

Abstract
Few things can be considered to be more important to a cell than its threshold for apoptotic cell death, which can be modulated up or down, but rarely in both directions, by a single enzyme. Therefore, it came as quite a surprise to find that one enzyme, glycogen synthase kinase-3 (GSK3), has the perplexing capacity to either increase or decrease the apoptotic threshold. These apparently paradoxical effects now are known to be due to GSK3 oppositely regulating the two major apoptotic signaling pathways. GSK3 promotes cell death caused by the mitochondrial intrinsic apoptotic pathway, but inhibits the death receptor-mediated extrinsic apoptotic signaling pathway. Intrinsic apoptotic signaling, activated by cell damage, is promoted by GSK3 by facilitation of signals that cause disruption of mitochondria and by regulation of transcription factors that control the expression of anti- or pro-apoptotic proteins. The extrinsic apoptotic pathway entails extracellular ligands stimulating cell-surface death receptors that initiate apoptosis by activating caspase-8, and this early step in extrinsic apoptotic signaling is inhibited by GSK3. Thus, GSK3 modulates key steps in each of the two major pathways of apoptosis, but in opposite directions. Consequently, inhibitors of GSK3 provide protection from intrinsic apoptosis signaling but potentiate extrinsic apoptosis signaling. Studies of this eccentric ability of GSK3 to oppositely influence two types of apoptotic signaling have shed light on important regulatory mechanisms in apoptosis and provide the foundation for designing the rational use of GSK3 inhibitors for therapeutic interventions.
Keywords: Apoptosis, Glycogen synthase kinase-3, Death receptors, Caspase, Programmed cell death
1. Introduction
Why would a single enzyme have opposing actions so that it both promotes and inhibits apoptosis? That is the paradox raised by recent findings of the regulatory influences of glycogen synthase kinase-3 (GSK3) on apoptosis. Analysis of this puzzling capacity of GSK3 reveals that it stems from two different apoptosis signaling pathways being regulated in opposite directions by GSK3. Thus, GSK3 promotes the mitochondria-mediated intrinsic apoptotic signaling pathway, but it inhibits the death receptor-mediated extrinsic apoptotic signaling pathway. Why, and how, does this enzyme associated with glycogen metabolism have opposite effects on the two major pathways of apoptosis?
GSK3 is an example of the way in which perceptions about an enzyme’s full range of functions can be constrained by how it was initially discovered and named. GSK3 is an ubiquitous serine/threonine kinase that is present in mammals in two isoforms: a and b (Woodgett, 1990). Although GSK3 was first identified as an enzyme capable of phosphorylating glycogen synthase to inhibit glycogen synthesis (Embi et al., 1980), since then it has been found to phosphorylate nearly 50 substrates (Jope and Johnson, 2004). Phosphorylating these substrates allows GSK3 to modulate many fundamental processes including cell structure, metabolism, gene expression, and, in a seemingly paradoxical manner, apoptosis (Grimes and Jope, 2001; Frame et al., 2001).
Carefully regulated cell death by apoptosis is crucial in the development and homeostasis of all multicellular organisms. This is emphasized by the prevalence of diseases associated with abnormal apoptosis. For example, deficient apoptosis is a hallmark of cancer and autoimmune diseases, whereas excessive cell death occurs in several neurodegenerative diseases. The two most common apoptotic pathways are the ‘intrinsic’ pathway in which cellular stresses disrupt mitochondrial integrity, and the ‘extrinsic’ pathway that is initiated by stimulation of ‘death receptors’ in the plasma membrane (Hengartner, 2000). Both apoptotic pathways culminate in the activation of a family of intracellular cysteine proteases called caspases. These are classified as initiator caspases (caspases-8, -9, and -10) or effector caspases (caspases-3, -6, and -7) (Riedl and Shi, 2004), which can disrupt entire cells within a few minutes of their activation (Earnshaw et al., 1999).
The paradoxical apoptosis-regulating actions of GSK3 came to light after the puzzling observations that GSK3 can have opposite actions on apoptosis, either strongly inhibiting or promoting apoptotic signaling. The concept that GSK3 inhibits apoptosis came from the discovery that GSK3β knockout mice died during embryonic development due to massive hepatocyte apoptosis (Hoeflich et al., 2000), which demonstrated that GSK3β is an important inhibitor of apoptosis. However, this observation appears to be in direct opposition to the finding that overexpression of GSK3β is sufficient to induce apoptosis (Pap and Cooper, 1998). These opposite effects of GSK3β on apoptosis have been reinforced by studies with inhibitors of GSK3, including the first known selective inhibitor, lithium (Klein and Melton, 1996), and many new synthetic inhibitors of GSK3 (Eldar-Finkelman, 2002; Martinez et al., 2002). GSK3 inhibitors promote apoptosis induced by stimulation of death domain-containing receptors but provide protection from many other insults that induce apoptosis. Recent studies have clarified that these apparent conflicting findings are due to the ability of GSK3 to have opposite effects on apoptosis depending on the apoptotic signaling pathway that is involved. We review here the substantial evidence that GSK3 is pro-apoptotic with insults that activate the intrinsic mitochondrial apoptotic pathway, and is anti-apoptotic with stimulation of death domain-containing receptors that activate the extrinsic apoptotic pathway.
2. Regulation of GSK3
In order to understand the physiological roles of GSK3 in apoptosis, it is important to consider the mechanisms that regulate GSK3. Both isoforms of GSK3 are constitutively active in resting cells, but their actions are tightly controlled by several mechanisms (Fig. 1). The most well-defined regulatory mechanism is by phosphorylation of serine-9 in GSK3β or serine-21 in GSK3a, which inhibits activity. The phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathway often is a major regulator of GSK3 because Akt phosphorylates GSK3 on these inhibitory serine residues, but several other kinases also can phosphorylate them (Grimes and Jope, 2001). Conversely, enzymatic activity is enhanced by phosphorylation of tyrosine-216 in GSK3β and tyrosine-279 in GSK3a, although mechanisms regulating this modification are not well-defined.
Because of its many substrates, additional mechanisms besides phosphorylation are needed to regulate GSK3. These include control of its intracellular localization and of its association in protein complexes, mechanisms that serve to selectively direct its actions towards specific substrates (Jope and Johnson, 2004). Additionally, the action of GSK3 is usually indirectly controlled by the phosphorylation state of its substrate. This is because most substrates of GSK3 must be ‘primed’, i.e., prephosphorylated at a residue 4-amino acids C-terminal to the GSK3 phosphorylation site. This necessitates temporal coordination of the activity of the priming kinase along with GSK3 activity for the latter to phosphorylate the primed substrate.
These complexities in the mechanisms controlling the actions of GSK3, involving phosphorylation, distribution, protein complexes, and substrate priming, combine to provide substrate-specific control of its actions, an especially important capacity for an enzyme that can regulate cell survival. Consequently, dysregulation of GSK3 can contribute to a diverse variety of pathologies. For example, dysregulation of GSK3 has been linked to mood disorders, neurodegenerative diseases such as Alzheimer’s disease, cancer, inflammation, cardiovascular disease, and diabetes (Manoukian and Woodgett, 2002; Eldar-Finkelman and Ilouz, 2003; Woodgett, 2003; Jope and Johnson, 2004; Patel et al., 2004; Fuentealba et al., 2004; Ferrer et al., 2005; Martin et al., 2005). Thus, GSK3 must be tightly regulated, its dysregulation is associated with a wide variety of diseases, and some of the detrimental actions of dysregulated GSK3 in these diseases may be linked to apoptosis.
3. GSK3 promotes the mitochondrial intrinsic apoptosis pathway
The most widely reported association between GSK3 and apoptosis involves the mitochondria-mediated intrinsic apoptotic pathway (Fig. 2). The intrinsic apoptotic signaling cascade can be induced by numerous stimuli that cause cell damage, such as DNA damage, oxidative stress, endoplasmic reticulum (ER) stress, and many other insults. These conditions that activate intrinsic apoptotic signaling cause the disruption of mitochondria, leading to cell destruction, processes that are promoted by GSK3.
3.1. Intrinsic apoptotic signaling is facilitated by GSK3
Intrinsic apoptotic signaling has been shown to be amplified by GSK3 following many types of cellular insults, as summarized in Table 1. Initially, GSK3 was found to be an important component of the apoptotic response occurring after growth factor withdrawal or inhibition of the PI3K/Akt signaling pathway, which is often coupled to growth factor receptors (Pap and Cooper, 1998, 2002; Hetman et al., 2000; Crowder and Freeman, 2000; Bhat et al., 2000; Li et al., 2000; Cross et al., 2001; Somervaille et al., 2001; Culbert et al., 2001; Bhat et al., 2003; Hongisto et al., 2003; Sanchez et al., 2003; Linseman et al., 2004; Chin et al., 2005; Enguita et al., 2005; Jin et al., 2005b; Sinha et al., 2005). GSK3 also promotes apoptosis caused by DNA damage (Watcharasit et al., 2002, 2003; Beurel et al., 2004, 2005; Jin et al., 2005a; Tan et al., 2006), ER stress (Song et al., 2002; Chen et al., 2004; Kim et al., 2005; Macanas-Pirard et al., 2005; Srinivasan et al., 2005), mitochondrial toxins (King et al., 2001; King and Jope, 2005), hypoxia/ischemia (Loberg et al., 2002; Mottet et al., 2003; Juhaszova et al., 2004; Kelly et al., 2004; Brywe et al., 2005; Cappuccio et al., 2005; Kaga et al., 2005; Rho et al., 2005; Yin et al., 2005), glutamate excitotoxicity (Hashimoto et al., 2002; Facci et al., 2003; Kelly et al., 2004; Takadera et al., 2004), platelet activating factor (Maggirwar et al., 1999; Tong et al., 2001), hypertonic stress (Rao et al., 2004), ceramide (Mora et al., 2002), ethanol (Takadera and Ohyashiki, 2004), heat shock (Bijur et al., 2000), staurosporine (Bijur et al., 2000), and oxidative stress (Shin et al., 2004; King and Jope, 2005). Many additional studies have reported that the GSK3 inhibitor lithium provides protection from other apoptotic conditions, but since GSK3 was not directly examined, these protective effects cannot yet unequivocally be attributed to inhibition of GSK3, as discussed in previous reviews (Jope, 2003, 2004; Chuang, 2005). Overall, these many studies have unequivocally demonstrated that GSK3 promotes the intrinsic apoptotic signaling cascade induced by a diverse array of insults, indicating that GSK3 regulates fundamental processes in this cascade.
3.2. Mitochondria-related targets of GSK3 during apoptosis
3.2.1. Intrinsic apoptotic signaling
The many apoptotic conditions that are facilitated by GSK3 raise the question of which components of intrinsic apoptotic signaling are subject to regulation by GSK3. One of these is disruption of mitochondrial function, a central feature of the intrinsic apoptotic pathway (Fig. 2). Pro-apoptotic members of the Bcl-2 family of proteins are key messengers for delivering the apoptotic signal to the mitochondria (Akhtar et al., 2004). For example, one of these, Bax, undergoes an activating conformational change following cellular insults that causes it to translocate from the cytoplasm to the mitochondria where it can both sequester anti-apoptotic Bcl-2 family proteins and oligomerize within the mitochondrial membrane (Martinou and Green, 2001). This oligomerization of Bax in the outer mitochondrial membrane contributes to the disruption of the mitochondrial membrane potential and the release of apoptotic proteins from the mitochondrial intermembrane space into the cytoplasm (Armstrong, 2006). Perhaps the most intriguing pro-apoptotic protein that is released is cytochrome c, which binds to the protein apoptotic protease activating factor-1 (APAF-1), ATP/dATP, and procaspase-9 to form the apoptosome in the cytoplasm. This causes the activation of caspase-9, thereby triggering the activation of the caspase cascade (Armstrong, 2006).
3.2.2. Mitochondrial disruption is promoted by GSK3 during apoptosis
GSK3 is positioned to be associated with intrinsic apoptotic events associated with mitochondrial function as it is present within mitochondria, and mitochondrial GSK3 activity is increased during intrinsic apoptotic signaling induced by DNA damage or ER stress (Bijur and Jope, 2003a,b). Although direct intramitochondrial substrates of GSK3 involved in apoptosis have not yet been identified, GSK3 targets several key proteins that regulate signals leading to the disruption of mitochondria. Investigations of the target of GSK3 showed that its apoptotic action was upstream of caspase-3 in many of the studies cited above, of caspase-9 (Bijur et al., 2000), of cytochrome c release from mitochondria (Watcharasit et al., 2003), of the permeability transition pore complex in mitochondria (Juhaszova et al., 2004), and of the activation of the pro-apoptotic Bcl-2 family members Bax and Bcl-2 interacting mediator of cell death (Bim) (Somervaille et al., 2001; Linseman et al., 2004; Hongisto et al., 2003). GSK3 can directly phosphorylate Bax on Ser-163, which results in the activation of Bax (Linseman et al., 2004), and GSK3 is required for the stress-induced expression of Bim (Hongisto et al., 2003). Additionally, a recent report found that phosphorylation by GSK3 enhanced the degradation of the anti-apoptotic Bcl-2 family member MCL-1 (Maurer et al., 2006). GSK3 also phosphorylates the voltage-dependent anion channel (VDAC), an abundant outer mitochondrial membrane protein implicated in maintaining the mitochondrial membrane potential. This phosphorylation prevents hexokinase II from associating with VDAC (Pastorino et al., 2005), which may facilitate the mitochondrial association of pro-apoptotic Bcl-2 family proteins to promote apoptosis (Pastorino et al., 2002; Cheng et al., 2003; Majewski et al., 2004). Thus, one way GSK3 promotes the intrinsic apoptotic signaling pathway is to expedite signals that contribute to the disruption of mitochondria, and several candidate targets of GSK3 have been identified that may contribute to accomplishing this outcome.
3.2.3. GSK3 modulates expression of apoptotic intermediates acting on mitochondria
Mitochondrial function during intrinsic apoptotic signaling is also influenced by GSK3-mediated regulation of the expression of proteins participating in the mitochondrial stage of the apoptotic cascade since GSK3 modulates the activity of a large number of transcription factors that control gene expression. Some of these are regulated by nuclear GSK3 where a small, but dynamic, portion of cellular GSK3β is located. Nuclear GSK3β is in a relatively greater activation state than cytosolic GSK3β, and apoptotic conditions can increase the level and/or activity of GSK3β in the nucleus (Bijur and Jope, 2001, 2003b; Watcharasit et al., 2002; Elyaman et al., 2002). Therefore, both nuclear GSK3 and targets of GSK3 that act in the nucleus, particularly transcription factors, may contribute to its promotion of the intrinsic apoptotic pathway. Conspicuous examples of transcription factors that encode apoptosis-regulating proteins targeted to the mitochondria and that are regulated by GSK3 in the nucleus include p53 and cyclic AMP response element binding protein (CREB). GSK3β activity promotes p53-induced expression of Bax in response to DNA damage (Watcharasit et al., 2003; Tan et al., 2005) and inhibits CREB (Bullock and Habener, 1998; Grimes and Jope, 2001), which can block the CREB-dependent expression of the anti-apoptotic protein Bcl-2 (Lonze and Ginty, 2002). This action is supported by evidence that treatment with the GSK3 inhibitor lithium increases Bcl-2 levels (Chen and Chuang, 1999; Chen et al., 1999; Kaga et al., 2005). Therefore, GSK3 can regulate the expression levels of proteins that are key mitochondrial components of the intrinsic apoptotic signaling pathway and by oppositely regulating the expression of proapoptotic and anti-apoptotic proteins GSK3 can change the balance to lower the threshold for intrinsic apoptotic signaling. Thus, GSK3 both ’sets the stage’ for intrinsic apoptotic signaling through its regulation of gene expression, and also facilitates signaling that leads to disruption of mitochondria.
3.3. GSK3 regulates transcription and translation factors
In addition to regulating the expression of proteins directly involved in mitochondrial function during apoptosis, GSK3 also regulates the expression of other components of apoptotic signaling as a result of its phosphorylation of transcription and translation factors.
3.3.1. p53
The important tumor suppressor transcription factor p53, which is a key mediator of cell cycle arrest, senescence, and apoptosis, has been implicated in the pro-apoptotic actions of GSK3 in several studies. p53 was first linked to GSK3 by the finding that expression of dominant-negative p53 blocked apoptosis induced by overexpression of GSK3β (Pap and Cooper, 1998). Following DNA damage, the normally short-lived p53 protein is stabilized and modified by a complex array of posttranslational modifications, such as phosphorylation, acetylation, ubiquitination, and sumoylation, and a large number of proteins interact with p53 to regulate its actions (Oren, 2003; Haupt et al., 2002).
One of these regulatory proteins is GSK3β, which forms a complex with nuclear p53 to promote p53-induced apoptosis (Watcharasit et al., 2002, 2003; Beurel et al., 2004). GSK3β also interacts with p53 in the nucleus during cellular senescence (Zmijewski and Jope, 2004). GSK3β binds directly to p53, and the C-terminal region of p53 is necessary for this interaction (Watcharasit et al., 2003). Although an interaction between GSK3β and p53 has been confirmed in several studies, the functional consequences are controversial, possibly because of the many other regulatory influences on p53 and the context- and cell-specific regulation and actions of p53. Individual laboratories have reported that GSK3 can phosphorylate Ser33-p53 (Turenne and Price, 2001) or Ser315-p53 and Ser376-p53 (Qu et al., 2004; Pluquet et al., 2005). Moreover, GSK3 can regulate p53 levels through the phosphorylation of the p53-regulating protein MDM2 (Kulikov et al., 2005; Pluquet et al., 2005) or other mechanisms (Ghosh and Altieri, 2005). GSK3 promotes p53-mediated transcription of specific genes (Watcharasit et al., 2003; Beurel et al., 2004) and regulates the intracellular localization of p53 (Beurel et al., 2004; Qu et al., 2004; Pluquet et al., 2005). p53 is also able to activate apoptosis independently of its transcription function by acting directly on mitochondrial proteins (Sansome et al., 2001; Marchenko et al., 2000; Mihara et al., 2003), and GSK3β binds p53 in the mitochondria, which may contribute to p53-induced apoptosis (Watcharasit et al., 2003).
Thus, there are multiple influences of GSK3 on p53, but control of the dominant outcomes by other regulatory mechanisms associated with specific conditions that activate p53 and cell type-selective effects remain to be more fully defined. In addition to GSK3β regulating p53, GSK3β is also regulated by p53. The activity of GSK3β is increased by a phosphorylation-independent mechanism by direct binding of p53 to GSK3β (Watcharasit et al., 2002). Thus, cooperative interactions involving activation of both GSK3 and p53 contribute to the promotion of intrinsic apoptosis signaling under conditions where p53 is activated.
3.3.2. β-Catenin
GSK3 also influences a number of transcription factors that regulate the expression of anti-apoptotic proteins, which has generated evidence, although largely correlative, that inhibition of these transcription factor targets of GSK3 may contribute to its promotion of intrinsic apoptotic signaling. In the canonical Wnt signaling pathway, the transcriptional co-activator β-catenin promotes growth and survival, but phosphorylation of β-catenin by GSK3 targets it for degradation by the proteasome (Ciani and Salinas, 2005). Activation of Wnt signaling inhibits GSK3 selectively in the Wnt signaling protein complex, causing accumulation of β-catenin and its translocation to the nucleus where it interacts with the TCF/LEF transcription factors to induce gene expression, several of which encode proteins that support cell survival (Polakis, 1999). Thus, apoptosis was inhibited by activation of β-catenin/TCF/LEF-mediated transcription by overexpression of Wnt (Chen et al., 2001; Alvarez et al., 2004) or expression of a dominant-stable β-catenin (Longo et al., 2002; Ueda et al., 2002). Furthermore, overexpressing β-catenin reduced apoptosis similarly to the protection provided by inhibitors of GSK3 (Yuan et al., 2005), and the accumulation of β-catenin caused by GSK3 inhibitors has been correlated with an increase of the expression of the anti-apoptotic proteins Bcl-2 and survivin (Kaga et al., 2005). Consistent with these findings, loss of β-catenin signaling caused by mutations of the presenilin-1 gene increased neuronal vulnerability to apoptosis (Zhang et al., 1998). These studies indicate that β-catenin may be an important target that is inhibited by GSK3 to promote apoptosis.
3.3.3. HSF-1
Heat shock factor-1 (HSF-1) is another survival-promoting transcription factor that is inhibited by GSK3. HSF-1 induces the expression of several heat shock proteins that protect cells from many insults. GSK3 phosphorylates HSF-1 to inhibit its transcriptional activity, thereby reducing expression of heat shock proteins, an action that can facilitate apoptosis (Chu et al., 1996; He et al., 1998a; Bijur and Jope, 2000; Khaleque et al., 2005). Evidence for this interaction in vivo was obtained in studies of ischemia, which showed that increased activation of HSF-1 was correlated with robust neuroprotection from ischemia provided by treatment with the GSK3 inhibitor lithium (Nonaka and Chuang, 1998; Ren et al., 2003).
3.3.4. Myc
There is also some evidence that regulation of the transcription factor Myc by GSK3 may modulate apoptosis. Myc is a transcription factor capable of promoting either proliferation or apoptosis, as well as other outcomes (Adhikary and Eilers, 2005). Myc expression can be increased by activation of β-catenin-TCF/LEF, which occurs when GSK3 in the Wnt system is inhibited, and active GSK3 phosphorylates Myc to promote its degradation (He et al., 1998b; Saksela et al., 1992). Inhibition of GSK3 by activation of Wnt signaling inhibited c-Myc-induced apoptosis (You et al., 2002), indicating multiple levels of regulation of Myc by GSK3, which may be involved in some apoptotic conditions, although this has not been widely studied.
3.3.5. NF-κB
Finally, the most studied, and also most ambiguous, effect of GSK3 on a transcription factor involved in regulating apoptosis is its regulation of nuclear factor-kB (NF-κB). As discussed in the following section about extrinsic apoptotic signaling, multiple effects of GSK3 have been reported on the signaling steps leading to transcriptionally active NF-κB, and modulation of NF-κB by GSK3 has been reported to either promote (Bournat et al., 2000; Sanchez et al., 2003; Buss et al., 2004; Rao et al., 2004; Viatour et al., 2004) or inhibit (Bournat et al., 2000; Sanchez et al., 2003; Buss et al., 2004; Rao et al., 2004; Viatour et al., 2004) apoptosis. This ambiguity has arisen in part because NF-κB can influence both intrinsic and extrinsic apoptotic signaling, systems where GSK3 itself has opposite effects, and from variations among cell types and stimuli in the role that NF-κB plays in apoptosis.
3.3.6. Protein synthesis
In addition to regulating gene expression, GSK3 also regulates translation, and Pap and Cooper (2002) showed that inhibition of protein synthesis, which GSK3 achieves by phosphorylating and inhibiting eIF2B, contributes an important component to GSK3-induced apoptosis.
In summary, with actions at both the transcriptional and translational levels, GSK3 can have broad regulatory effects on the expression of proteins involved in apoptotic signaling. Thus, it is now evident that GSK3 performs coordinated actions in cells that promote disruption of mitochondria and increases the expression of pro-apoptotic proteins while reducing the expression of anti-apoptotic proteins to lower the threshold necessary for intrinsic apoptotic signaling to culminate in cell death.
3.4. GSK3 regulates cell structure
In addition to GSK3-regulating cell survival by phosphorylating proteins in the apoptosis cascade and transcription/translation factors, phosphorylation of proteins that dynamically regulate cell structure also likely contributes to the facilitation of apoptosis by GSK3. Several proteins associated with microtubules are substrates of GSK3, and phosphorylation of these by GSK3 contributes to destabilization of microtubules (Jope and Johnson, 2004). Destabilization and the subsequent collapse of microtubules permits some of the cellular structural changes that occur during apoptosis. For example, the microtubule-associated protein tau is one of the most well-characterized substrates of GSK3 (Johnson and Bailey, 2002). Phosphorylation of tau by GSK3 causes it to dissociate from microtubules, promoting destabilization of microtubules. In addition to this structural component of apoptosis, tau can form oligomers and aggregates that include neurofibrillary tangles, preceded by hyperphosphorylation of tau to which GSK3 likely contributes (Hong et al., 1997; Perez et al., 2003β; Noble et al., 2005), and these complexes of tau themselves can be toxic (Avila et al., 2004; Santacruz et al., 2005). Another example of a structural substrate of GSK3 is kinesin, which is a molecular motor responsible for intracellular transport of cargo proteins. Phosphorylation of kinesin by GSK3 disrupts this intracellular transport, which can contribute to apoptotic cell death (Morfini et al., 2002; Mudher et al., 2004). Thus, actions of GSK3 on cellular structural proteins likely underlie a portion of the morphological changes that occur during the apoptosis process.
3.5. Overexpression of GSK3 induces apoptosis
Although studies of the pro-apoptotic actions of GSK3 were first stimulated by the seminal finding that overexpression of GSK3β is sufficient to induce apoptosis, it remains to be determined how well this condition models intrinsic apoptotic signaling mechanisms. By overexpressing GSK3β in PC12 cells and Rat1 fibroblasts it was first discovered that GSK3β alone, without additional toxic insults, is sufficient to induce apoptosis (Pap and Cooper, 1998). Subsequent studies showed that this is a widespread effect, as overexpression of GSK3β induced apoptosis in cortical neurons (Hetman et al., 2000, 2002), sympathetic neurons (Crowder and Freeman, 2000), vascular smooth muscle cells (Hall et al., 2001), endothelial cells (Kim et al., 2002), astrocytes (Sanchez et al., 2003) and human umbilical vein endothelial cells (Suhara et al., 2003). Although apoptosis induced by overexpressed GSK3β has provided valuable information about its pro-apoptotic actions, the mechanism by which overexpressed GSK3β causes apoptosis has not been identified. This remains an important question because overexpressed GSK3β likely involves increases in cellular GSK3β activity many-fold above that attainable by activation of endogenous GSK3β, and thus different targets and apoptotic signals may be recruited by overexpressed GSK3β than those employed by endogenous GSK3 during apoptosis. This concept that GSK3 at physiological levels is a facilitator, rather than an inducer, of apoptosis is illustrated by the observation that double knock in GSK3a/b mice, in which both isoforms were constitutively activated by changing the codons for serine-21 of GSK3a and serine-9 of GSK3β to encode nonphosphorylatable alanines, displayed no overt phenotype (McManus et al., 2005). This shows that constitutively active GSK3 is not alone sufficient to cause excessive apoptosis in these transgenic mice, strengthening the conclusion that the role of GSK3 in apoptosis lies in its capacity to modulate the threshold for apoptosis.
3.6. Summary
In summary, promotion by GSK3 of the intrinsic apoptotic signaling pathway is now well-established. This action of GSK3 appears to be the culmination of orchestrated events that include facilitating the disruption of mitochondrial function and regulating the expression of proteins that are capable of modulating apoptosis signaling. The critical events causing disruption of the mitochondria leading to caspase activation are clearly facilitated by GSK3, although the precise substrate, or set of substrates, of GSK3 that accounts for this action remains to be more completely clarified. Concurrently, GSK3 regulates the expression of proteins involved in apoptosis, generally inhibiting the expression of anti-apoptotic proteins and facilitating expression of pro-apoptotic proteins to allow efficient progression of the apoptosis signal. However, much of the expression data remains correlative, as seldom have alterations in the expression levels of particular proteins regulated by GSK3 been shown to specifically contribute to the promotion of intrinsic apoptotic signaling caused by GSK3 activity. Although these expression changes may only be correlative, the known participation of such proteins as Bax, Bcl-2, and heat shock proteins, among others, that are regulated by GSK3 is highly suggestive that changes in their levels under the regulation of GSK3 contributes to the promotion of apoptotic signaling. These effects suggest that GSK3 generally sets the stage for apoptosis, by both promoting expression of pro-apoptotic proteins and inhibiting the expression of anti-apoptotic proteins, actions that can lower the threshold for the induction of apoptosis. It is likely that the most influential of these many actions of GSK3 on apoptosis vary depending upon the cellular insult and the cell type. Taken together, enhancing apoptotic disruption of mitochondrial function while regulating the expression of proteins involved in the apoptotic cascade makes GSK3 a powerful regulator of apoptosis. These key actions likely underlie the ability of GSK3 to promote the intrinsic apoptotic signaling pathway following such a wide array of different types of cellular insults that have been linked to GSK3.
Promotion of the intrinsic apoptotic signaling pathway by GSK3 may be particularly important in the neuronal loss and apoptosis that occurs in a number of neurodegenerative diseases. The most well-documented of these is Alzheimer’s disease where GSK3 has been shown to promote the toxicity of both the amyloid β-peptide and other neurodegenerative processes (Takashima et al., 1993; Alvarez et al., 1999, 2004; Wei et al., 2000; Garrido et al., 2002; Bhat et al., 2003; De Ferrari et al., 2003; Hoshi et al., 2003; Phiel et al., 2003; Suhara et al., 2003; Zhang et al., 2003; Su et al., 2004; Noble et al., 2005). There is also evidence that GSK3 contributes to the promotion of apoptosis caused by Parkinson’s disease-related toxic agents such as 6-hydroxydopamine, rotenone, and MPTP (King et al., 2001; Chen et al., 2004; Bai et al., 2004; Avraham et al., 2005), by prion peptide (Perez et al., 2003a), by mutations in superoxide dismutase linked to amyotrophic lateral sclerosis (Koh et al., 2005), by HIV-associated conditions (Maggirwar et al., 1999; Tong et al., 2001; Everall et al., 2002; Dou et al., 2003, 2005), by polyglutamine toxicity associated with Huntington’s disease (Carmichael et al., 2002), and by ischemia in the brain (Chuang, 2005). These findings raise the possibility that inhibitors of GSK3 may ameliorate some of the apoptosis and neuronal loss occurring in a number of neurodegenerative diseases. Furthermore, consideration of this broad pro-apoptotic influence of GSK3 during intrinsic apoptosis makes even more surprising the finding that GSK3 has an opposite, anti-apoptotic, effect on extrinsic apoptosis induced by activation of death receptors, as discussed in the following section.
4. GSK3 inhibits the death receptor-mediated extrinsic apoptosis pathway
4.1. Extrinsic apoptotic signaling
Death domain-containing receptors, commonly called death receptors (DR), belong to the tumor necrosis factor (TNF) family of receptors that contain conserved intracellular death domains that are critical for the initiation of extrinsic apoptotic signaling (Ashkenazi and Dixit, 1998). Among the most well known death receptors are TNF-R1 (also called p55 or CD120a), Fas (also called CD95 or Apol), DR4 (also called TRAIL-R1 for TNF-related apoptosis-inducing ligand receptor-1) and DR5 (also called TRAIL-R2, Apo2, TRICK2, or KILLER). These receptors share common signaling mechanisms (Fig. 3) although each receptor is stimulated by its own specific ligand, for example TNF, Fas Ligand, and TRAIL activate TNF-R1, Fas and DR4/DR5, respectively (Wajant et al., 2003). A common feature of death receptors is that stimulation causes receptor homo-trimerization followed by the recruitment of cytoplasmic adaptor and effector proteins (Krammer, 2000). The activated trimerized death receptors bind to the cytoplasmic proteins FADD (Fas-associated death domain protein) and procaspase-8 (or procaspase-10) to form a complex known as the death-inducing signaling complex (DISC) (Peter and Krammer, 2003). DISC formation can allow autoactivation of caspase-8, which then leads to the activation of effector caspases, primarily caspases-3, -6, and -7 (Kaufmann and Hengartner, 2001). Downstream from DISC formation, two pathways can be used to complete the activation of apoptosis. In “type I” cells, such as lymphocytes, DISC-activated caspase-8 directly mobilizes downstream effector caspases to trigger apoptosis. However, the extrinsic apoptotic pathway also can incorporate portions of the intrinsic apoptotic pathway through caspase-8 mediated cleavage of the BH3-only protein Bid, which then interacts with mitochondria to facilitate the release of mitochondrial proteins that promote apoptosis (Luo et al., 1998; Li et al., 1998). Thus, in the majority of cells (“type II”), DISC-induced activation of caspase-8 is insufficient to kill cells without recruiting the mitochondrial apoptotic program through cleavage and activation of Bid (Scaffidi et al., 1998).
4.2. GSK3 protects from TNF-mediated cytotoxicity
As early as 1989, lithium had been identified as a factor that increased TNF-mediated cytotoxicity in several types of cells in vitro and in mouse tumors in vivo (Beyaert et al., 1989). Although these findings were extended in subsequent reports (Beyaert et al., 1992, 1993; Schotte et al., 2001), mechanistic studies were difficult at that time due to the limited understanding of lithium’s targets and the little amount of information available about GSK3. Two major findings were critical for further progress. First, the discovery in 1996 that lithium inhibits GSK3 (Klein and Melton, 1996) was critical to allow later studies to examine if lithium’s cytotoxic interaction with TNF involved GSK3. Second, the finding that knocking out GSK3β caused mouse embryonic lethality on day E14 due to TNF hypersensitivity in the liver (Hoeflich et al., 2000) provided the key insight that GSK3β inhibits TNF-induced apoptosis. This was supported by studies in mouse embryonic fibroblasts (MEFs) from GSK3β knockout and normal mice, and lithium was shown to potentiate TNF-induced cytotoxicity in MEFs from wild-type mice (Hoeflich et al., 2000). That inhibition of GSK3 by lithium accounted for potentiation of TNF-induced apoptosis in hepatocytes was further confirmed by Schwabe and Brenner (2002). Thus, knocking out GSK3β or lithium treatment potentiated TNF-induced apoptosis, showing that the mechanism underlying the cytotoxic interaction of lithium and TNF results from lithium’s inhibition of GSK3.
4.3. GSK3 protects from death receptor-mediated apoptosis
The inhibitory effect of GSK3 on TNF-induced apoptosis has been extended to other death receptors, demonstrating that this is a generalized action regulating extrinsic apoptosis. Liao et al. (2003) showed in human prostrate cancer cell lines that TRAIL-induced apoptosis was potentiated by both lithium and another highly selective inhibitor of GSK3, SB216763, as well as by knockdown of the GSK3β protein level using RNA interference. More recently, apoptosis induced by selective stimulation of DR5 with an agonistic antibody was shown to be potentiated by inhibition of GSK3 (Rottmann et al., 2005). Apoptosis signaling induced by the other major member of the death receptor family, Fas, also was recently found to be regulated by the anti-apoptotic action of GSK3 (Song et al., 2004). Several inhibitors of GSK3 potentiated Fas-induced apoptosis in Jurkat cells and also in differentiated hippocampal neurons, extending this interaction between GSK3 and death receptor-induced apoptosis simultaneously to non-proliferating cells and to neurons (Song et al., 2004). These results indicate that the beneficial effect that might be achieved in cancer cells by inhibition of GSK3 through potentiation of death receptor-mediated apoptosis contrasts with its detrimental effects for neurons subjected to death receptor-mediated apoptosis. Overall, it is now clear that GSK3 inhibits TNF-, TRAIL- or Fas-mediated apoptosis and that the potentiation of death receptor-induced cytotoxicity by lithium is due to inhibition of GSK3.
4.4. Mechanisms of extrinsic apoptotic signaling promotion by GSK3
Several steps upstream of cell death have been examined in order to identify the site of action of GSK3 that accounts for its anti-apoptotic effect in death receptor-mediated apoptotic signaling. By identifying steps in the apoptotic cascade that are potentiated by inhibitors of GSK3, several studies have shown that the inhibition of GSK3 enhances the extrinsic apoptotic signaling pathway upstream of the activation of caspase-3 (Schwabe and Brenner, 2002; Liao et al., 2003; Aza-Blanc et al., 2003; Song et al., 2004), of Bid cleavage (Aza-Blanc et al., 2003; Liao et al., 2003), and of the activation of caspase-8 (Schwabe and Brenner, 2002; Aza-Blanc et al., 2003; Liao et al., 2003). Thus the anti-apoptotic effect of GSK3 is directed at a very early stage in death receptor-induced signaling that precedes the initiating step of caspase-8 activation.
Although the anti-apoptotic effect of GSK3 in death receptor-stimulated apoptosis is now well-established, the mechanistic basis for this action of GSK3 remains to be identified. Many studies of this mechanism have focused on the NF-κB transcription factor (Karin and Ben-Neriah, 2000), which, although regulated by GSK3, appears unlikely to account for the protective action of GSK3 against death receptor-induced apoptosis. NF-κB was studied because it is activated by TNF and it provides an anti-apoptotic balance to the pro-apoptotic signaling induced by TNF, so promotion of NF-κB activity by GSK3 represented a potential mechanism by which GSK3 could strengthen the anti-apoptotic response of cells to TNF. The connection between GSK3 and NF-κB was first established by the finding that TNF-induced NF-κB activity was greatly diminished by knocking out or inhibiting GSK3β (Hoeflich et al., 2000). Furthermore, these investigators showed that GSK3 did not modulate signaling leading to NF-κB activation but that it was necessary for the transcriptional activity of NF-κB (Hoeflich et al., 2000). This conclusion was recently extended in an elegant study demonstrating that this modulatory effect of GSK3 was promoter-selective so that GSK3 promotes the transcription of only a subset of NF-κB-induced genes (Steinbrecher et al., 2005). This may explain the ambiguous regulatory effect of GSK3 on NF-κB that appears to be context- and cell type-specific. A number of studies concerning this regulation, although not always in the context of death receptor signaling, have reported targets of GSK3 within the signaling pathway leading to NF-κB activation as well as on its transcriptional activity and have shown that GSK3 has inhibitory (Nemeth et al., 2002; Demarchi et al., 2003; Liao et al., 2003; Sanchez et al., 2003; Buss et al., 2004; Viatour et al., 2004) as well as stimulatory (Schwabe and Brenner, 2002; Guha and Mackman, 2002; De Ketelaere et al., 2004; Takada et al., 2004; Eto et al., 2005) effects on NF-κB. As noted by Steinbrecher et al. (2005) some of these conflicting reports may stem from the fact that inhibition of GSK3 can increase levels of β-catenin and β-catenin itself can inhibit NF-κB activity (Deng et al., 2002, 2004). Taken together, these studies indicate that the modulation of NF-κB by GSK3 is highly complex and likely depends on cell type, the particular stimulus used to activate NF-κB, and the NF-κB-regulated gene being studied. Overall, this absence of consensus mechanisms of NF-κB regulation by GSK3 makes it evident that although NF-κB can be regulated by GSK3 in a complex manner, it is not likely to account completely for the protective effect of GSK3 in apoptosis induced by activation of all death receptors. Thus, the primary target of GSK3 located upstream of caspase-8 activation by stimulated death receptors that is responsible for the attenuation of death receptor-induced apoptosis by GSK3 remains to be identified.
Considerations of the anti-apoptotic effect of GSK3 on death receptor-induced apoptosis also must take into account the possibility that the two isoforms of GSK3 may have differential effects. This was first indicated by the work of Hoeflich et al. (2000) who noted the selective importance of GSK3β in regulating toxicity induced by TNF because endogenous GSK3a was unable to compensate for the loss of GSK3β and the ensuing increase in TNF-induced apoptosis. In agreement with the selective role of GSK3β, knockdown of GSK3β, but not GSK3a, potentiated TRAIL-induced apoptosis in human prostrate cancer PC-3 cells (Liao et al., 2003) and promoted apoptosis induced by activation of DR5 (Rottmann et al., 2005). However, in contrast to these findings, in the only report that showed that inhibition of GSK3 did not potentiate death receptor-induced apoptosis, Aza-Blanc et al. (2003) found that GSK3a knockdown, but not GSK3β knockdown, by RNA interference reduced, rather than enhanced, TRAIL-induced apoptosis in HeLa cells. These findings raise the possibility that there may be differences in the effects of each GSK3 isoform in different types of cells, so the balance between GSK3 isoforms may be important in regulating the extrinsic apoptosis signaling pathway.
In summary, it is now well-established that GSK3 has robust anti-apoptotic actions in the extrinsic apoptosis signaling pathway stimulated by all death receptors, so inhibitors of GSK3 strongly potentiate this type of programmed cell death. However, much remains to be learned about this important regulatory interaction, especially the targets of GSK3 that account for its attenuation of the extrinsic apoptotic signaling pathway. The identification of the substrates of GSK3 that account for its inhibition of extrinsic apoptosis may provide targets for clinically directed research to selectively modify death receptor-induced apoptosis.
5. Resolution of the paradoxical actions of GSK3 in apoptotic signaling
Paradoxical findings often can be attributed to an incomplete understanding of a system rather than being manifestations of incongruent properties. This has proven to be the case with studies of GSK3 in which both strong pro-apoptotic and anti-apoptotic effects were found in a wide variety of conditions and cell types. Now it is evident that GSK3 possesses both attributes and that these seemingly paradoxical effects are in fact the result of GSK3 having opposite effects on the two major apoptotic signaling pathways. Thus, GSK3 promotes the intrinsic apoptotic signaling pathway but, conversely, GSK3 inhibits death receptor-induced extrinsic apoptotic signaling.
Promotion of intrinsic apoptotic signaling by GSK3 has been linked to its facilitation of signaling that leads to disruption of mitochondria in conjunction with regulation of the expression of proteins involved in apoptotic signaling. It is important to emphasize that the role of GSK3 in intrinsic apoptotic signaling is not that of an initiator but as a facilitator, promoting the signaling responses to insults that initiate this pathway. Thus, in the intrinsic signaling pathway, GSK3 facilitates, but under physiological conditions does not cause, apoptotic signaling by regulating both the phosphorylation and the expression of apoptotic proteins. The phosphorylation of pro-apoptotic proteins by GSK3 promotes intrinsic mediated apoptosis while its phosphorylation of anti-apoptotic molecules neutralizes their action as blockers of apoptosis. Similarly, in a coordinated fashion, GSK3 promotes increased expression of pro-apoptotic molecules induced by intrinsic stimuli while reducing the expression of anti-apoptotic molecules. Together, these actions underlie the ability of GSK3 to facilitate cell death induced by the intrinsic apoptotic pathway.
In contrast to the intrinsic pathway, in extrinsic apoptotic signaling GSK3 inhibits inception of the cascade by impeding activation of the initiator caspase-8. This upstream anti-apoptotic site of action of GSK3 in extrinsic apoptotic signaling thwarts the pro-apoptotic actions of GSK3 that are targeted downstream at the level of the mitochondria by blocking the signal from reaching that stage of the apoptosis cascade. It is particularly interesting that this avoids any overlapping and conflicting signals in the opposite actions of GSK3 on the two apoptotic pathways, a distinction made possible because of the target sites of GSK3 in each apoptotic signaling pathway. Thus, in the extrinsic pathway the target of GSK3 is upstream in the cascade to inhibit its initiation by blocking activation of caspase-8 thereby impeding the signal before it reaches the mitochondrial level (in type II cells involving caspase-8 signaling through Bid to the mitochondria) where GSK3 has the opposite action of promoting apoptotic signaling. Conversely, caspase-8 generally is not involved in intrinsic apoptotic signaling, so inhibition of caspase-8 activation by GSK3 does not influence its facilitation of the intrinsic apoptotic cascade. Thus there is a clear segregation of the actions of GSK3 on these two apoptotic signaling cascades, which allows GSK3 to effectively regulate each pathway individually and in opposite directions. Taken together, the apparent paradoxical effects of GSK3 on apoptosis now are understood as being compatible because they are due to differences in its effects on these two pathways of apoptotic signaling.
Therefore, physiologically the dual actions of GSK3 on intrinsic and extrinsic apoptotic signaling indicate that GSK3 contributes to cellular sensing mechanisms that set the tolerance threshold for incoming apoptotic signals. This responsibility returns us to the concept that GSK3 has to be a tightly regulated enzyme and that this regulation needs to be substrate-selective. The convergence of multiple intracellular signals impinging on GSK3 regulate it in such a way that the activity of GSK3 contributes to shifts in the balance between survival and apoptosis. For example, in healthy cells many receptor-coupled intracellular signaling pathways dynamically signal to GSK3 by regulating the activities of GSK3-serine-phosphorylating enzymes and phosphatases to maintain GSK3 within a tolerable range of oscillating activity associated with cycles of its phosphorylation and dephosphorylation. In such cells, this convergence of signals on GSK3 maintains a strong anti-apoptosis signal, strengthening the ability of cells to ward off the detrimental effects of potentially lethal conditions that can activate the intrinsic apoptosis signaling pathway. However, if the survival signals converging on GSK3 are weakened, GSK3 becomes more active due to decreased serine-phosphorylation, allowing intrinsic apoptosis to proceed in damaged cells containing weakened defenses. On the other hand, death receptor-induced extrinsic apoptosis often involves the killing of cells that have adequate growth factor signaling. In this case, inhibition of GSK3 is the optimal condition to allow extrinsic apoptosis to proceed, so death receptor stimulation is capable of killing cells even though such cells are protected from intrinsic apoptosis. Thus the apparent paradoxical effects of GSK3 on apoptosis are resolved and now can be understood as functioning to allow GSK3 to be a central integrator and sensor of signals that impact on cellular survival.
Understanding these opposite influences of GSK3 on the two apoptosis signaling cascades may facilitate the rational design of strategies to modulate apoptosis for therapeutic applications. Intrinsic and extrinsic apoptotic signaling are normal mechanisms used to rid organisms of unwanted cells, the former in response to cell damage and the latter in response to external signals. Aberrant apoptosis can occur in association with a wide variety of diseases, especially well-documented for abnormally high apoptosis in neurodegenerative diseases and deficient apoptosis in cancer. Thus, modulating apoptosis by GSK3-regulating drugs may provide a powerful approach to attenuate apoptosis in neurodegenerative diseases or to eliminate proliferative cancer cells. Since GSK3 appears to be an important component of both apoptosis mechanisms, in these conditions GSK3 provides a potential therapeutic target to reduce detrimental apoptosis or to increase beneficial apoptosis, and the rapidly burgeoning number of GSK3 inhibitors provides an arsenal of agents that may contribute to regulating disease-associated apoptosis. For example, the use of lithium to inhibit GSK3, which is already used in humans for the treatment of mood disorders (Jope, 1999), may prove to be beneficial for the protection of neuronal cells from intrinsic apoptotic signaling in neurodegenerative diseases. On the other hand, potentiation of death receptor-induced extrinsic apoptotic signaling is enhanced by lithium and other GSK3 inhibitors, and these may be useful in cancer therapies in conjunction with agents that activate death receptors, for example by employing GSK3 inhibitors in combination with the DR4/DR5 activating ligand TRAIL, since these death receptors are preferentially expressed in cancer cells so this combination may prove to be particularly efficient for killing tumor cells. Thus, this fuller understanding of the differential effects of GSK3 on the two apoptotic signaling pathways provides a basis for designing new therapeutic approaches for a number of diseases by incorporating the judicious use of inhibitors of GSK3.
6. Conclusions
Among the large number of substrates and signaling pathways impacted by GSK3, apoptosis regulation is of particular interest because of the intriguing dual nature of the regulatory effects of GSK3 and the potential therapeutic applications of inhibitors of GSK3. The maintenance of appropriate levels of GSK3 activity is crucial, because either too little or too much GSK3 activity can promote cell death in certain conditions. Increased GSK3 activity lowers the threshold for activating the intrinsic apoptotic pathway, so pharmacological inhibition of GSK3 raises the threshold for signals that activate this mechanism of programmed cell death. This reduction of the intrinsic apoptotic threshold by GSK3 is mediated by the regulatory actions of GSK3 on proteins involved in causing mitochondrial disruption and the expression levels of apoptotic-regulating proteins. However, over-expression of supraphysiological levels of GSK3 is sufficient to modulate these, and perhaps other, targets to a large enough extent to allow apoptosis to proceed in the absence of an additional apoptotic insult. In contrast to these conditions, GSK3 increases the threshold for extrinsic apoptotic signaling by repressing initiation of the death receptor-mediated activation upstream of caspase-8, an early event in this pathway, so GSK3 inhibitors facilitate signaling by the extrinsic apoptotic pathway. Thus, studies of the role of GSK3 as a cellular sensor that integrates stimuli that activate apoptosis have revealed its dual functions to promote intrinsic, and inhibit extrinsic, apoptotic signaling. The understanding of such mechanisms has important implications in the therapeutic applications of GSK3 inhibitors in conditions associated with intrinsic or extrinsic apoptotic signaling.
Fig. 1.
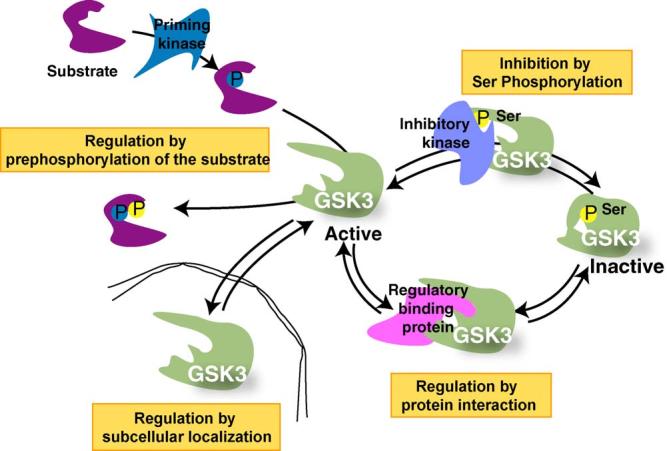
Mechanisms that regulate the actions of GSK3. Four mechanisms act in concert to regulate the phosphorylation of substrates by GSK3. Substrate phosphorylation by GSK3 is limited by the activity of the priming kinase which prepares the substrate for GSK3 because GSK3 most often phosphorylates primed substrates that are prephosphorylated four residues C-terminal to the GSK3 phosphorylation site. A major mechanism for inhibiting the activity of GSK3 is via serine-phosphorylation, so activity is inhibited when serine-9 of GSK3β or serine-21 of GSK3a is phosphorylated. Conversely, the activity of GSK3 is optimal when phosphorylated on tyrosine-216 of GSK3β or tyrosine-279 of GSK3a (not shown). When the substrate is prephosphorylated and GSK3 is active, with the regulatory serine dephosphorylated, two spatial restrictions also contribute to regulating the actions of GSK3, its subcellular localization and its association with other proteins in regulatory complexes. GSK3 is considered to be largely a cytosolic enzyme, but it is also associated with, or internalized in, subcellular compartments such as the nucleus, mitochondria, and growth cones, so dynamic regulation of the subcellular localization of GSK3 can regulate its access to substrates within subcellular compartments. Besides this gross cellular distribution of GSK3, its distribution in the cell is constrained by its propensity to be associated in protein complexes which provides an important mechanism for regulating its phosphorylation of specific substrates that are colocalized in such complexes. Thus, substrate-specific regulation of phosphorylation by GSK3 is achieved by regulation of the priming kinase activity, phosphorylation of GSK3, the subcellular localization of GSK3, and assembly of GSK3 in protein complexes.
Fig. 2.

The intrinsic apoptotic signaling pathway. (A) Depiction of the intrinsic apoptotic signaling pathway. In response to cell damaging insults, pro-apoptotic members of the bcl-2 family (e.g., Bax, Bim) are activated and they translocate to the mitochondria to neutralize anti-apoptotic proteins (e.g., bcl-2, Mcl-1, Bcl-xL). This results in disruption of mitochondria which triggers the release of pro-apoptotic molecules from the mitochondrial intermembrane space. Released cytochrome c clusters with APAF-1 and procaspase-9 in the presence of dATP to form the apoptosome to activate caspase-9. Activated caspase-9 cleaves and activates caspase-3, triggering a caspase cascade which ultimately results in the death of the cell. (B) GSK3 promotes the intrinsic apoptotic signaling pathway be regulating transcription factors that control the expression of pro- and anti-apoptotic proteins, by promoting microtubule disruption and cell structural changes that occur during apoptosis, and by promoting disruption of mitochondria.
Fig. 3.

The extrinsic apoptotic signaling pathway. (A) Depiction of the extrinsic apoptotic signaling pathway. The four major apoptosis-inducing death receptors are activated when Fas ligand (FasL) activates Fas, TRAIL activates DR4 or DR5, or TNF activates TNF-R1 (left inset). The binding of the ligand to the death receptor induces trimerization of the receptor which produces a conformation that recruits FADD and procaspase-8 to the cytoplasmic tail of the receptor, altogether forming a protein complex known as the DISC. Within the DISC, caspase-8 is activated by auto-cleavage. In type I cells, sufficient active caspase-8 is generated to directly activate caspase-3 to carry out the apoptotic program. In type II cells, activation of caspase-8 leads to activation of caspase-3 indirectly through cleavage and activation of Bid, forming tBid, which activates the mitochondrial apoptotic mechanisms that are involved in the intrinsic apoptotic pathway. (B) GSK3 inhibits the extrinsic apoptotic pathway by impairing transduction of the signal from activated death receptors to the activation of caspase-8.
Table 1.
Apoptosis-inducing conditions promoted by GSK3
| Stimulus | References |
|---|---|
| Growth factor withdrawal and PI3K inhibition | Pap and Cooper (1998, 2002), Hetman et al. (2000), Crowder and Freeman (2000), Bhat et al. (2000, 2003), Li et al. (2000), Cross et al. (2001), Somervaille et al. (2001), Culbert et al. (2001), Hongisto et al. (2003), Sanchez et al. (2003), Linseman et al. (2004), Chin et al. (2005), Enguita et al. (2005), Jin et al. (2005b), and Sinha et al. (2005) |
| DNA damage | Watcharasit et al. (2002, 2003), Beurel et al. (2004, 2005), Jin et al. (2005a), and Tan et al. (2006) |
| ER stress | Song et al. (2002), Chen et al. (2004), Kim et al. (2005), Macanas-Pirard et al. (2005), and Srinivasan et al. (2005) |
| Mitochondrial toxins | King et al. (2001) and King and Jope (2005) |
| Hypoxia/ischemia | Loberg et al. (2002), Mottet et al. (2003), Juhaszova et al. (2004), Kelly et al. (2004), Brywe et al. (2005), Cappuccio et al. (2005), Chuang (2005), Kaga et al. (2005); Rho et al. (2005), and Yin et al. (2005) |
| Glutamate excitotoxicity | Hashimoto et al. (2002), Facci et al. (2003), Kelly et al. (2004), and Takadera et al. (2004) Maggirwar et al. (1999) and Tong et al. (2001) |
| Platelet activating factor | Maggirwar et al. (1999) and Tong et al. (2001) |
| Hypertonic stress | Rao et al. (2004) |
| Ceramide | Mora et al. (2002) |
| Ethanol | Takadera and Ohyashiki (2004) |
| Oxidative stress | Shin et al. (2004) and King and Jope (2005) |
| Alzheimer’s disease-related toxicity | Takashima et al. (1993), Alvarez et al. (1999), Wei et al. (2000), Garrido et al. (2002), Bhat et al. (2003), De Ferrari et al. (2003), Hoshi et al. (2003), Phiel et al. (2003), Suhara et al. (2003), Zhang et al. (2003), Alvarez et al. (2004), Su et al. (2004), and Noble et al. (2005) |
| Parkinsons disease-related toxicity | King et al. (2001), Chen et al. (2004), Bai et al. (2004), and Avraham et al. (2005) |
| Prion peptide | Perez et al. (2003a) |
| Amyotrophic lateral sclerosis-related toxicity | Koh et al. (2005) |
| HIV-related toxicity | Maggirwar et al. (1999), Tong et al. (2001), Everall et al. (2002), and Dou et al. (2003, 2005) |
Acknowledgement
Research in the authors’ laboratory was supported by grants from the National Institute of Health.
Footnotes
- APAF-1
- apoptotic protease activating factor-1
- CREB
- cyclic AMP response element binding protein
- DISC
- death-inducing signaling complex
- DR
- death receptor
- ER
- endoplasmic reticulum
- FADD
- Fas-associated death domain protein
- GSK3
- glycogen synthase kinase-3
- HSF-1
- heat shock factor-1
- MEF
- mouse embryonic fibroblast
- NF-κB
- nuclear factor-kB
- PI3K
- phosphatidylinositol 3-kinase
- TNF
- tumor necrosis factor
- TRAIL
- TNF-related apoptosis-inducing ligand
- VDAC
- voltage-dependent anion channel
References
- Adhikary S, Eilers M. Transcriptional regulation and transformation by Myc proteins. Nat. Rev. Mol. Cell. Biol. 2005;6:635–645. doi: 10.1038/nrm1703. [DOI] [PubMed] [Google Scholar]
- Akhtar RS, Ness JM, Roth KA. Bcl-2 family regulation of neuronal development and neurodegeneration. Biochim. Biophys. Acta. 2004;1644:189–203. doi: 10.1016/j.bbamcr.2003.10.013. [DOI] [PubMed] [Google Scholar]
- Alvarez G, Munoz-Montano JR, Satrustegui J, Avila J, Bogonez E, Diaz-Nido J. Lithium protects cultured neurons against β-amyloid-induced neurodegeneration. FEBS Lett. 1999;453:260–264. doi: 10.1016/s0014-5793(99)00685-7. [DOI] [PubMed] [Google Scholar]
- Alvarez AR, Godoy JA, Mullendorff K, Olivares GH, Bronfman M, Inestrosa NC. Wnt-3a overcomes β-amyloid toxicity in rat hippocampal neurons. Exp. Cell Res. 2004;297:186–196. doi: 10.1016/j.yexcr.2004.02.028. [DOI] [PubMed] [Google Scholar]
- Armstrong JS. Mitochondrial membrane permeabilization: the sine qua non for cell death. Bioessays. 2006;28:253–260. doi: 10.1002/bies.20370. [DOI] [PubMed] [Google Scholar]
- Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281:1305–1308. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- Avila J, Lucas JJ, Perez M, Hernandez F. Role of tau protein in both physiological and pathological conditions. Physiol. Rev. 2004;84:361–384. doi: 10.1152/physrev.00024.2003. [DOI] [PubMed] [Google Scholar]
- Avraham E, Szargel R, Eyal A, Rott R, Engelender S. Glycogen synthase kinase 3 β modulates synphilin-1 ubiquitylation and cellular inclusion formation by SIAH: implications for proteasomal function and Lewy body formation. J. Biol. Chem. 2005;280:42877–42886. doi: 10.1074/jbc.M505608200. [DOI] [PubMed] [Google Scholar]
- Aza-Blanc P, Cooper CL, Wagner K, Batalov S, Deveraux QL, Cooke MP. Identification of modulators of TRAIL-induced apoptosis via RNAi-based phenotypic screening. Mol. Cell. 2003;12:627–637. doi: 10.1016/s1097-2765(03)00348-4. [DOI] [PubMed] [Google Scholar]
- Bai L, Nakamura H, Ueda S, Kwon YW, Tanaka T, Ban S, Yodoi J. Proteasome-dependent degradation of cyclin Dl in l-methyl-4-phe-nylpyridinium ion (MPP+)-induced cell cycle arrest. J. Biol. Chem. 2004;279:38710–38714. doi: 10.1074/jbc.M403329200. [DOI] [PubMed] [Google Scholar]
- Beurel E, Kornprobst M, Blivet-Van Eggelpoel MJ, Ruiz-Ruiz C, Cadoret A, Capeau J, Desbois-Mouthon C. GSK3β inhibition by lithium confers resistance to chemotherapy-induced apoptosis through the repression of CD95 (Fas/APO-1) expression. Exp. Cell Res. 2004;300:354–364. doi: 10.1016/j.yexcr.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Beurel E, Kornprobst M, Blivet-Van Eggelpoel MJ, Cadoret A, Capeau J, Desbois-Mouthon C. GSK-3β reactivation with LY294002 sensitizes hepatoma cells to chemotherapy-induced apoptosis. Int. J. Oncol. 2005;27:215–222. [PubMed] [Google Scholar]
- Beyaert R, Vanhaesebroeck B, Suffys P, Van Roy F, Fiers W. Lithium chloride potentiates tumor necrosis factor-mediated cytotoxicity in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 1989;86:9494–9498. doi: 10.1073/pnas.86.23.9494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyaert R, Schulze-Osthoff K, Van Roy F, Fiers W. Synergistic induction of interleukin-6 by tumor necrosis factor and lithium chloride in mice: possible role in the triggering and exacerbation of psoriasis by lithium treatment. Eur. J. Immunol. 1992;22:2181–2184. doi: 10.1002/eji.1830220835. [DOI] [PubMed] [Google Scholar]
- Beyaert R, Heyninck K, De Valck D, Boeykens F, van Roy F, Fiers W. Enhancement of tumor necrosis factor cytotoxicity by lithium chloride is associated with increased inositol phosphate accumulation. J. Immunol. 1993;151:291–300. [PubMed] [Google Scholar]
- Bhat RV, Shanley J, Correll MP, Fieles WE, Keith RA, Scott CW, Lee CM. Regulation and localization of tyrosine216 phosphorylation of glycogen synthase kinase-3β in cellular and animal models of neuronal degeneration. Proc. Natl. Acad. Sci. U.S.A. 2000;97:11074–11079. doi: 10.1073/pnas.190297597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat R, Xue Y, Berg S, Hellberg S, Ormo M, Nilsson Y, Radesater AC, Jerning E, Markgren PO, Borgegard T, Nylof M, Gimenez-Cassina A, Hernandez F, Lucas JJ, Diaz-Nido J, Avila J. Structural insights and biological effects of glycogen synthase kinase 3-specific inhibitor AR-A014418. J. Biol. Chem. 2003;278:45937–45945. doi: 10.1074/jbc.M306268200. [DOI] [PubMed] [Google Scholar]
- Bijur GN, Jope RS. Opposing actions of phosphatidylinositol 3-kinase and glycogen synthase kinase-3β in the regulation of HSF-1 activity. J. Neurochem. 2000;75:2401–2408. doi: 10.1046/j.1471-4159.2000.0752401.x. [DOI] [PubMed] [Google Scholar]
- Bijur GN, De Sarno P, Jope RS. Glycogen synthase kinase-3β facilitates staurosporine- and heat shock-induced apoptosis: protection by lithium. J. Biol. Chem. 2000;275:7583–7590. doi: 10.1074/jbc.275.11.7583. [DOI] [PubMed] [Google Scholar]
- Bijur GN, Jope RS. Proapoptotic stimuli induce nuclear accumulation of glycogen synthase kinase-3β. J. Biol. Chem. 2001;276:37436–37442. doi: 10.1074/jbc.M105725200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bijur GN, Jope RS. Rapid accumulation of Akt in mitochondria following phosphatidylinositol 3-kinase activation. J. Neurochem. 2003a;87:1427–1435. doi: 10.1046/j.1471-4159.2003.02113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bijur GN, Jope RS. Glycogen synthase kinase-3β is highly activated in nuclei and mitochondria. Neuroreport. 2003b;14:2415–2419. doi: 10.1097/00001756-200312190-00025. [DOI] [PubMed] [Google Scholar]
- Bournat JC, Brown AM, Soler AP. Wnt-1 dependent activation of the survival factor NF-κB in PC12 cells. J. Neurosci. Res. 2000;61:21–32. doi: 10.1002/1097-4547(20000701)61:1<21::AID-JNR3>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Brywe KG, Mallard C, Gustavsson M, Hedtjarn M, Leverin AL, Wang X, Blomgren K, Isgaard J, Hagberg H. IGF-I neuroprotection in the immature brain after hypoxia-ischemia, involvement of Akt and GSK3β? Eur. J. Neurosci. 2005;21:1489–1502. doi: 10.1111/j.1460-9568.2005.03982.x. [DOI] [PubMed] [Google Scholar]
- Bullock BP, Habener JF. Phosphorylation of the cAMP response element binding protein CREB by cAMP-dependent protein kinase A and glycogen synthase kinase-3 alters DNA-binding affinity, conformation, and increases net charge. Biochemistry. 1998;37:3795–3809. doi: 10.1021/bi970982t. [DOI] [PubMed] [Google Scholar]
- Buss H, Dorrie A, Schmitz ML, Frank R, Livingstone M, Resch K, Kracht M. Phosphorylation of serine 468 by GSK3β negatively regulates basal p65 NF-κB activity. J. Biol. Chem. 2004;279:49571–49574. doi: 10.1074/jbc.C400442200. [DOI] [PubMed] [Google Scholar]
- Cappuccio I, Calderone A, Busceti CL, Biagioni F, Pontarelli F, Bruno V, Storto M, Terstappen GT, Gaviraghi G, Fornai F, Battaglia G, Melchiorri D, Zukin S, Nicoletti F, Caricasole A. Induction of Dickkopf-1, a negative modulator of the Wnt pathway, is required for the development of ischemic neuronal death. J. Neurosci. 2005;25:2647–2657. doi: 10.1523/JNEUROSCI.5230-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmichael J, Sugars KL, Bao YP, Rubinsztein DC. Glycogen synthase kinase-3β inhibitors prevent cellular polyglutamine toxicity caused by the Huntington’s disease mutation. J. Biol. Chem. 2002;277:33791–33798. doi: 10.1074/jbc.M204861200. [DOI] [PubMed] [Google Scholar]
- Chen RW, Chuang DM. Long term lithium treatment suppresses p53 and Bax expression but increases Bcl-2 expression. A prominent role in neuroprotection against excitotoxicity. J. Biol. Chem. 1999;274:6039–6042. doi: 10.1074/jbc.274.10.6039. [DOI] [PubMed] [Google Scholar]
- Chen G, Zeng WZ, Yuan PX, Huang LD, Jiang YM, Zhao ZH, Manji HK. The mood-stabilizing agents lithium and valproate robustly increase the levels of the neuroprotective protein bcl-2 in the CNS. J. Neurochem. 1999;72:879–882. doi: 10.1046/j.1471-4159.1999.720879.x. [DOI] [PubMed] [Google Scholar]
- Chen G, Bower KA, Ma C, Fang S, Thiele CJ, Luo J. Glycogen synthase kinase 3β (GSK3β) mediates 6-hydroxydopamine-induced neuronal death. FASEB J. 2004;18:1162–1164. doi: 10.1096/fj.04-1551fje. [DOI] [PubMed] [Google Scholar]
- Chen S, Guttridge DC, You Z, Zhang Z, Fribley A, Mayo MW, Kitajewski J, Wang CY. Wnt-1 signaling inhibits apoptosis by activating β-catenin/T cell factor-mediated transcription. J. Cell Biol. 2001;8:87–96. doi: 10.1083/jcb.152.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng EH, Sheiko TV, Fisher JK, Craigen WL, Korsmeyer SJ. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science. 2003;301:513–517. doi: 10.1126/science.1083995. [DOI] [PubMed] [Google Scholar]
- Chin PC, Majdzadeh N, D’Mello SR. Inhibition of GSK3β is a common event in neuroprotection by different survival factors. Mol. Brain Res. 2005;137:193–201. doi: 10.1016/j.molbrainres.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Chu B, Soncin F, Price BD, Stevenson MA, Calderwood SK. Sequential phosphorylation by mitogen-activated protein kinase and glycogen synthase kinase 3 represses transcriptional activation by heat shock factor-1. J. Biol. Chem. 1996;271:30847–30857. doi: 10.1074/jbc.271.48.30847. [DOI] [PubMed] [Google Scholar]
- Chuang DM. The antiapoptotic actions of mood stabilizers: molecular mechanisms and therapeutic potentials. Ann. N.Y. Acad. Sci. 2005;1053:195–204. doi: 10.1196/annals.1344.018. [DOI] [PubMed] [Google Scholar]
- Ciani L, Salinas PC. WNTs in the vertebrate nervous system: from patterning to neuronal connectivity. Nat. Rev. Neurosci. 2005;6:351–362. doi: 10.1038/nrn1665. [DOI] [PubMed] [Google Scholar]
- Cross DA, Culbert AA, Chalmers KA, Facci L, Skaper SD, Reith AD. Selective small-molecule inhibitors of glycogen synthase kinase-3 activity protect primary neurones from death. J. Neurochem. 2001;77:94–102. doi: 10.1046/j.1471-4159.2001.t01-1-00251.x. [DOI] [PubMed] [Google Scholar]
- Crowder RJ, Freeman RS. Glycogen synthase kinase-3β activity is critical for neuronal death caused by inhibiting PI 3-kinase or Akt but not for death caused by NGF withdrawal. J. Biol. Chem. 2000;275:34266–34271. doi: 10.1074/jbc.M006160200. [DOI] [PubMed] [Google Scholar]
- Culbert AA, Brown MJ, Frame S, Hagen T, Cross DA, Bax B, Reith AD. GSK3 inhibition by adenoviral FRAT1 overexpression is neuroprotective and induces Tau dephosphorylation and β-catenin stabilisation without elevation of glycogen synthase activity. FEBS Lett. 2001;507:288–294. doi: 10.1016/s0014-5793(01)02990-8. [DOI] [PubMed] [Google Scholar]
- De Ferrari GV, Chacon MA, Barria MI, Garrido JL, Godoy JA, Olivares G, Reyes AE, Alvarez A, Bronfman M, Inestrosa NC. Activation of Wnt signaling rescues neurodegeneration and behavioral impairments induced by β-amyloid fibrils. Mol. Psychiatry. 2003;8:195–208. doi: 10.1038/sj.mp.4001208. [DOI] [PubMed] [Google Scholar]
- De Ketelaere A, Vermeulen L, Vialard J, Van De Weyer I, Van Wauwe J, Haegeman G, Moelans I. Involvement of GSK3β in TWEAK-mediated NF-κB activation. FEBS Lett. 2004;566:60–64. doi: 10.1016/j.febslet.2004.04.041. [DOI] [PubMed] [Google Scholar]
- Demarchi F, Bertoli C, Sandy P, Schneider C. Glycogen synthase kinase-3β regulates NF-κB1/p105 stability. J. Biol. Chem. 2003;278:39583–39590. doi: 10.1074/jbc.M305676200. [DOI] [PubMed] [Google Scholar]
- Deng J, Miller SA, Wang HY, Xia W, Wen Y, Zhou BP, Li Y, Lin SY, Hung MC. β-Catenin interacts with and inhibits NF-κB in human colon and breast cancer. Cancer Cell. 2002;2:323–334. doi: 10.1016/s1535-6108(02)00154-x. [DOI] [PubMed] [Google Scholar]
- Deng J, Xia W, Miller SA, Wen Y, Wang HY, Hung MC. Crossregulation of NF-κB by the APC/GSK3β/β-catenin pathway. Mol. Carcinog. 2004;39:139–146. doi: 10.1002/mc.10169. [DOI] [PubMed] [Google Scholar]
- Dou H, Birusingh K, Faraci J, Gorantla S, Poluektova LY, Maggirwar SB, Dewhurst S, Gelbard HA, Gendelman HE. Neuroprotective activities of sodium valproate in a murine model of human immunodeficiency virus-1 encephalitis. J. Neurosci. 2003;23:9162–9170. doi: 10.1523/JNEUROSCI.23-27-09162.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou H, Ellison B, Bradley J, Kasiyanov A, Poluektova LY, Xiong H, Maggirwar S, Dewhurst S, Gelbard HA, Gendelman HE. Neuroprotective mechanisms of lithium in murine human immunodeficiency virus-1 encephalitis. J. Neurosci. 2005;25:8375–8385. doi: 10.1523/JNEUROSCI.2164-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earnshaw WC, Martins LM, Kaufmann SH. Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu. Rev. Biochem. 1999;68:383–424. doi: 10.1146/annurev.biochem.68.1.383. [DOI] [PubMed] [Google Scholar]
- Eldar-Finkelman H. Glycogen synthase kinase 3: an emerging therapeutic target. Trends Mol. Med. 2002;8:126–132. doi: 10.1016/s1471-4914(01)02266-3. [DOI] [PubMed] [Google Scholar]
- Eldar-Finkelman H, Ilouz R. Challenges and opportunities with glycogen synthase kinase-3 inhibitors for insulin resistance and Type 2 diabetes treatment. Expert Opin. Investig. Drugs. 2003;12:1511–1519. doi: 10.1517/13543784.12.9.1511. [DOI] [PubMed] [Google Scholar]
- Elyaman W, Terro F, Wong NS, Hugon J. In vivo activation and nuclear translocation of phosphorylated glycogen synthase kinase-3β in neuronal apoptosis: links to tau phosphorylation. Eur. J. Neurosci. 2002;15:651–660. doi: 10.1046/j.1460-9568.2002.01899.x. [DOI] [PubMed] [Google Scholar]
- Embi N, Rylatt DB, Cohen P. Glycogen synthase kinase-3 from rabbit skeletal muscle. Separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur. J. Biochem. 1980;107:519–527. [PubMed] [Google Scholar]
- Enguita M, DeGregorio-Rocasolano N, Abad A, Trullas R. Glycogen synthase kinase 3 activity mediates neuronal pentraxin 1 expression and cell death induced by potassium deprivation in cerebellar granule cells. Mol. Pharmacol. 2005;67:1237–1246. doi: 10.1124/mol.104.007062. [DOI] [PubMed] [Google Scholar]
- Eto M, Kouroedov A, Cosentino F, Luscher TF. Glycogen synthase kinase-3 mediates endothelial cell activation by tumor necrosis factor-α. Circulation. 2005;112:1316–1322. doi: 10.1161/CIRCULATIONAHA.105.564112. [DOI] [PubMed] [Google Scholar]
- Everall IP, Bell C, Mallory M, Langford D, Adame A, Rockestein E, Masliah E. Lithium ameliorates HIV-gpl20-mediated neurotoxicity. Mol. Cell. Neurosci. 2002;493:501. doi: 10.1006/mcne.2002.1196. [DOI] [PubMed] [Google Scholar]
- Facci L, Stevens DA, Skaper SD. Glycogen synthase kinase-3 inhibitors protect central neurons against excitotoxicity. Neuroreport. 2003;14:1467–1470. doi: 10.1097/00001756-200308060-00012. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Gomez-Isla T, Puig B, Freixes M, Ribe E, Dalfo E, Avila J. Current advances on different kinases involved in tau phosphorylation, and implications in Alzheimer’s disease and tauopathies. Curr. Alzheimer Res. 2005;2:3–18. doi: 10.2174/1567205052772713. [DOI] [PubMed] [Google Scholar]
- Frame S, Cohen P, Biondi RM. A common phosphate binding site explains the unique substrate specificity of GSK3 and its inactivation by phosphorylation. Mol. Cell. 2001;7:1321–1327. doi: 10.1016/s1097-2765(01)00253-2. [DOI] [PubMed] [Google Scholar]
- Fuentealba RA, Farias G, Scheu J, Bronfman M, Marzolo MP, Inestrosa NC. Signal transduction during amyloid-β-peptide neurotoxicity: role in Alzheimer disease. Brain Res. Rev. 2004;47:275–289. doi: 10.1016/j.brainresrev.2004.07.018. [DOI] [PubMed] [Google Scholar]
- Garrido JL, Godoy JA, Alvarez A, Bronfman M, Inestrosa NC. Protein kinase C inhibits amyloid b peptide neurotoxicity by acting on members of the Wnt pathway. FASEB J. 2002;16:1982–1984. doi: 10.1096/fj.02-0327fje. [DOI] [PubMed] [Google Scholar]
- Ghosh JC, Altieri DC. Activation of p53-dependent apoptosis by acute ablation of glycogen synthase kinase-3β in colorectal cancer cells. Clin. Cancer Res. 2005;11:4580–4588. doi: 10.1158/1078-0432.CCR-04-2624. [DOI] [PubMed] [Google Scholar]
- Grimes CA, Jope RS. The multi-faceted roles of glycogen synthase kinase-3β in cellular signaling. Prog. Neurobiol. 2001;65:391–426. doi: 10.1016/s0301-0082(01)00011-9. [DOI] [PubMed] [Google Scholar]
- Guha M, Mackman N. The phosphatidylinositol 3-kinase-Akt pathway limits lipopolysaccharide activation of signaling pathways and expression of inflammatory mediators in human monocytic cells. J. Biol. Chem. 2002;277:32124–32132. doi: 10.1074/jbc.M203298200. [DOI] [PubMed] [Google Scholar]
- Hall JL, Chatham JC, Eldar-Finkelman H, Gibbons GH. Upregulation of glucose metabolism during intimal lesion formation is coupled to the inhibition of vascular smooth muscle cell apoptosis. Role of GSK3β. Diabetes. 2001;50:1171–1179. doi: 10.2337/diabetes.50.5.1171. [DOI] [PubMed] [Google Scholar]
- Hashimoto M, Sagara Y, Langford D, Everall IP, Mallory M, Everson A, Digicaylioglu M, Masliah E. Fibroblast growth factor 1 regulates signaling via the glycogen synthase kinase-3β pathway. Implications for neuroprotection. J. Biol. Chem. 2002;277:32985–32991. doi: 10.1074/jbc.M202803200. [DOI] [PubMed] [Google Scholar]
- Haupt Y, Robles AI, Prives C, Rotter V. Deconstruction of p53 functions and regulation. Oncogene. 2002;21:8223–8231. doi: 10.1038/sj.onc.1206137. [DOI] [PubMed] [Google Scholar]
- He B, Meng YH, Mivechi NF. Glycogen synthase kinase 3β and extracellular signal-regulated kinase inactivate heat shock transcription factor 1 by facilitating the disappearance of transcriptionally active granules after heat shock. Mol. Cell. Biol. 1998a;18:6624–6633. doi: 10.1128/mcb.18.11.6624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Identification of c-MYC as a target of the APC pathway. Science. 1998b;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- Hetman M, Cavanaugh JE, Kimelman D, Xia Z. Role of glycogen synthase kinase-3β in neuronal apoptosis induced by trophic withdrawal. J. Neurosci. 2000;20:2567–2674. doi: 10.1523/JNEUROSCI.20-07-02567.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetman M, Hsuan SL, Habas A, Higgins MJ, Xia Z. ERK1/2 antagonizes glycogen synthase kinase-3β-induced apoptosis in cortical neurons. J. Biol. Chem. 2002;277:49577–49584. doi: 10.1074/jbc.M111227200. [DOI] [PubMed] [Google Scholar]
- Hoeflich KP, Luo J, Rubie EA, Tsao M-S, Jin O, Woodgett JR. Requirement for glycogen synthase kinase-3β in cell survival and NF-κB activation. Nature. 2000;406:86–90. doi: 10.1038/35017574. [DOI] [PubMed] [Google Scholar]
- Hong M, Chen DC, Klein PS, Lee VM. Lithium reduces tau phosphorylation by inhibition of glycogen synthase kinase-3. J. Biol. Chem. 1997;272:25326–25332. doi: 10.1074/jbc.272.40.25326. [DOI] [PubMed] [Google Scholar]
- Hongisto V, Smeds N, Brecht S, Herdegen T, Courtney MJ, Coffey ET. Lithium blocks the c-Jun stress response and protects neurons via its action on glycogen synthase kinase 3. Mol. Cell. Biol. 2003;23:6027–6036. doi: 10.1128/MCB.23.17.6027-6036.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi M, Sato M, Matsumoto S, Noguchi A, Yasutake K, Yoshida N, Sato K. Spherical aggregates of beta-amyloid (amylospheroid) show high neurotoxicity and activate tau protein kinase I/glycogen synthase kinase-3β. Proc. Natl. Acad. Sci. U.S.A. 2003;100:6370–6375. doi: 10.1073/pnas.1237107100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin ZH, Kurosu T, Yamaguchi M, Arai A, Miura O. Hematopoietic cytokines enhance Chk1-dependent G2/M checkpoint activation by etoposide through the Akt/GSK3 pathway to inhibit apoptosis. Oncogene. 2005a;24:1973–1981. doi: 10.1038/sj.onc.1208408. [DOI] [PubMed] [Google Scholar]
- Jin N, Kovacs AD, Sui Z, Dewhurst S, Maggirwar SB. Opposite effects of lithium and valproic acid on trophic factor deprivation-induced glycogen synthase kinase-3 activation, c-Jun expression and neuronal cell death. Neuropharmacology. 2005b;48:576–583. doi: 10.1016/j.neuropharm.2004.11.010. [DOI] [PubMed] [Google Scholar]
- Johnson GVW, Bailey CD. Tau, where are we now? J. Alzheimers Dis. 2002;4:375–398. doi: 10.3233/jad-2002-4505. [DOI] [PubMed] [Google Scholar]
- Jope RS. Anti-bipolar therapy: mechanism of action of lithium. Mol. Psychiatry. 1999;4:117–128. doi: 10.1038/sj.mp.4000494. [DOI] [PubMed] [Google Scholar]
- Jope RS. Lithium and GSK3: one inhibitor, two inhibitory actions, multiple outcomes. Trends Pharmacol. Sci. 2003;24:441–443. doi: 10.1016/S0165-6147(03)00206-2. [DOI] [PubMed] [Google Scholar]
- Jope RS, Johnson GVW. The glamour and gloom of glycogen synthase kinase-3 (GSK3) Trends Biochem. Sci. 2004;29:95–102. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Jope RS. Inhibition of glycogen synthase kinase-3: a potential therapeutic target of lithium. Clin. Neurosci. Res. 2004;4:171–179. [Google Scholar]
- Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW, Ziman BD, Wang S, Ytrehus K, Antos CL, Olson EN, Sollott SJ. Glycogen synthase kinase-3β mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J. Clin. Invest. 2004;113:1535–1549. doi: 10.1172/JCI19906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaga S, Zhan L, Altaf E, Maulik N. Glycogen synthase-3β/β-catenin promotes angiogenic and anti-apoptotic signaling through the induction of VEGF, Bcl-2 and survivin expression in rat ischemic pre-conditioned myocardium. J. Mol. Cell. Cardiol. 2005;40:138–147. doi: 10.1016/j.yjmcc.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-κB activity. Annu. Rev. Immunol. 2000;18:621–663. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- Kaufmann SH, Hengartner MO. Programmed cell death: alive and well in the new millennium. Trends Cell Biol. 2001;11:526–534. doi: 10.1016/s0962-8924(01)02173-0. [DOI] [PubMed] [Google Scholar]
- Kelly S, Zhao H, Hua Sun G, Cheng D, Qiao Y, Luo J, Martin K, Steinberg GK, Harrison SD, Yenari MA. Glycogen synthase kinase 3β inhibitor Chir025 reduces neuronal death resulting from oxygenglucose deprivation, glutamate excitotoxicity, and cerebral ischemia. Exp. Neurol. 2004;188:378–386. doi: 10.1016/j.expneurol.2004.04.004. [DOI] [PubMed] [Google Scholar]
- Khaleque MA, Bharti A, Sawyer D, Gong J, Benjamin IJ, Stevenson MA, Calderwood SK. Induction of heat shock proteins by heregulin β1 leads to protection from apoptosis and anchorage-independent growth. Oncogene. 2005;24:6564–6573. doi: 10.1038/sj.onc.1208798. [DOI] [PubMed] [Google Scholar]
- Kim HS, Skurk C, Thomas SR, Bialik A, Suhara T, Kureishi Y, Birnbaum M, Keaney JF, Jr., Walsh K. Regulation of angiogenesis by glycogen synthase kinase-3β. J. Biol. Chem. 2002;277:41888–41896. doi: 10.1074/jbc.M206657200. [DOI] [PubMed] [Google Scholar]
- Kim AJ, Shi Y, Austin RC, Werstuck GH. Valproate protects cells from ER stress-induced lipid accumulation and apoptosis by inhibiting glycogen synthase kinase-3. J. Cell Sci. 2005;118:89–99. doi: 10.1242/jcs.01562. [DOI] [PubMed] [Google Scholar]
- King TD, Bijur GN, Jope RS. Caspase-3 activation induced by inhibition of mitochondrial complex I is facilitated by glycogen synthase kinase-3β and attenuated by lithium. Brain Res. 2001;919:106–114. doi: 10.1016/s0006-8993(01)03005-0. [DOI] [PubMed] [Google Scholar]
- King TD, Jope RS. Inhibition of GSK3 protects cells from intrinsic but not extrinsic oxidative stress. Neuroreport. 2005;16:597–601. doi: 10.1097/00001756-200504250-00016. [DOI] [PubMed] [Google Scholar]
- Klein PS, Melton DA. A molecular mechanism for the effect of lithium on development. Proc. Natl. Acad. Sci. U.S.A. 1996;93:8455–8459. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh SH, Lee YB, Kim KS, Kim HJ, Kim M, Lee YJ, Kim J, Lee KW, Kim SH. Role of GSK-3β activity in motor neuronal cell death induced by G93A or A4V mutant hSOD1 gene. Eur. J. Neurosci. 2005;22:301–309. doi: 10.1111/j.1460-9568.2005.04191.x. [DOI] [PubMed] [Google Scholar]
- Krammer PH. CD95’s deadly mission in the immune system. Nature. 2000;407:789–795. doi: 10.1038/35037728. [DOI] [PubMed] [Google Scholar]
- Kulikov R, Boehme KA, Blattner C. Glycogen synthase kinase 3-dependent phosphorylation of Mdm2 regulates p53 abundance. Mol. Cell. Biol. 2005;25:7170–7180. doi: 10.1128/MCB.25.16.7170-7180.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- Li M, Wang X, Meintzer MK, Laessig T, Birnbaum MJ, Heidenreich KA. Cyclic AMP promotes neuronal survival by phosphorylation of glycogen synthase kinase 3β. Mol. Cell. Biol. 2000;20:9356–9363. doi: 10.1128/mcb.20.24.9356-9363.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao X, Zhang L, Thrasher JB, Du J, Li B. Glycogen synthase kinase-3β suppression eliminates tumor necrosis factor-related apoptosis-inducing ligand resistance in prostate cancer. Mol. Cancer Ther. 2003;2:1215–1222. [PubMed] [Google Scholar]
- Linseman DA, Butts BD, Precht TA, Phelps RA, Le SS, Laessig TA, Bouchard RJ, Florez-McClure ML, Heidenreich KA. Glycogen synthase kinase-3β phosphorylates Bax and promotes its mitochondrial localization during neuronal apoptosis. J. Neurosci. 2004;24:9993–10002. doi: 10.1523/JNEUROSCI.2057-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loberg RD, Vesely E, Brosius FC., III Enhanced glycogen synthase kinase-3β activity mediates hypoxia-induced apoptosis of vascular smooth muscle cells and is prevented by glucose transport and metabolism. J. Biol. Chem. 2002;277:41667–41673. doi: 10.1074/jbc.M206405200. [DOI] [PubMed] [Google Scholar]
- Longo KA, Kennell JA, Ochocinska MJ, Ross SE, Wright WS, MacDougald OA. Wnt signaling protects 3T3-L1 preadipocytes from apoptosis through induction of insulin-like growth factors. J. Biol. Chem. 2002;277:38239–38244. doi: 10.1074/jbc.M206402200. [DOI] [PubMed] [Google Scholar]
- Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35:605–623. doi: 10.1016/s0896-6273(02)00828-0. [DOI] [PubMed] [Google Scholar]
- Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–490. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- Macanas-Pirard P, Yaacob NS, Lee PC, Holder JC, Hinton RH, Kass GE. Glycogen synthase kinase-3 mediates acetaminophen-induced apoptosis in human hepatoma cells. J. Pharmacol. Exp. Ther. 2005;313:780–789. doi: 10.1124/jpet.104.081364. [DOI] [PubMed] [Google Scholar]
- Maggirwar SB, Tong N, Ramirez S, Gelbard HA, Dewhurst S. HIV-1 Tat-mediated activation of glycogen synthase kinase-3β contributes to Tat-mediated neurotoxicity. J. Neurochem. 1999;73:578–586. doi: 10.1046/j.1471-4159.1999.0730578.x. [DOI] [PubMed] [Google Scholar]
- Majewski N, Nogueira V, Robey RB, Hay N. Akt inhibits apoptosis downstream of BID cleavage via a glucose-dependent mechanism involving mitochondrial hexokinases. Mol. Cell. Biol. 2004;24:730–740. doi: 10.1128/MCB.24.2.730-740.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manoukian AS, Woodgett JR. Role of glycogen synthase kinase-3 in cancer: regulation by Wnts and other signaling pathways. Adv. Cancer Res. 2002;84:203–229. doi: 10.1016/s0065-230x(02)84007-6. [DOI] [PubMed] [Google Scholar]
- Marchenko ND, Zaika A, Moll UM. Death signal-induced localization of p53 protein to mitochondria. A potential role in apoptotic signaling. J. Biol. Chem. 2000;275:16202–16212. doi: 10.1074/jbc.275.21.16202. [DOI] [PubMed] [Google Scholar]
- Martin M, Rehani K, Jope RS, Michalek SM. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat. Immunol. 2005;6:777–784. doi: 10.1038/ni1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez A, Castro A, Dorronsoro I, Alonso M. Glycogen synthase kinase 3 (GSK-3) inhibitors as new promising drugs for diabetes, neuro-degeneration, cancer, and inflammation. Med. Res. Rev. 2002;22:373–384. doi: 10.1002/med.10011. [DOI] [PubMed] [Google Scholar]
- Martinou JC, Green DR. Breaking the mitochondrial barrier. Nat. Rev. Mol. Cell. Biol. 2001;2:63–67. doi: 10.1038/35048069. [DOI] [PubMed] [Google Scholar]
- Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR. Glycogen synthase kinase-3 regulates mitochondrial outer membrane per-meabilization and apoptosis by destabilization of MCL-1. Mol. Cell. 2006;21:749–760. doi: 10.1016/j.molcel.2006.02.009. [DOI] [PubMed] [Google Scholar]
- McManus EJ, Sakamoto K, Armit LJ, Ronaldson L, Shpiro N, Marquez R, Alessi DR. Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J. 2005;24:1571–1583. doi: 10.1038/sj.emboj.7600633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, Moll UM. p53 has a direct apoptogenic role at the mitochondria. Mol. Cell. 2003;11:577–590. doi: 10.1016/s1097-2765(03)00050-9. [DOI] [PubMed] [Google Scholar]
- Mora A, Sabio G, Risco AM, Cuenda A, Alonso JC, Soler G, Centeno F. Lithium blocks the PKB and GSK3 dephosphorylation induced by ceramide through protein phosphatase-2A. Cell. Signal. 2002;14:557–562. doi: 10.1016/s0898-6568(01)00282-0. [DOI] [PubMed] [Google Scholar]
- Morfini G, Szebenyi G, Elluru R, Ratner N, Brady ST. Glycogen synthase kinase 3 phosphorylates kinesin light chains and negatively regulates kinesin-based motility. EMBO J. 2002;21:281–293. doi: 10.1093/emboj/21.3.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mottet D, Dumont V, Deccache Y, Demazy C, Ninane N, Raes M, Michiels C. Regulation of hypoxia-inducible factor-la protein level during hypoxic conditions by the phosphatidylinositol 3-kinase/Akt/glycogen synthase kinase 3β pathway in HepG2 cells. J. Biol. Chem. 2003;278:31277–31285. doi: 10.1074/jbc.M300763200. [DOI] [PubMed] [Google Scholar]
- Mudher A, Shepherd D, Newman TA, Mildren P, Jukes JP, Squire A, Mears A, Drummond JA, Berg S, MacKay D, Asuni AA, Bhat R, Lovestone S. GSK-3β inhibition reverses axonal transport defects and behavioural phenotypes in Drosophila. Mol. Psychiatry. 2004;9:522–530. doi: 10.1038/sj.mp.4001483. [DOI] [PubMed] [Google Scholar]
- Nemeth ZH, Deitch EA, Szabo C, Fekete Z, Hauser CJ, Hasko G. Lithium induces NF-κB activation and interleukin-8 production in human intestinal epithelial cells. J. Biol. Chem. 2002;277:7713–7719. doi: 10.1074/jbc.M109711200. [DOI] [PubMed] [Google Scholar]
- Noble W, Planel E, Zehr C, Olm V, Meyerson J, Suleman F, Gaynor K, Wang L, LaFrancois J, Feinstein B, Burns M, Krishnamurthy P, Wen Y, Bhat R, Lewis J, Dickson D, Duff K. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc. Natl. Acad. Sci. U.S.A. 2005;102:6990–6995. doi: 10.1073/pnas.0500466102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka S, Chuang DM. Neuroprotective effects of chronic lithium on focal cerebral ischemia in rats. Neuroreport. 1998;9:2081–2084. doi: 10.1097/00001756-199806220-00031. [DOI] [PubMed] [Google Scholar]
- Oren M. Decision making by p53: life, death and cancer. Cell Death Differ. 2003;10:431–442. doi: 10.1038/sj.cdd.4401183. [DOI] [PubMed] [Google Scholar]
- Pap M, Cooper GM. Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-kinase/Akt cell survival pathway. J. Biol. Chem. 1998;273:19929–19932. doi: 10.1074/jbc.273.32.19929. [DOI] [PubMed] [Google Scholar]
- Pap M, Cooper GM. Role of translation initiation factor 2B in control of cell survival by the phosphatidylinositol 3-kinase/Akt/glycogen synthase kinase 3β signaling pathway. Mol. Cell. Biol. 2002;22:578–586. doi: 10.1128/MCB.22.2.578-586.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastorino JG, Shulga N, Hoek JB. Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J. Biol. Chem. 2002;277:7610–7618. doi: 10.1074/jbc.M109950200. [DOI] [PubMed] [Google Scholar]
- Pastorino JG, Hoek JB, Shulga N. Activation of glycogen synthase kinase 3β disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res. 2005;65:10545–10554. doi: 10.1158/0008-5472.CAN-05-1925. [DOI] [PubMed] [Google Scholar]
- Patel S, Doble B, Woodgett JR. Glycogen synthase kinase-3 in insulin and Wnt signalling: a double-edged sword? Biochem. Soc. Trans. 2004;32:803–808. doi: 10.1042/BST0320803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez M, Rojo AI, Wandosell F, Diaz-Nido J, Avila J. Prion peptide induces neuronal cell death through a pathway involving glycogen synthase kinase 3. Biochem. J. 2003a;372:129–136. doi: 10.1042/BJ20021596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez M, Hernandez F, Lim F, Diaz-Nido J, Avila J. Chronic lithium treatment decreases mutant tau protein aggregation in a transgenic mouse model. J. Alzheimers Dis. 2003β;5:301–308. doi: 10.3233/jad-2003-5405. [DOI] [PubMed] [Google Scholar]
- Peter ME, Krammer PH. The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ. 2003;10:26–35. doi: 10.1038/sj.cdd.4401186. [DOI] [PubMed] [Google Scholar]
- Phiel CJ, Wilson CA, Lee VM, Klein PS. GSK3a regulates production of Alzheimer’s disease amyloid-β peptides. Nature. 2003;423:435–439. doi: 10.1038/nature01640. [DOI] [PubMed] [Google Scholar]
- Pluquet O, Qu LK, Baltzis D, Koromilas AE. Endoplasmic reticulum stress accelerates p53 degradation by the cooperative actions of Hdm2 and glycogen synthase kinase 3β. Mol. Cell. Biol. 2005;25:9392–9405. doi: 10.1128/MCB.25.21.9392-9405.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polakis P. The oncogenic activation of β-catenin. Curr. Opin. Genet. Dev. 1999;9:15–21. doi: 10.1016/s0959-437x(99)80003-3. [DOI] [PubMed] [Google Scholar]
- Qu L, Huang S, Baltzis D, Rivas-Estilla AM, Pluquet O, Hatzoglou M, Koumenis C, Taya Y, Yoshimura A, Koromilas AE. Endoplasmic reticulum stress induces p53 cytoplasmic localization and prevents p53-dependent apoptosis by a pathway involving glycogen synthase kinase-3β. Genes Dev. 2004;18:261–277. doi: 10.1101/gad.1165804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao R, Hao CM, Breyer MD. Hypertonic stress activates glycogen synthase kinase 3β-mediated apoptosis of renal medullary interstitial cells, suppressing an NFkB-driven cyclooxygenase-2-dependent survival path-way. J. Biol. Chem. 2004;279:3949–3955. doi: 10.1074/jbc.M309325200. [DOI] [PubMed] [Google Scholar]
- Ren M, Senatorov VV, Chen RW, Chuang DM. Postinsult treatment with lithium reduces brain damage and facilitates neurological recovery in a rat ischemia/reperfusion model. Proc. Natl. Acad. Sci. U.S.A. 2003;100:6210–6215. doi: 10.1073/pnas.0937423100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rho MS, Eom TY, Zmijewska AA, De Sarno P, Roth KA, Jope RS. Hypoxia activates glycogen synthase kinase-3 in mouse brain in vivo: protection by mood stabilizers and imipramine. Biol. Psychiatry. 2005;57:278–286. doi: 10.1016/j.biopsych.2004.10.039. [DOI] [PubMed] [Google Scholar]
- Riedl SJ, Shi Y. Molecular mechanisms of caspase regulation during apoptosis. Nat. Rev. Mol. Cell. Biol. 2004;5:897–907. doi: 10.1038/nrm1496. [DOI] [PubMed] [Google Scholar]
- Rottmann S, Wang Y, Nasoff M, Deveraux QL, Quon KC. A TRAIL receptor-dependent synthetic lethal relationship between MYC activation and GSK3β/FBW7 loss of function. Proc. Natl. Acad. Sci. U.S.A. 2005;102:15195–15200. doi: 10.1073/pnas.0505114102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saksela K, Makela TP, Hughes K, Woodgett JR, Alitalo K. Activation of protein kinase C increases phosphorylation of the L-Myc trans-activator domain at a GSK-3 target site. Oncogene. 1992;7:347–353. [PubMed] [Google Scholar]
- Sanchez JF, Sniderhan LF, Williamson AL, Fan S, Chakraborty-Sett S, Maggirwar SB. Glycogen synthase kinase 3β-mediated apoptosis of primary cortical astrocytes involves inhibition of nuclear factor κB signaling. Mol. Cell. Biol. 2003;23:4649–4662. doi: 10.1128/MCB.23.13.4649-4662.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansome C, Zaika A, Marchenko ND, Moll UM. Hypoxia death stimulus induces translocation of p53 protein to mitochondria. Detection by immunofluorescence on whole cells. FEBS Lett. 2001;488:110–115. doi: 10.1016/s0014-5793(00)02368-1. [DOI] [PubMed] [Google Scholar]
- Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi C, Fulda S, Srinivasan A, Friesen C, Li F, Tomaselli KJ, Debatin KM, Krammer PH, Peter ME. Two CD95 (APO-I/Fas) signaling pathways. EMBO J. 1998;17:1675–1687. doi: 10.1093/emboj/17.6.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schotte P, Van Loo G, Carpentier I, Vandenabeele P, Beyaert R. Lithium sensitizes tumor cells in an NF-κB-independent way to caspase activation and apoptosis induced by tumor necrosis factor (TNF). Evidence for a role of the TNF receptor-associated death domain protein. J. Biol. Chem. 2001;276:25939–25945. doi: 10.1074/jbc.M104014200. [DOI] [PubMed] [Google Scholar]
- Schwabe RF, Brenner DA. Role of glycogen synthase kinase-3 in TNF-a-induced NF-κB activation and apoptosis in hepatocytes. Am. J. Physiol. Gastrointest. Liver Physiol. 2002;283:G204–G211. doi: 10.1152/ajpgi.00016.2002. [DOI] [PubMed] [Google Scholar]
- Shin SY, Kim CG, Jho EH, Rho MS, Kim YS, Kim YH, Lee YH. Hydrogen peroxide negatively modulates Wnt signaling through downregulation of β-catenin. Cancer Lett. 2004;212:225–231. doi: 10.1016/j.canlet.2004.03.003. [DOI] [PubMed] [Google Scholar]
- Sinha D, Wang Z, Ruchalski KL, Levine JS, Krishnan S, Lieberthal W, Schwartz JH, Borkan SC. Lithium activates the Wnt and phos-phatidylinositol 3-kinase Akt signaling pathways to promote cell survival in the absence of soluble survival factors. Am. J. Physiol. Renal Physiol. 2005;288:F703–F713. doi: 10.1152/ajprenal.00189.2004. [DOI] [PubMed] [Google Scholar]
- Somervaille TC, Linch DC, Khwaja A. Growth factor withdrawal from primary human erythroid progenitors induces apoptosis through a pathway involving glycogen synthase kinase-3 and Bax. Blood. 2001;98:1374–1381. doi: 10.1182/blood.v98.5.1374. [DOI] [PubMed] [Google Scholar]
- Song L, De Sarno P, Jope RS. Central role of glycogen synthase kinase-3β in endoplasmic reticulum stress-induced caspase-3 activation. J. Biol. Chem. 2002;277:44701–44708. doi: 10.1074/jbc.M206047200. [DOI] [PubMed] [Google Scholar]
- Song L, Zhou T, Jope RS. Lithium facilitates apoptotic signaling induced by activation of the Fas death domain-containing receptor. BMC Neurosci. 2004;5:20–27. doi: 10.1186/1471-2202-5-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan S, Ohsugi M, Liu Z, Fatrai S, Bernal-Mizrachi E, Permutt MA. Endoplasmic reticulum stress-induced apoptosis is partly mediated by reduced insulin signaling through phosphatidylinositol 3-kinase/Akt and increased glycogen synthase kinase-3β in mouse insulinoma cells. Diabetes. 2005;54:968–975. doi: 10.2337/diabetes.54.4.968. [DOI] [PubMed] [Google Scholar]
- Steinbrecher KA, Wilson W, III, Cogswell PC, Baldwin AS. Glycogen synthase kinase 3β functions to specify gene-specific, NF-κB-dependent transcription. Mol. Cell. Biol. 2005;25:8444–8455. doi: 10.1128/MCB.25.19.8444-8455.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, Ryder J, Li B, Wu X, Fox N, Solenberg P, Brune K, Paul S, Zhou Y, Liu F, Ni B. Lithium, a common drug for bipolar disorder treatment, regulates amyloid-β precursor protein processing. Biochemistry. 2004;43:6899–6908. doi: 10.1021/bi035627j. [DOI] [PubMed] [Google Scholar]
- Suhara T, Magrane J, Rosen K, Christensen R, Kim HS, Zheng B, McPhie DL, Walsh K, Querfurth H. Ab42 generation is toxic to endothelial cells and inhibits eNOS function through an Akt/GSK3β signaling-dependent mechanism. Neurobiol. Aging. 2003;24:437–451. doi: 10.1016/s0197-4580(02)00135-5. [DOI] [PubMed] [Google Scholar]
- Takada Y, Fang X, Jamaluddin MS, Boyd DD, Aggarwal BB. Genetic deletion of glycogen synthase kinase-3β abrogates activation of IkBa kinase, JNK, Akt, and p44/p42 MAPK but potentiates apoptosis induced by tumor necrosis factor. J. Biol. Chem. 2004;279:39541–39554. doi: 10.1074/jbc.M403449200. [DOI] [PubMed] [Google Scholar]
- Takadera T, Ohyashiki T. Glycogen synthase kinase-3 inhibitors prevent caspase-dependent apoptosis induced by ethanol in cultured rat cortical neurons. Eur. J. Pharmacol. 2004;499:239–245. doi: 10.1016/j.ejphar.2004.07.115. [DOI] [PubMed] [Google Scholar]
- Takadera T, Sakamoto Y, Ohyashiki T. NMDA receptor 2B-selective antagonist ifenprodil-induced apoptosis was prevented by glycogen synthase kinase-3 inhibitors in cultured rat cortical neurons. Brain Res. 2004;1020:196–203. doi: 10.1016/j.brainres.2004.06.035. [DOI] [PubMed] [Google Scholar]
- Takashima A, Noguchi K, Sato K, Hoshino T, Imahori K. Tau protein kinase I is essential for amyloid β-protein-induced neurotoxicity. Proc. Natl. Acad. Sci. U.S.A. 1993;90:7789–7793. doi: 10.1073/pnas.90.16.7789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J, Zhuang L, Leong HS, Iyer NG, Liu ET, Yu Q. Pharmacologic modulation of glycogen synthase kinase-3β promotes p53-dependent apoptosis through a direct Bax-mediated mitochondrial pathway in colorectal cancer cells. Cancer Res. 2005;65:9012–9020. doi: 10.1158/0008-5472.CAN-05-1226. [DOI] [PubMed] [Google Scholar]
- Tan J, Geng L, Yazlovitskaya EM, Hallahan DE. Protein kinase B/Akt-dependent phosphorylation of glycogen synthase kinase-3β in irradiated vascular endothelium. Cancer Res. 2006;66:2320–2327. doi: 10.1158/0008-5472.CAN-05-2700. [DOI] [PubMed] [Google Scholar]
- Tong N, Sanchez JF, Maggirwar SB, Ramirez SH, Guo H, Dewhurst S, Gelbard HA. Activation of glycogen synthase kinase 3β (GSK3β) by platelet activating factor mediates migration and cell death in cerebellar granule neurons. Eur. J. Neurosci. 2001;13:1913–1922. doi: 10.1046/j.0953-816x.2001.01572.x. [DOI] [PubMed] [Google Scholar]
- Turenne GA, Price BD. Glycogen synthase kinase3β phosphorylates serine 33 of p53 and activates p53’s transcriptional activity. BMC Cell Biol. 2001;2:12. doi: 10.1186/1471-2121-2-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda Y, Hijikata M, Takagi S, Takada R, Takada S, Chiba T, Shimotohno K. Wnt/β-catenin signaling suppress apoptosis in low serum medium and induces morphologic change in rodent fibroblasts. Int. J. Cancer. 2002;99:681–688. doi: 10.1002/ijc.10418. [DOI] [PubMed] [Google Scholar]
- Viatour P, Dejardin E, Warnier M, Lair F, Claudio E, Bureau F, Marine JC, Merville MP, Maurer U, Green D, Piette J, Siebenlist U, Bours V, Chariot A. GSK3-mediated BCL-3 phosphorylation modulates its degradation and its oncogenicity. Mol. Cell. 2004;16:35–45. doi: 10.1016/j.molcel.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10:45–65. doi: 10.1038/sj.cdd.4401189. [DOI] [PubMed] [Google Scholar]
- Watcharasit P, Bijur GN, Zmijewski JW, Song L, Zmijewska A, Chen X, Johnson GVW, Jope RS. Direct, activating interaction between glycogen synthase kinase-3β and p53 after DNA damage. Proc. Natl. Acad. Sci. U.S.A. 2002;99:7951–7955. doi: 10.1073/pnas.122062299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watcharasit P, Bijur GN, Song L, Zhu J, Chen X, Jope RS. Glycogen synthase kinase-3β (GSK3β) binds to and promotes the actions of p53. J. Biol. Chem. 2003;278:48872–48879. doi: 10.1074/jbc.M305870200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H, Leeds PR, Qian Y, Wei W, Chen R, Chuang D. β-Amyloid peptide-induced death of PC12 cells and cerebellar granule cell neurons is inhibited by long-term lithium treatment. Eur. J. Pharmacol. 2000;392:117–123. doi: 10.1016/s0014-2999(00)00127-8. [DOI] [PubMed] [Google Scholar]
- Woodgett JR. Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J. 1990;9:2431–2438. doi: 10.1002/j.1460-2075.1990.tb07419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodgett JR. Physiological roles of glycogen synthase kinase-3: potential as a therapeutic target for diabetes and other disorders. Curr. Drug Targets Immune Endocr. Metabol. Disord. 2003;3:281–290. doi: 10.2174/1568008033340153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Chao L, Chao J. Kallikrein/kinin protects against myocardial apoptosis after ischemia/reperfusion via Akt-glycogen synthase kinase-3 and Akt-Bad.14-3-3 signaling pathways. J. Biol. Chem. 2005;280:8022–8030. doi: 10.1074/jbc.M407179200. [DOI] [PubMed] [Google Scholar]
- You Z, Saims D, Chen S, Zhang Z, Guttridge DC, Guan KL, Mac-Dougald OA, Brown AM, Evan G, Kitajewski J, Wang CY. Wnt signaling promotes oncogenic transformation by inhibiting c-Myc-induced apoptosis. J. Cell Biol. 2002;157:429–440. doi: 10.1083/jcb.200201110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Zhang J, Wong BW, Si X, Wong J, Yang D, Luo H. Inhibition of glycogen synthase kinase 3β suppresses coxsackievirus-induced cytopathic effect and apoptosis via stabilization of β-catenin. Cell Death Differ. 2005;12:1097–1106. doi: 10.1038/sj.cdd.4401652. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Hartmann H, Do VM, Abramowski D, Sturchler-Pierrat C, Staufenbiel M, Sommer B, van de Wetering M, Clevers H, Saftig P, De Strooper B, He X, Yankner BA. Destabilization of β-catenin by mutations in presenilin-1 potentiates neuronal apoptosis. Nature. 1998;395:698–702. doi: 10.1038/27208. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Hong Y, Bounhar Y, Blacker M, Roucou X, Tounekti O, Vereker E, Bowers WJ, Federoff HJ, Goodyer CG, LeBlanc A. p75 neurotrophin receptor protects primary cultures of human neurons against extracellular amyloid b peptide cytotoxicity. J. Neurosci. 2003;23:7385–7394. doi: 10.1523/JNEUROSCI.23-19-07385.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zmijewski JW, Jope RS. Nuclear accumulation of glycogen synthase kinase-3 during replicative senescence of human fibroblasts. Aging Cell. 2004;3:309–317. doi: 10.1111/j.1474-9728.2004.00117.x. [DOI] [PMC free article] [PubMed] [Google Scholar]