cdk5) and the regulation of neurofilament metabolism. Eur J Biochem. 2001;268:1534–1546. [PubMed] [Google Scholar]
- Vincent I, Pae CI, Hallows JL. The cell cycle and human neurodegenerative disease. Prog Cell Cycle Res. 2003;5:31–34. [PubMed] [Google Scholar]
- Herrup K, Arendt T. Re-expression of cell cycle proteins induces neuronal cell death during Alzheimer’s disease. J Alzheimers Dis. 2002;4:243–247. doi: 10.3233/jad-2002-4315. [DOI] [PubMed] [Google Scholar]
- Raina AK, Zhu X, Rottkamp CA, Monteiro M, Takeda A, Smith MA. Cyclin’ toward dementia: cell cycle abnormalities and abortive oncogenesis in Alzheimer disease. J Neurosci Res. 2000;61:128–133. doi: 10.1002/1097-4547(20000715)61:2<128::AID-JNR2>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Lee MS, Tsai LH. Cdk5: one of the links between senile plaques and neurofibrillary tangles? J Alzheimers Dis. 2003;5:127–137. doi: 10.3233/jad-2003-5207. [DOI] [PubMed] [Google Scholar]
- Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 1999;402:615–622. doi: 10.1038/45159. [DOI] [PubMed] [Google Scholar]
- Nguyen MD, Boudreau M, Kriz J, Couillard-Despres S, Kaplan DR, Julien JP. Cell cycle regulators in the neuronal death pathway of amyotrophic lateral sclerosis caused by mutant superoxide dismutase 1. J Neurosci. 2003;23:2131–2140. doi: 10.1523/JNEUROSCI.23-06-02131.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu B, Klunemann H, Suzuki K, Li J, Bird T, Jin LW, Vincent I. Niemann-Pick disease type C yields possible clue for why cerebellar neurons do not form neurofibrillary tangles. Neurobiol Dis. 2002;11:285–297. doi: 10.1006/nbdi.2002.0551. [DOI] [PubMed] [Google Scholar]
- Park DS, Morris EJ, Padmanabhan J, Shelanski ML, Geller HM, Greene LA. Cyclin-dependent kinases participate in death of neurons evoked by DNA-damaging agents. J Cell Biol. 1998;143:457–467. doi: 10.1083/jcb.143.2.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi Y, Lehtinen M, Donovan N, Bonni A. Cdc2 phosphorylation of BAD links the cell cycle to the cell death machinery. Mol Cell. 2002;9:1005–1016. doi: 10.1016/s1097-2765(02)00524-5. [DOI] [PubMed] [Google Scholar]
- Rideout HJ, Wang Q, Park DS, Stefanis L. Cyclin-dependent kinase activity is required for apoptotic death but not inclusion formation in cortical neurons after proteasomal inhibition. J Neurosci. 2003;23:1237–1245. doi: 10.1523/JNEUROSCI.23-04-01237.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katchanov J, Harms C, Gertz K, Hauck L, Waeber C, Hirt L, Priller J, von Harsdorf R, Bruck W, Hortnagl H, Dirnagl U, Bhide PG, Endres M. Mild cerebral ischemia induces loss of cyclin-dependent kinase inhibitors and activation of cell cycle machinery before delayed neuronal cell death. J Neurosci. 2001;21:5045–5053. doi: 10.1523/JNEUROSCI.21-14-05045.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, O’Hare MJ, Park DS. Cyclin-dependent kinases and stroke. Expert Opin Ther Targets. 2001;5:557–567. doi: 10.1517/14728222.5.5.557. [DOI] [PubMed] [Google Scholar]
- El-Khodor BF, Oo TF, Kholodilov N, Burke RE. Ectopic expression of cell cycle markers in models of induced programmed cell death in dopamine neurons of the rat substantia nigra pars compacta. Exp Neurol. 2003;179:17–27. doi: 10.1006/exnr.2002.8047. [DOI] [PubMed] [Google Scholar]
- Lefevre K, Clarke PG, Danthe EE, Castagne V. Involvement of cyclin-dependent kinases in axotomy-induced retinal ganglion cell death. J Comp Neurol. 2002;447:72–81. doi: 10.1002/cne.10215. [DOI] [PubMed] [Google Scholar]
- Lee MS, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature. 2000;405:360–364. doi: 10.1038/35012636. [DOI] [PubMed] [Google Scholar]
- Alvarez A, Munoz JP, Maccioni RB. A Cdk5–p35 stable complex is involved in the β-amyloid-induced deregulation of Cdk5 activity in hippocampal neurons. Exp Cell Res. 2001;264:266–274. doi: 10.1006/excr.2001.5152. [DOI] [PubMed] [Google Scholar]
- Otth C, Concha II, Arendt T, Stieler J, Schliebs R, Gonzalez-Billault C, Maccioni RB. AβPP induces cdk5-dependent tau hyperphosphorylation in transgenic mice Tg2576. J Alzheimers Dis. 2002;4:417–430. doi: 10.3233/jad-2002-4508. [DOI] [PubMed] [Google Scholar]
- Noble W, Olm V, Takata K, Casey E, Mary O, Meyerson J, Gaynor K, LaFrancois J, Wang L, Kondo T, Davies P, Burns M, Veeranna, Nixon R, Dickson D, Matsuoka Y, Ahlijanian M, Lau LF, Duff K. Cdk5 is a key factor in tau aggregation and tangle formation in vivo. Neuron. 2003;38:555–565. doi: 10.1016/s0896-6273(03)00259-9. [DOI] [PubMed] [Google Scholar]
- Nguyen MD, Lariviere RC, Julien JP. Deregulation of Cdk5 in a mouse model of ALS: toxicity alleviated by perikaryal neurofilament inclusions. Neuron. 2001;30:135–147. doi: 10.1016/s0896-6273(01)00268-9. [DOI] [PubMed] [Google Scholar]
- Loftus SK, Morris JA, Carstea ED, Gu JZ, Cummings C, Brown A, Ellison J, Ohno K, Rosenfeld MA, Tagle DA, Pentchev PG, Pavan WJ. Murine model of Niemann-Pick C disease: mutation in a cholesterol homeostasis gene. Science. 1997;277:232–235. doi: 10.1126/science.277.5323.232. [DOI] [PubMed] [Google Scholar]
- Vincent I, Bu B, Erickson RP. Understanding Niemann-Pick type C disease: a fat problem. Curr Opin Neurol. 2003;16:155–161. doi: 10.1097/01.wco.0000063764.15877.1c. [DOI] [PubMed] [Google Scholar]
- Higashi Y, Murayama S, Pentchev PG, Suzuki K. Cerebellar degeneration in the Niemann-Pick type C mouse. Acta Neuropathol (Berl) 1993;85:175–184. doi: 10.1007/BF00227765. [DOI] [PubMed] [Google Scholar]
- Auer IA, Schmidt ML, Lee VM, Curry B, Suzuki K, Shin RW, Pentchev PG, Carstea ED, Trojanowski JQ. Paired helical filament tau (PHFtau) in Niemann-Pick type C disease is similar to PHFtau in Alzheimer’s disease. Acta Neuropathol (Berl) 1995;90:547–551. doi: 10.1007/BF00318566. [DOI] [PubMed] [Google Scholar]
- German DC, Quintero EM, Liang CL, Ng B, Punia S, Xie C, Dietschy JM. Selective neurodegeneration, without neurofibrillary tangles, in a mouse model of Niemann-Pick C disease. J Comp Neurol. 2001;433:415–425. doi: 10.1002/cne.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu B, Li J, Davies P, Vincent I. Deregulation of cdk5, hyperphosphorylation, and cytoskeletal pathology in the Niemann-Pick type C murine model. J Neurosci. 2002;22:6515–6525. doi: 10.1523/JNEUROSCI.22-15-06515.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer L, Borgne A, Mulner O, Chong JP, Blow JJ, Inagaki N, Inagaki M, Delcros JG, Moulinoux JP. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2, and cdk5. Eur J Biochem. 1997;243:527–536. doi: 10.1111/j.1432-1033.1997.t01-2-00527.x. [DOI] [PubMed] [Google Scholar]
- Meijer L, Raymond E. Roscovitine and other purines as kinase inhibitors: from starfish oocytes to clinical trials. Acc Chem Res. 2003;36:417–425. doi: 10.1021/ar0201198. [DOI] [PubMed] [Google Scholar]
- Bibb JA, Chen J, Taylor JR, Svenningsson P, Nishi A, Snyder GL, Yan Z, Sagawa ZK, Ouimet CC, Nairn AC, Nestler EJ, Greengard P. Effects of chronic exposure to cocaine are regulated by the neuronal protein Cdk5. Nature. 2001;410:376–380. doi: 10.1038/35066591. [DOI] [PubMed] [Google Scholar]
- Osuga H, Osuga S, Wang F, Fetni R, Hogan MJ, Slack RS, Hakim AM, Ikeda JE, Park DS. Cyclin-dependent kinases as a therapeutic target for stroke. Proc Natl Acad Sci USA. 2000;97:10254–10259. doi: 10.1073/pnas.170144197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voikar V, Rauvala H, Ikonen E. Cognitive deficit and development of motor impairment in a mouse model of Niemann-Pick type C disease. Behav Brain Res. 2002;132:1–10. doi: 10.1016/s0166-4328(01)00380-1. [DOI] [PubMed] [Google Scholar]
- De Azevedo WF, Leclerc S, Meijer L, Havlicek L, Strnad M, Kim SH. Inhibition of cyclin-dependent kinases by purine analogues: crystal structure of human cdk2 complexed with roscovitine. Eur J Biochem. 1997;243:518–526. doi: 10.1111/j.1432-1033.1997.0518a.x. [DOI] [PubMed] [Google Scholar]
- Husseman JW, Nochlin D, Vincent I. Mitotic activation: a convergent mechanism for a cohort of neurodegenerative diseases. Neurobiol Aging. 2000;21:815–828. doi: 10.1016/s0197-4580(00)00221-9. [DOI] [PubMed] [Google Scholar]
- Dranovsky A, Vincent I, Gregori L, Schwarzman A, Colflesh D, Enghild J, Strittmatter W, Davies P, Goldgaber D. Cdc2 phosphorylation of nucleolin demarcates mitotic stages and Alzheimer’s disease pathology. Neurobiol Aging. 2001;22:517–528. doi: 10.1016/s0197-4580(00)00248-7. [DOI] [PubMed] [Google Scholar]
- Pelech SL, Charest DL. MAP kinase-dependent pathways in cell cycle control. Prog Cell Cycle Res. 1995;1:33–52. doi: 10.1007/978-1-4615-1809-9_4. [DOI] [PubMed] [Google Scholar]
- Sananbenesi F, Fischer A, Schrick C, Spiess J, Radulovic J. Phosphorylation of hippocampal Erk-1/2, Elk-1, and p90-Rsk-1 during contextual fear conditioning: interactions between Erk-1/2 and Elk-1. Mol Cell Neurosci. 2002;21:463–476. doi: 10.1006/mcne.2002.1188. [DOI] [PubMed] [Google Scholar]
- Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science. 1999;286:1358–1362. doi: 10.1126/science.286.5443.1358. [DOI] [PubMed] [Google Scholar]
- Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116:1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vesely J, Havlicek L, Strnad M, Blow JJ, Donella-Deana A, Pinna L, Letham DS, Kato J, Detivaud L, Leclerc S. Inhibition of cyclin-dependent kinases by purine analogues. Eur J Biochem. 1994;224:771–786. doi: 10.1111/j.1432-1033.1994.00771.x. [DOI] [PubMed] [Google Scholar]
- Sarna JR, Larouche M, Marzban H, Sillitoe RV, Rancourt DE, Hawkes R. Patterned Purkinje cell degeneration in mouse models of Niemann-Pick type C disease. J Comp Neurol. 2003;456:279–291. doi: 10.1002/cne.10522. [DOI] [PubMed] [Google Scholar]
- Shea TB, Yabe JT, Ortiz D, Pimenta A, Loomis P, Goldman RD, Amin N, Pant HC. Cdk5 regulates axonal transport and phosphorylation of neurofilaments in cultured neurons. J Cell Sci. 2004;117:933–941. doi: 10.1242/jcs.00785. [DOI] [PubMed] [Google Scholar]
- Smith DS, Tsai LH. Cdk5 behind the wheel: a role in trafficking and transport? Trends Cell Biol. 2002;12:28–36. doi: 10.1016/s0962-8924(01)02181-x. [DOI] [PubMed] [Google Scholar]
- Morfini G, Pigino G, Beffert U, Busciglio J, Brady ST. Fast axonal transport misregulation and Alzheimer’s disease. Neuromolecular Med. 2002;2:89–99. doi: 10.1385/NMM:2:2:089. [DOI] [PubMed] [Google Scholar]
- Rao MV, Nixon RA. Defective neurofilament transport in mouse models of amyotrophic lateral sclerosis: a review. Neurochem Res. 2003;28:1041–1047. doi: 10.1023/a:1023259207015. [DOI] [PubMed] [Google Scholar]
- Bian F, Nath R, Sobocinski G, Booher RN, Lipinski WJ, Callahan MJ, Pack A, Wang KK, Walker LC. Axonopathy, tau abnormalities, and dyskinesia, but no neurofibrillary tangles in p25-transgenic mice. J Comp Neurol. 2002;446:257–266. doi: 10.1002/cne.10186. [DOI] [PubMed] [Google Scholar]
- Ahlijanian MK, Barrezueta NX, Williams RD, Jakowski A, Kowsz KP, McCarthy S, Coskran T, Carlo A, Seymour PA, Burkhardt JE, Nelson RB, McNeish JD. Hyperphosphorylated tau and neurofilament and cytoskeletal disruptions in mice overexpressing human p25, an activator of cdk5. Proc Natl Acad Sci USA. 2000;97:2910–2915. doi: 10.1073/pnas.040577797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz JC, Tseng HC, Goldman JA, Shih H, Tsai LH. Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron. 2003;40:471–483. doi: 10.1016/s0896-6273(03)00627-5. [DOI] [PubMed] [Google Scholar]
- Athanasiou MC, Yunis W, Coleman N, Ehlenfeldt R, Clark HB, Orr HT, Feddersen RM. The transcription factor E2F-1 in SV40 T antigen-induced cerebellar Purkinje cell degeneration. Mol Cell Neurosci. 1998;12:16–28. doi: 10.1006/mcne.1998.0699. [DOI] [PubMed] [Google Scholar]
- Sawamura N, Gong JS, Garver WS, Heidenreich RA, Ninomiya H, Ohno K, Yanagisawa K, Michikawa M. Site-specific phosphorylation of tau accompanied by activation of mitogen-activated protein kinase (MAPK) in brains of Niemann-Pick type C mice. J Biol Chem. 2001;276:10314–10319. doi: 10.1074/jbc.M009733200. [DOI] [PubMed] [Google Scholar]
- Karten B, Vance DE, Campenot RB, Vance JE. Cholesterol accumulates in cell bodies, but is decreased in distal axons, of Niemann-Pick C1-deficient neurons. J Neurochem. 2002;83:1154–1163. doi: 10.1046/j.1471-4159.2002.01220.x. [DOI] [PubMed] [Google Scholar]
- Muller U, Kins S. APP on the move. Trends Mol Med. 2002;8:152–155. doi: 10.1016/s1471-4914(02)02320-1. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Wong PC, Becher MW, Pardo CA, Lee MK, Xu ZS, Thinakaran G, Jenkins NA, Copeland NG, Sisodia SS, Cleveland DW, Price DL, Hoffman PN. Axonal transport of mutant superoxide dismutase 1 and focal axonal abnormalities in the proximal axons of transgenic mice. Neurobiol Dis. 1998;5:27–35. doi: 10.1006/nbdi.1998.0178. [DOI] [PubMed] [Google Scholar]
- Maller JL. Mitotic control. Curr Opin Cell Biol. 1991;3:269–275. doi: 10.1016/0955-0674(91)90151-n. [DOI] [PubMed] [Google Scholar]
- Yang Y, Geldmacher DS, Herrup K. DNA replication precedes neuronal cell death in Alzheimer’s disease. J Neurosci. 2001;21:2661–2668. doi: 10.1523/JNEUROSCI.21-08-02661.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi Y, Bonni A. The E2F-Cdc2 cell-cycle pathway specifically mediates activity deprivation-induced apoptosis of post-mitotic neurons. J Neurosci. 2003;23:1649–1658. doi: 10.1523/JNEUROSCI.23-05-01649.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Rottkamp CA, Raina AK, Brewer GJ, Ghanbari HA, Boux H, Smith MA. Neuronal CDK7 in hippocampus is related to aging and Alzheimer disease. Neurobiol Aging. 2000;21:807–813. doi: 10.1016/s0197-4580(00)00217-7. [DOI] [PubMed] [Google Scholar]
- Zhu X, McShea A, Harris PL, Raina AK, Castellani RJ, Funk JO, Shah S, Atwood C, Bowen R, Bowser R, Morelli L, Perry G, Smith MA. Elevated expression of a regulator of the G2/M phase of the cell cycle, neuronal CIP-1-associated regulator of cyclin B, in Alzheimer’s disease. J Neurosci Res. 2004;75:698–703. doi: 10.1002/jnr.20028. [DOI] [PubMed] [Google Scholar]
- Patterson MC, Di Bisceglie AM, Higgins JJ, Abel RB, Schiffmann R, Parker CC, Argoff CE, Grewal RP, Yu K, Pentchev PG. The effect of cholesterol-lowering agents on hepatic and plasma cholesterol in Niemann-Pick disease type C. Neurology. 1993;43:61–64. doi: 10.1212/wnl.43.1_part_1.61. [DOI] [PubMed] [Google Scholar]
- Erickson RP, Garver WS, Camargo F, Hossain GS, Heidenreich RA. Pharmacological and genetic modifications of somatic cholesterol do not substantially alter the course of CNS disease in Niemann-Pick C mice. J Inherit Metab Dis. 2000;23:54–62. doi: 10.1023/a:1005650930330. [DOI] [PubMed] [Google Scholar]
- Camargo F, Erickson RP, Garver WS, Hossain GS, Carbone PN, Heidenreich RA, Blanchard J. Cyclodextrins in the treatment of a mouse model of Niemann-Pick C disease. Life Sci. 2001;70:131–142. doi: 10.1016/s0024-3205(01)01384-4. [DOI] [PubMed] [Google Scholar]