

Abstract
Glycogen synthase kinase-3 (GSK3), which is inhibited by serine-phosphorylation, is involved in the neuropathology of Alzheimer's disease (AD). We tested if the two therapeutic strategies used for AD, inhibition of acetylcholinesterase and of N-methyl-d-aspartate (NMDA) receptors, modulate the phosphorylation state of the two isoforms of GSK3 in mouse brain. Large, rapid increases in the levels of phospho-Ser21-GSK3α and phospho-Ser9-GSK3β occurred in mouse hippocampus, cerebral cortex, and striatum after treatment of mice with the muscarinic agonist pilocarpine or the acetylcholinesterase inhibitor physostigmine. Treatment with memantine, an NMDA receptor antagonist, also increased the serine-phosphorylation of both GSK3 isoforms in mouse brain. Co-administration of physostigmine and memantine increased serine-phosphorylated GSK3 levels equally to that achieved by either agent alone, indicating that the actions of these two drugs converge on overlapping pools of GSK3. Thus, drugs in each class of therapeutic agents used for AD have the common property of increasing the regulatory serine-phosphorylation of GSK3 within common pools of the enzyme.
Keywords: Cholinergic, GSK3 phosphorylation, Mice, Pilocarpine, Physostigmine, Memantine, Alzheimer's disease
1. Introduction
Alzheimer's disease (AD) is a progressively debilitating neurodegenerative disease involving several characteristic pathological features. Prominent among these are amyloid plaques, neurofibrillary tangles, and neuronal dysfunction and loss [3]. Glycogen synthase kinase-3 (GSK3) is an enzyme with a diverse number of actions in intracellular signaling systems, regulating neuronal plasticity, gene expression and cell survival, and it is closely associated with each of the prominent features of AD, as previously reviewed in detail [21]. For example, amyloid β-peptide (Aβ), the primary component of plaques, activates GSK3 [51] and causes cell death in a GSK3-dependent manner [1,2,11,19,27,50,55,56]. Furthermore, GSK3 activity promotes Aβ production [41,47]. Presenilin-1, a protein that contributes to Aβ production and that is mutated in one form of familial AD, also binds and can regulate GSK3 [17,31,49]. Neurofibrillary tangles consist primarily of hyperphosphorylated tau. GSK3 is a prime candidate kinase for contributing to this phosphorylation which results in decreased microtubule binding, perhaps enhancing paired helical filament formation, a possible prelude to neurofibrillary tangle formation [29]. Finally, the structure, function, and death of neurons is regulated by GSK3 in numerous ways that may be related to the neuropathology of AD [30]. For example endoplasmic reticulum (ER) stress and the accumulation of misfolded proteins contributes to the neurotoxicity evident in Alzheimer's disease [25,34,37,39], and endoplasmic reticulum stress causes activation of GSK3β which promotes apoptotic cell death [46]. Thus, through these and other mechanisms GSK3 is intimately associated with many features of AD, raising the possibility that inhibition of GSK3 may be therapeutic for the disease.
The activity of GSK3 is primarily regulated by phosphorylation, although of importance for specific substrates are other regulatory mechanisms that include modulation of its subcellular distribution, complex formation with GSK3-binding proteins, and regulation of the phosphorylation state of certain substrates [30]. Phosphorylation-dependent regulation of GSK3 primarily involves an N-terminal serine in each of the two isoforms of GSK3, serine-9 in GSK3β and serine-21 in GSK3α. It is firmly established that this modification acts as an internal pseudosubstrate that binds the substrate-binding pocket of GSK3 to inhibit its activity [10,16]. Conversely, decreased serine-phosphorylation increases the activity of GSK3. Thus, measurements of changes in the phosphorylation state of the regulatory N-terminal serine of GSK3 reflect modulation of its activity. GSK3 is also phosphorylated on a tyrosine, a modulation that may be regulatory or may be an autophosphorylation event [9,16].
Although there is no known cure for AD, two types of agents that provide some degree of therapeutic benefit are currently approved for treatment in the United States: agents that inhibit acetylcholinesterase [45] or that block the NMDA receptor [42,43]. Additionally, initial evidence indicates that both types of agents can be administered concurrently [24,53]. Since GSK3 is so closely linked to the prominent pathological features of AD, the present study examined the effects of these therapeutic agents on the in vivo phosphorylation state of GSK3 in mouse brain. The results show that administration of agents that increase cholinergic stimulation or block NMDA receptors increase the inhibitory serine-phosphorylation of GSK3 in mouse brain in vivo. Thus, agents belonging to each of the two classes of drugs used to treat AD share the common outcome of increasing the inhibitory serine-phosphorylation of GSK3.
2. Experimental procedures
2.1. Tissue preparation
Adult (2–4 months of age), male C57BL/6 mice (Frederick Cancer Research, Frederick, MD) were treated with pilocarpine, physostigmine, or memantine (Sigma–Aldrich, St. Louis, MO) by intraperitoneal injection. After the indicated intervals, mice were decapitated, and brain regions (hippocampus, cerebral cortex, striatum) were rapidly dissected in ice-cold saline. Brain regions were homogenized in ice-cold lysis buffer containing 10 mM Tris–HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 0.5% NP-40, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 5 μg/ml pepstatin, 0.1 mM β-glycerophosphate, 1 mM phenylmethanesulfonyl fluoride, 1 mM sodium vanadate, 50 mM NaF, and 100 nM okadaic acid. The lysates were centrifuged at 20,800 × g for 15 min. Protein concentrations in the supernatants were determined using the Bradford protein assay [8]. For subcellular fractionation, cytosolic, nuclear, and mitochondrial extracts were prepared from cerebral cortices exactly as described previously [7].
2.2. Western blotting
Samples were mixed with Laemmli sample buffer (2% SDS) and placed in a boiling water bath for 5 min. Proteins were resolved in 7% SDS-polyacrylamide gels, and transferred to nitrocellulose. Membranes were probed with antibodies to phospho-Ser21-GSK3α, phospho-Ser9-GSK3β, phospho-Tyr279/216-GSK3α/β, total GSK3α/β, phospho-Ser473-Akt, phospho-Thr308-Akt (Cell Signaling Technology, Beverly, MA) and total Akt (Sigma Chemical Co., St. Louis, MO). Immunoblots were developed using horseradish peroxidase-conjugated goat anti-mouse, or goat anti-rabbit IgG, followed by detection with enhanced chemiluminescence, and the protein bands were quantitated with a densitometer. Data were analyzed by a t-test and values of p < 0.05 were considered to be statistically significant.
3. Results
3.1. Muscarinic receptor stimulation increases the in vivo serine-phosphorylation of GSK3 in mouse brain
The first goal of this study was to test if activation of cholinergic muscarinic receptors modulates the serine-phosphorylation state of either isoform of GSK3 in vivo. Therefore, mice were treated with the muscarinic receptor-specific agonist pilocarpine for 5–120 min. Phospho-serine-selective antibodies were used to determine the serine-phosphorylation states of GSK3α and GSK3β in immunoblots of extracts from three brain regions. In the hippocampus, treatment with pilocarpine induced a rapid and robust increase in the phospho-serine levels of both GSK3 isoforms (Fig. 1). The increases in both phospho-Ser21-GSK3α and phospho-Ser9-GSK3β were time-dependent and maximal increases occurred between 15 and 30 min after pilocarpine administration. The rapid and transient nature of the responses likely result from the pharmacokinetics of pilocarpine which acts rapidly in the brain following peripheral administration and then it is rapidly cleared. Examination of the peak effect of pilocarpine in multiple mice demonstrated that there was relatively small interindividual variance, as 15 min after pilocarpine administration the levels of phospho-Ser21-GSK3α and phospho-Ser9-GSK3β were increased to 654 ± 97 and 1046 ± 135%, respectively, of the control levels (means ± S.E.M.; n=3; p < 0.05).
Fig. 1.
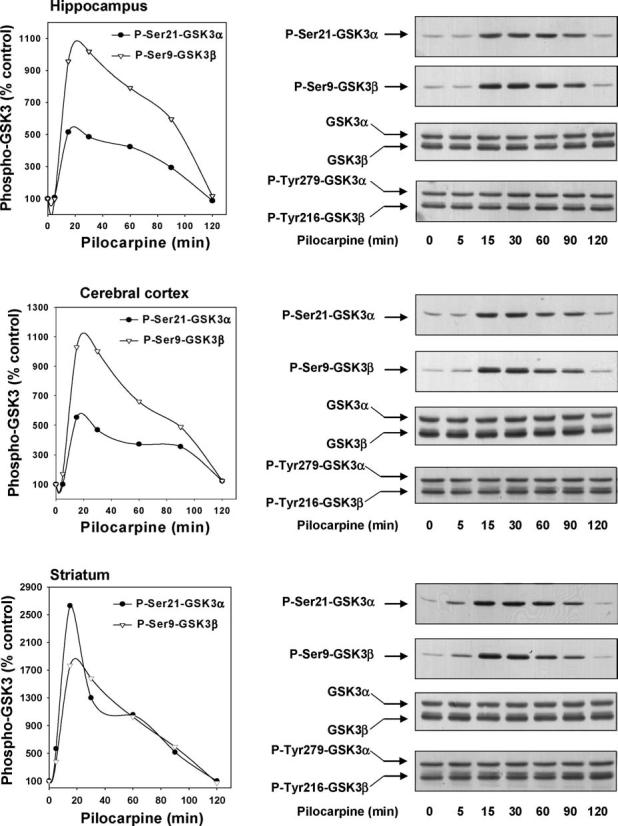
Pilocarpine administration stimulates serine-phosphorylation of GSK3 in mouse brain. Pilocarpine (30 mg/kg; 5, 15, 30, 60, 90 and 120 min) was administered to mice and protein extracts from the hippocampus, cerebral cortex, and striatum were immunoblotted with antibodies for phospho-Ser21-GSK3α, phospho-Ser9-GSK3β, total GSK3α/β, and phospho-Tyr279/216-GSK3α/β. Quantitative values are expressed as a percentage of values from control, saline-treated, mice. The number of mice tested at 5, 15, 30, 60, 90, and 120 min after pilocarpine administration were 1, 3, 2, 3, 1, and 1, respectively.
Similarly large increases in the serine-phosphorylation of both GSK3 isoforms occurred in the cerebral cortex and the striatum following pilocarpine administration (Fig. 1). As in the hippocampus, the increases in serine-phosphorylation were rapid and reached maximal levels between 15 and 30 min after treatment with pilocarpine. Measurements in multiple animals revealed that the peak effect at 15 min after pilocarpine administration increased the levels of phospho-Ser21-GSK3α to 542 ± 21% of the control levels in the cerebral cortex, and 2849 ± 143% in the striatum, and the levels of phospho-Ser9-GSK3β to 673 ± 184% in the cerebral cortex, and 2138 ± 249% in the striatum (means ± S.E.M.; n=3; p < 0.05). The pilocarpine-induced increases in serine-phosphorylated GSK3 were independent of any change in the total level of GSK3, which remained unaltered following administration of pilocarpine (Fig. 1). Additionally, pilocarpine treatment did not cause any changes in phospho-Tyr279-GSK3α or phospho-Tyr216-GSK3β.
GSK3 is distributed throughout cells and its phosphorylation state can be regulated selectively within subcellular domains, such as nuclei or mitochondria [7]. Therefore, we examined which pools of GSK3 were subjected to regulation by serine-phosphorylation following pilocarpine administration. The cerebral cortices of control and pilocarpine-treated mice were separated into cytosolic, nuclear, and mitochondrial fractions, and the serine-phosphorylated and total levels of both GSK3 isoforms were measured in each fraction. The levels of phospho-Ser21-GSK3α and phospho-Ser9-GSK3β increased in all three subcellular fractions after pilocarpine treatment (Fig. 2). No changes in the total levels of GSK3 were observed in any fraction, indicating that muscarinic receptor stimulation with pilocarpine did not change the subcellular distribution of GSK3. Thus, muscarinic receptor stimulation increased the serine-phosphorylation of GSK3 throughout the cell.
Fig. 2.
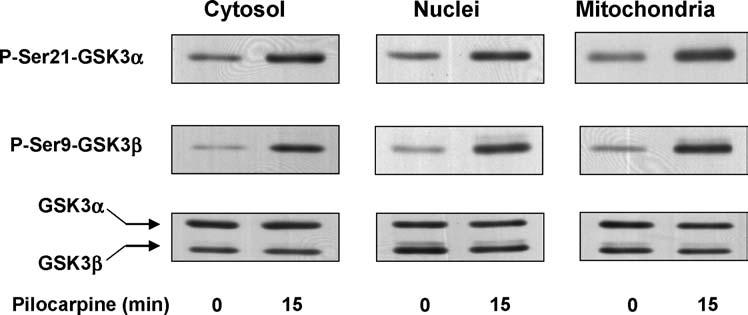
Pilocarpine administration stimulates serine-phosphorylation of GSK3 in subcellular fractions of mouse cerebral cortex. Pilocarpine (30 mg/kg; 15 min) was administered to mice and the cytosol, nuclear, and mitochondrial fractions from the cerebral cortex were immunoblotted with antibodies for phospho-Ser21-GSK3α, phospho-Ser9-GSK3β, and total GSK3α/β. Immunoblots shown are representative of results obtained from three mice.
3.2. Serine-phosphorylation of GSK3 is increased in vivo following inhibition of acetylcholinesterase
To test if enhanced endogenous cholinergic activity attained by inhibition of acetylcholinesterase was sufficient to modulate the serine-phosphorylation of GSK3, mice were treated with a low dose (0.1 mg/kg) of the acetylcholinesterase inhibitor physostigmine. Physostigmine is a rapid and short-acting inhibitor of acetylcholinesterase, with maximal inhibition attained approximately 15 min after administration [20,23]. Therefore, 15, 30, and 60 min after physostigmine administration the levels of phospho-Ser-GSK3 and total GSK3 were measured. Treatment with physostigmine caused rapid increases of both phospho-Ser21-GSK3α and phospho-Ser9-GSK3β in the hippocampus, cerebral cortex, and striatum, with a maximal effect occurring after 15 min (Fig. 3). Total levels of GSK3 did not change. Therefore, further experiments utilized the 15 min time point after physostigmine administration.
Fig. 3.
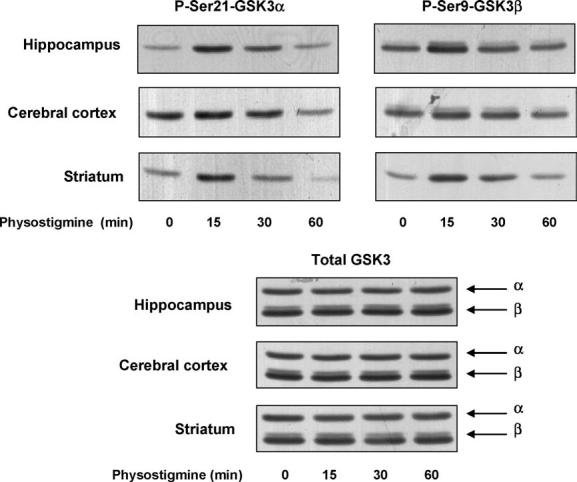
Physostigmine administration stimulates serine-phosphorylation of GSK3 in mouse brain. Physostigmine (0.1 mg/kg; 15, 30, 60 min) was administered to mice and protein extracts from the hippocampus, cerebral cortex, and striatum were immunoblotted with antibodies for phospho-Ser21-GSK3α, phospho-Ser9-GSK3β, and total GSK3α/β.
The dose–response of physostigmine-induced serine-phosphorylation of GSK3 was measured in all three brain regions (Fig. 4). Small increases in the serine-phosphorylation of both GSK3 isoforms were induced by 0.1 mg/kg physostigmine, and larger increases occurred following higher doses of physostigmine. Measurements made in multiple animals 15 min after physostigmine (0.4 mg/kg) administration showed that the levels of phospho-Ser21-GSK3α were increased to 350 ± 64% of the control levels in the hippocampus, 252 ± 38% in the cerebral cortex, and 425 ± 56% in the striatum, and the levels of phospho-Ser9-GSK3β were 313 ± 73% in the hippocampus, 164 ± 53% in the cerebral cortex, and 397 ± 27% in the striatum (means ± S.E.M.; n=4; p < 0.05). As with other treatments, no changes in the total levels of GSK3α and GSK3β were detected following physostigmine treatment.
Fig. 4.
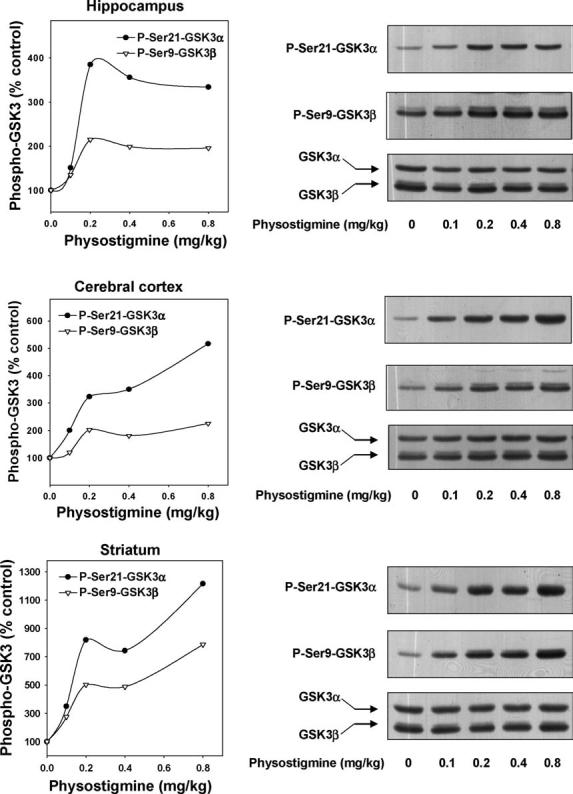
Dose-dependence of physostigmine-stimulated serine-phosphorylation of GSK3 in mouse brain. Physostigmine (0.1, 0.2, 0.4, 0.8 mg/kg; 15 min) was administered to mice and protein extracts from the hippocampus, cerebral cortex, and striatum were immunoblotted with antibodies for phospho-Ser21-GSK3α, phospho-Ser9-GSK3β, and total GSK3α/β. Quantitative values are given as a percentage of values from control, saline-treated, mice. The number of mice tested with 0.1, 0.2, 0.4, and 0.8 mg/kg pilocarpine were 2, 1, 4, and 1, respectively.
3.3. NMDA receptor inhibition increases the in vivo serine-phosphorylation of GSK3 in mouse brain
To test if inhibition of NMDA receptors had a similar or different effect on the phosphorylation of GSK3 compared with stimulation of muscarinic receptors, the NMDA receptor antagonist memantine was administered to mice and the serine-phosphorylation levels of both isoforms of GSK3 were measured. Measurements of the serine-phosphorylation of GSK3α and GSK3β in the cerebral cortex after administration of 50 mg/kg memantine to mice revealed increases in phosphorylation of both isoforms between 1 and 2 h and a plateau was maintained 3 h after treatment (Fig. 5A). In contrast to the increases in serine-phosphorylation, no changes in total levels of GSK3α or GSK3β occurred following the administration of memantine. Increases in the serine-phosphorylation levels of both GSK3α and GSK3β also were observed in the hippocampus and striatum 2 h after treatment with memantine (Fig. 5B), so this time period was used in further experiments.
Fig. 5.
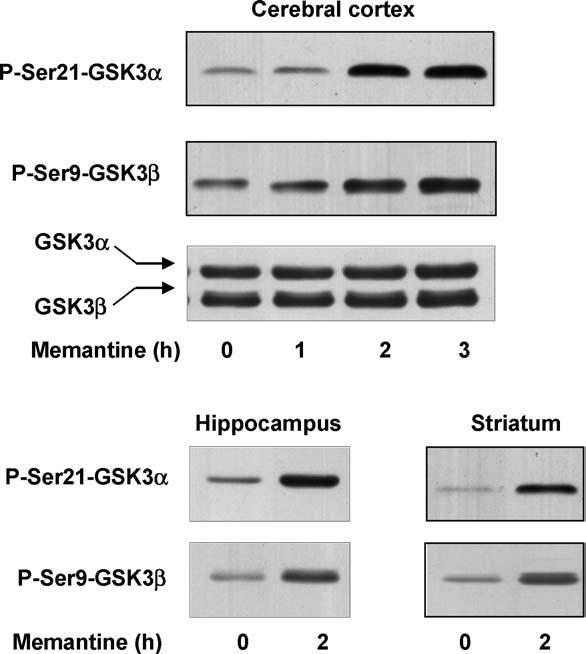
Memantine administration stimulates serine-phosphorylation of GSK3 in mouse brain. (A) Memantine (50 mg/kg; 1, 2 and 3 h) was administered to mice and protein extracts from the cerebral cortex were immunoblotted with antibodies for phospho-Ser21-GSK3α, phospho-Ser9-GSK3β, and total GSK3α/β. (B) Memantine (50 mg/kg; 2 h) was administered to mice and protein extracts from the hippocampus and striatum were immunoblotted with antibodies for phospho-Ser21-GSK3α and phospho-Ser9-GSK3β. The number of mice tested 1, 2, and 3 h after memantine administration were 3, 4, and 2, respectively.
3.4. Cholinergic stimulation and NMDA receptor inhibition converge to increase the serine-phosphorylation of GSK3
The next goal was to test if additive effects on serine-phosphorylation of GSK3 were obtained with combined cholinergic stimulation with physostigmine and NMDA receptor inhibition with memantine. For these experiments, measurements were taken in multiple animals at the times of peak effect of each drug, 2 h after memantine (50 mg/kg) treatment and 15 min after administration of physostigmine (0.4 mg/kg). Quantitative evaluations of the serine-phosphorylation levels revealed that after co-administration of both agents there was no greater increase in the serine-phosphorylation of GSK3α or GSK3β than was attained by each drug individually (Fig. 6), indicating that they converge on the same pools of GSK3. The levels of phospho-Tyr279-GSK3α or phospho-Tyr216-GSK3β and total levels of each isoform were unaltered after administration of each drug alone or after combined treatment.
Fig. 6.

Physostigmine and memantine stimulation converge on serine-phosphorylation of GSK3 in mouse brain. Mice were treated with physostigmine (0.4 mg/kg; 15 min), memantine (50 mg/kg; 2 h), or both drugs (physostigmine was given 105 min after memantine, for 15 min), and protein extracts from the hippocampus, cerebral cortex, and striatum were immunoblotted with antibodies for phospho-Ser21-GSK3α, phospho-Ser9-GSK3β, total GSK3α/β, and phospho-Tyr279/216-GSK3α/β. Quantitative values were obtained by densitometric scans of immunoblots (means ± S.E.M.; n=4). No statistical differences (p > 0.05) were observed comparing the results from treatment with physostigmine alone to those following treatment with memantine plus physostigmine.
3.5. Phosphorylation of Akt following cholinergic stimulation and NMDA receptor inhibition
Likely the most widely studied kinase responsible for serine-phosphorylation of GSK3 is Akt, and Akt has been reported to be activated via dual phosphorylation on serine-473 and threonine-308 in cultured cells following stimulation of muscarinic receptors [22,36,52]. Therefore, we examined in the same mouse brain samples if increased Akt phosphorylation correlated with the increases in serine-phosphorylated GSK3 that we observed. Fig. 7A shows results obtained 5–120 min after pilocarpine administration. In the hippocampus only slight increases in Akt phosphorylation occurred following pilocarpine administration, with the largest effect being an increase in phospho-Thr308-Akt after 15 min but less of an increase in phospho-Ser473-Akt. These increases were much less than the increases in phospho-Ser-GSK3, suggesting that Akt may not mediate this effect in mouse hippocampus after pilocarpine treatment. Mouse cerebral cortex displayed similar weak phosphorylation of Akt whereas larger increases were evident in the striatum, raising the possibility that different signaling systems may be involved in different brain regions. Administration of physostigmine caused a relatively strong dose-dependent increase in phospho-Thr308-Akt in all three brain regions, but the increase in phospho-Ser473-Akt was weaker and more variable (Fig. 7B). After administration of 0.4 mg/kg physostigmine, phospho-Thr308-Akt was 440 ± 31% (p < 0.01), 1053 ± 150% (p < 0.01), and 616 ± 84% (p < 0.01) of control (n=4) and phospho-Ser473-Akt was 141 ± 19% (p > 0.05), 231 ± 28% (p < 0.05), and 400 ± 67% (p < 0.01) of control, in the hippocampus, cerebral cortex, and striatum, respectively. Administration of memantine caused relatively weak increases in the dual phosphorylation of Akt, and in combination with physostigmine treatment the increases in Akt phosphorylation were equivalent to those induced by administration of physostigmine alone (Fig. 7C). Overall, although these agents caused some increases in Akt phosphorylation, notably more at threonine-308 than at serine-473, the changes did not closely mirror the increases in the serine-phosphorylation of GSK3, indicating that activation of Akt may contribute a part of the signal to GSK3 but it is not likely to be the sole intermediate.
Fig. 7.
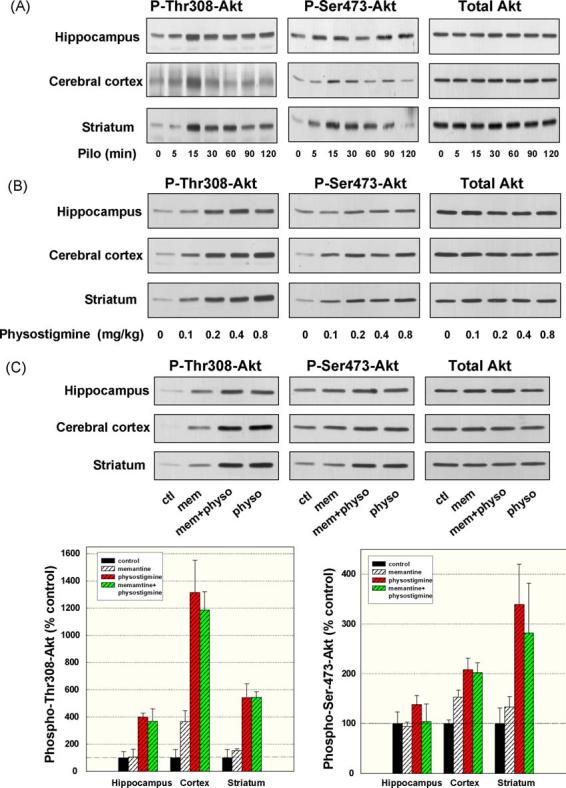
Pilocarpine, physostigmine, and memantine stimulation of Akt phosphorylation. Mice were treated with (A) pilocarpine (30 mg/kg; 5, 15, 30, 60, 90 and 120 min), (B) physostigmine (0.1, 0.2, 0.4 and 0.8 mg/kg; 15 min), or (C) memantine (50 mg/kg; 2 h), physostigmine (0.4 mg/kg; 15 min), or both drugs (physostigmine was given 105 min after memantine, for 15 min). Protein extracts from the hippocampus, cerebral cortex, and striatum were immunoblotted with antibodies for phospho-Thr308-Akt, phospho-Ser473-Akt, and total Akt. Quantitative values were obtained by densitometric scans of immunoblots (means ± S.E.M.; n=4). No statistically significant differences (p > 0.05) were observed comparing the results from treatment with physostigmine alone to those following treatment with memantine plus physostigmine.
4. Discussion
Therapeutic treatments currently used for AD include agents that inhibit acetylcholinesterase [45] or that block the NMDA receptor [42,43], and the combination of both types of agents [24,53]. Each of these classes of drugs was developed to target a specific component of the neuropathology of AD, with acetylcholinesterase inhibitors being used to bolster deficiencies in the actions of acetylcholine and NMDA blockade meant to attenuate toxicity mediated by abnormal hyperactivation of these receptors. Here we report that actions on these two disparate targets converge on GSK3, increasing the level of the inhibitory N-terminal serine-phosphorylation on both isoforms of GSK3. This reveals a previously unknown convergent point for these two classes of therapeutic agents and raises the possibility that inhibition of GSK3 may contribute to the therapeutic benefits derived from these drugs.
This is the first investigation to study if cholinergic activity regulates the phosphorylation of GSK3 in vivo, and the results showed that cholinergic stimulation rapidly increased the serine-phosphorylation of GSK3 by several-fold in three brain regions. This response was greater following administration of the direct-acting muscarinic agonist pilocarpine than after treatment with the acetylcholinesterase inhibitor physostigmine, as would be expected by the greater activation achieved by direct receptor stimulation. The effects of both pilocarpine and physostigmine on GSK3 phosphorylation were rapid and large. Within 15 min of administration both agents increased GSK3 phosphorylation, and the maximal increases caused by pilocarpine administration in three brain regions all involved more than five-fold increases above the control level of serine-phosphorylation of both GSK3α and GSK3β. The more transient effect of physostigmine compared with pilocarpine correlates with its rapidly reversible inhibition of acetylcholinesterase [20,23]. Thus, cholinergic stimulation can contribute to the inhibitory control of GSK3 in vivo by promoting the serine-phosphorylation of both GSK3 isoforms. This raises the possibility that impaired cholinergic activity which is known to occur in AD [54] may contribute to inadequate inhibitory control of GSK3 which can be restored by stimulation of muscarinic receptors.
Previous reports have linked signaling by NMDA receptors to changes in the serine-phosphorylation of GSK3, and those were confirmed here. NMDA treatment of cultured hippocampal neurons caused a rapid and nearly complete dephosphorylation of phospho-Ser9-GSK3, indicating that GSK3 is activated by NMDA receptor signaling [32]. In accordance with that conclusion, in vivo blockade of NMDA receptors by administration of the antagonist phencyclidine increased mouse brain serine-phosphorylation [48], a response we also observed in this study following memantine administration. A conflicting report showed that in immature rats blockade of NMDA receptors by in vivo administration of the antagonist MK-801 transiently decreased the serine-phosphorylation of GSK3 [13], a difference that could be due to age-dependent differences in responses to NMDA receptor modulation. Thus, although still few, the majority of studies indicate that NMDA receptor stimulation dephosphorylates GSK3 and that blockade of NMDA receptors in vivo is sufficient to cause increased levels of serine-phosphorylated GSK3.
Following our finding that either cholinergic stimulation or NMDA receptor blockade increased serine-phosphorylated GSK3 in vivo, we tested if the responses to these two therapeutic strategies used in AD were additive. This is of interest both because GSK3 appears to be a common target of the two therapeutic strategies and because recent studies suggest that both acetylcholinesterase inhibitors and memantine may be administered concurrently to patients with AD [24,53]. These experiments showed that co-administration of physostigmine and memantine increased serine-phosphorylated GSK3 to levels attained by administration of either agent alone, indicating that the actions of these two drugs converge on overlapping pools of GSK3. Activation of Akt may contribute a part of the drug-induced increases in serine-phosphorylation of GSK3, but it appears not to be sufficiently robust to account entirely for the increased phosphorylation of GSK3. Thus, drugs in each class of therapeutic agents used for AD share the capacity to increase the regulatory serine-phosphorylation of GSK3, and this occurs within the same pools of the enzyme, raising the possibility that inhibition of GSK3 contributes to the therapeutic actions of these agents.
These results add indirect support to previous suggestions that inhibition of GSK3 may provide therapeutic benefits in AD [12,21,33,41]. Increased serine-phosphorylation of GSK3 induced by muscarinic receptor stimulation or NMDA receptor inhibition could have multiple ramifications on neuropathological processes associated with AD. For example, two recent reports demonstrated that GSK3 activity promotes the production of Aβ [41,47]. Therefore, the reported reduction by cholinergic stimulation of Aβ in animal brain [5,6,14,18] or Aβ in cerebrospinal fluid from AD patients [4,26,38] may result, at least partially, from our observed inhibition of GSK3 caused by cholinergic stimulation. Similarly, serine-phosphorylation of GSK3 also may contribute partially to the reported protection from Aβ toxicity provided by NMDA receptor inhibition [28,35] since Aβ-induced cell death is attenuated by inhibitors of GSK3 [1,2,11,19,27,50,55,56]. The present results also raise the possibility that tau phosphorylation, which may be mediated by GSK3, which was reduced in cultured cells following cholinergic stimulation [15,44] also may be derived from inhibition of GSK3 caused by muscarinic receptor stimulation. It is notable that recent in vivo data concerning the influence of muscarinic activity on AD-type neuropathology revealed increased density of amyloid plaque and neurofibrillary tangles in patients after long-term anti-muscarinic medication, further supporting the hypothesis that cholinergic activity attenuates these events [40]. Additionally, GSK3 has many actions that can impair neuronal plasticity and promote cell death [21], suggesting that inhibition of GSK3 may bolster neuronal function and survival following exposure to potentially lethal insults.
In summary, administration of agents used in the treatment of AD that stimulate muscarinic receptors or block NMDA receptors caused large, widespread increases in the serine-phosphorylation levels of GSK3 in mouse brain in vivo. Furthermore, the actions of these agents converged on overlapping pools of GSK3. Considering the many known interactions of GSK3 with the major neuropathological characteristics of AD, these findings raise the possibility that modulation of GSK3 phosphorylation state contributes to the therapeutic actions of agents used in the treatment of the disease.
Acknowledgments
This research was supported by grants NS37768 and AG21045 from the National Institutes of Health.
References
- 1.Alvarez G, Munoz-Montano JR, Satrustegui J, Avila J, Bogonez E, Diaz-Nido J. Lithium protects cultured neurons against β-amyloid-induced neurodegeneration. FEBS Lett. 1999;453(3):260–4. doi: 10.1016/s0014-5793(99)00685-7. [DOI] [PubMed] [Google Scholar]
- 2.Alvarez G, Munoz-Montano JR, Satrustegui J, Avila J, Bogonez E, Diaz-Nido J. Regulation of tau phosphorylation and protection against β-amyloid-induced neurodegeneration by lithium. Possible implications for Alzheimer's disease. Bipolar Disord. 2002;4(3):153–65. doi: 10.1034/j.1399-5618.2002.01150.x. [DOI] [PubMed] [Google Scholar]
- 3.Annaert W, De Strooper B. A cell biological perspective on Alzheimer's disease. Annu Rev Cell Develop Biol. 2002;18:25–51. doi: 10.1146/annurev.cellbio.18.020402.142302. [DOI] [PubMed] [Google Scholar]
- 4.Beach TG. Muscarinic agonists as preventative therapy for Alzheimer's disease. Curr Opin Invest Drugs. 2002;3(11):1633–6. [PubMed] [Google Scholar]
- 5.Beach TG, Kuo YM, Schwab C, Walker DG, Roher AE. Reduction of cortical amyloid β levels in guinea pig brain after systemic administration of physostigmine. Neurosci Lett. 2001;310(1):21–4. doi: 10.1016/s0304-3940(01)02076-6. [DOI] [PubMed] [Google Scholar]
- 6.Beach TG, Walker DG, Potter PE, Sue LI, Fisher A. Reduction of cerebrospinal fluid amyloid β after systemic administration of M1 muscarinic agonists. Brain Res. 2001;905(1–2):220–3. doi: 10.1016/s0006-8993(01)02484-2. [DOI] [PubMed] [Google Scholar]
- 7.Bijur GN, Jope RS. Glycogen synthase kinase-3β is highly activated in nuclei and mitochondria. Neuroreport. 2003;14(18):2415–9. doi: 10.1097/00001756-200312190-00025. [DOI] [PubMed] [Google Scholar]
- 8.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein–dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 9.Cole A, Frame S, Cohen P. Further evidence that the tyrosine phosphorylation of glycogen synthase kinase-3 (GSK3) in mammalian cells is an autophosphorylation event. Biochem J. 2004;377(Pt 1):249–55. doi: 10.1042/BJ20031259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dajani R, Fraser E, Roe SM, Young N, Good V, Dale TC, et al. Crystal structure of glycogen synthase kinase 3β: structural basis for phosphate-primed substrate specificity and autoinhibition. Cell. 2001;105(6):721–32. doi: 10.1016/s0092-8674(01)00374-9. [DOI] [PubMed] [Google Scholar]
- 11.De Ferrari GV, Chacon MA, Barria MI, Garrido JL, Godoy JA, Olivares G, et al. Activation of Wnt signaling rescues neurodegeneration and behavioral impairments induced by β-amyloid fibrils. Mol Psychiatry. 2003;8(2):195–208. doi: 10.1038/sj.mp.4001208. [DOI] [PubMed] [Google Scholar]
- 12.Eldar-Finkelman H. Glycogen synthase kinase 3: an emerging therapeutic target. Trends Mol Med. 2002;8(3):126–32. doi: 10.1016/s1471-4914(01)02266-3. [DOI] [PubMed] [Google Scholar]
- 13.Elyaman W, Terro F, Wong NS, Hugon J. In vivo activation and nuclear translocation of phosphorylated glycogen synthase kinase-3β in neuronal apoptosis: links to tau phosphorylation. Eur J Neurosci. 2002;15(4):651–60. doi: 10.1046/j.1460-9568.2002.01899.x. [DOI] [PubMed] [Google Scholar]
- 14.Fisher A, Pittel Z, Haring R, Bar-Ner N, Kliger-Spatz M, Natan N, et al. M1 muscarinic agonists can modulate some of the hallmarks in Alzheimer's disease: implications in future therapy. J Mol Neurosci. 2003;20(3):349–56. doi: 10.1385/JMN:20:3:349. [DOI] [PubMed] [Google Scholar]
- 15.Forlenza OV, Spink JM, Dayanandan R, Anderton BH, Olesen OF, Lovestone S. Muscarinic agonists reduce tau phosphorylation in non-neuronal cells via GSK-3β inhibition and in neurons. J Neural Transm. 2000;107(10):1201–12. doi: 10.1007/s007020070034. [DOI] [PubMed] [Google Scholar]
- 16.Frame S, Cohen P, Biondi RM. A common phosphate binding site explains the unique substrate specificity of GSK3 and its inactivation by phosphorylation. Mol Cell. 2001;7(6):1321–7. doi: 10.1016/s1097-2765(01)00253-2. [DOI] [PubMed] [Google Scholar]
- 17.Gantier R, Gilbert D, Dumanchin C, Campion D, Davoust D, Toma F, et al. The pathogenic L392V mutation of presenilin 1 decreases the affinity to glycogen synthase kinase-3β. Neurosci Lett. 2000;283(3):217–20. doi: 10.1016/s0304-3940(00)00949-6. [DOI] [PubMed] [Google Scholar]
- 18.Genis I, Fisher A, Michaelson DM. Site-specific dephosphorylation of tau of apolipoprotein E-deficient and control mice by M1 muscarinic agonist treatment. J Neurochem. 1999;72(1):206–13. doi: 10.1046/j.1471-4159.1999.0720206.x. [DOI] [PubMed] [Google Scholar]
- 19.Ghribi O, Herman MM, Savory J. Lithium inhibits Aβ-induced stress in endoplasmic reticulum of rabbit hippocampus but does not prevent oxidative damage and tau phosphorylation. J Neurosci Res. 2003;71(6):853–62. doi: 10.1002/jnr.10511. [DOI] [PubMed] [Google Scholar]
- 20.Gottwald MD, Rozanski RI. Rivastigmine, a brain-region selective acetylcholinesterase inhibitor for treating Alzheimer's disease: review and current status. Expert Opin Invest Drugs. 1999;8(10):1673–82. doi: 10.1517/13543784.8.10.1673. [DOI] [PubMed] [Google Scholar]
- 21.Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3β in cellular signaling. Prog Neurobiol. 2001;65(4):391–426. doi: 10.1016/s0301-0082(01)00011-9. [DOI] [PubMed] [Google Scholar]
- 22.Guizzetti M, Costa LG. Activation of phosphatidylinositol 3 kinase by muscarinic receptors in astrocytoma cells. Neuroreport. 2001;12(8):1639–42. doi: 10.1097/00001756-200106130-00025. [DOI] [PubMed] [Google Scholar]
- 23.Harris LW, Anderson DR, Pastelak AM, Bowersox SL, Vanderpool BA, Lennox WJ. Acetylcholinesterase inhibition and antisoman efficacy of homologs of physostigmine. Drug Chem Toxicol. 1992;15(2):127–43. doi: 10.3109/01480549209032295. [DOI] [PubMed] [Google Scholar]
- 24.Hartmann S, Mobius HJ. Tolerability of memantine in combination with cholinesterase inhibitors in dementia therapy. Int Clin Psychopharmacol. 2003;18(2):81–115. doi: 10.1097/01.yic.0000054279.38655.74. [DOI] [PubMed] [Google Scholar]
- 25.Hitomi J, Katayama T, Eguchi Y, Kudo T, Taniguchi M, Koyama Y, et al. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Aβ-induced cell death. J Cell Biol. 2004;165(3):347–56. doi: 10.1083/jcb.200310015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hock C, Maddalena A, Raschig A, Muller-Spahn F, Eschweiler G, Hager K, et al. Treatment with the selective muscarinic m1 agonist talsaclidine decreases cerebrospinal fluid levels of Aβ 42 in patients with Alzheimer's disease. Amyloid. 2003;10(1):1–6. doi: 10.3109/13506120308995249. [DOI] [PubMed] [Google Scholar]
- 27.Hoshi M, Sato M, Matsumoto S, Noguchi A, Yasutake K, Yoshida N, et al. Spherical aggregates of β-amyloid (amylospheroid) show high neurotoxicity and activate tau protein kinase I/glycogen synthase kinase-3β. Proc Natl Acad Sci USA. 2003;100(11):6370–5. doi: 10.1073/pnas.1237107100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ikezu T, Luo X, Weber GA, Zhao J, McCabe L, Buescher JL, et al. Amyloid precursor protein-processing products affect mononuclear phagocyte activation: pathways for sAPP- and Aβ-mediated neurotoxicity. J Neurochem. 2003;85(4):925–34. doi: 10.1046/j.1471-4159.2003.01739.x. [DOI] [PubMed] [Google Scholar]
- 29.Johnson GV, Bailey CD. Tau, where are we now? J Alzheimer's Dis. 2002;4(5):375–98. doi: 10.3233/jad-2002-4505. [DOI] [PubMed] [Google Scholar]
- 30.Jope RS, Johnson GV. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci. 2004;29(2):95–102. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 31.Kang DE, Soriano S, Frosch MP, Collins T, Naruse S, Sisodia SS, et al. Presenilin 1 facilitates the constitutive turnover of β-catenin: differential activity of Alzheimer's disease-linked PS1 mutants in the β-catenin-signaling pathway. J Neurosci. 1999;19(11):4229–37. doi: 10.1523/JNEUROSCI.19-11-04229.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luo HR, Hattori H, Hossain MA, Hester L, Huang Y, Lee-Kwon W, et al. Akt as a mediator of cell death. Proc Natl Acad Sci USA. 2003;100(20):11712–7. doi: 10.1073/pnas.1634990100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martinez A, Castro A, Dorronsoro I, Alonso M. Glycogen synthase kinase 3 (GSK-3) inhibitors as new promising drugs for diabetes, neurodegeneration, cancer, and inflammation. Med Res Rev. 2002;22(4):373–84. doi: 10.1002/med.10011. [DOI] [PubMed] [Google Scholar]
- 34.Mattson MP, LaFerla FM, Chan SL, Leissring MA, Shepel PN, Geiger JD. Calcium signaling in the ER: its role in neuronal plasticity and neurodegenerative disorders. Trends Neurosci. 2000;23(5):222–9. doi: 10.1016/s0166-2236(00)01548-4. [DOI] [PubMed] [Google Scholar]
- 35.Miguel-Hidalgo JJ, Alvarez XA, Cacabelos R, Quack G. Neuro-protection by memantine against neurodegeneration induced by β-amyloid(1–40) Brain Res. 2002;958(1):210–21. doi: 10.1016/s0006-8993(02)03731-9. [DOI] [PubMed] [Google Scholar]
- 36.Murga C, Laguinge L, Wetzker R, Cuadrado A, Gutkind JS. Activation of Akt/protein kinase B by G protein-coupled receptors. A role for α and βγ subunits of heterotrimeric G proteins acting through phosphatidylinositol-3-OH kinaseγ. J Biol Chem. 1998;273(30):19080–5. doi: 10.1074/jbc.273.30.19080. [DOI] [PubMed] [Google Scholar]
- 37.Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature. 2000;403(6765):98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- 38.Nitsch RM, Deng M, Tennis M, Schoenfeld D, Growdon JH. The selective muscarinic M1 agonist AF102B decreases levels of total Aβ in cerebrospinal fluid of patients with Alzheimer's disease. Ann Neurol. 2000;48(6):913–8. [PubMed] [Google Scholar]
- 39.Pereira C, Ferreiro E, Cardoso SM, de Oliveira CR. Cell degeneration induced by amyloid-β peptides: implications for Alzheimer's disease. J Mol Neurosci. 2004;23(1–2):97–104. doi: 10.1385/JMN:23:1-2:097. [DOI] [PubMed] [Google Scholar]
- 40.Perry EK, Kilford L, Lees AJ, Burn DJ, Perry RH. Increased Alzheimer pathology in Parkinson's disease related to antimuscarinic drugs. Ann Neurol. 2003;54(2):235–8. doi: 10.1002/ana.10639. [DOI] [PubMed] [Google Scholar]
- 41.Phiel CJ, Wilson CA, Lee VM, Klein PS. GSK-3α regulates production of Alzheimer's disease amyloid-β peptides. Nature. 2003;423(6938):435–9. doi: 10.1038/nature01640. [DOI] [PubMed] [Google Scholar]
- 42.Reisberg B, Doody R, Stoffler A, Schmitt F, Ferris S, Mobius HJ. Memantine in moderate-to-severe Alzheimer's disease. N Engl J Med. 2003;348(14):1333–41. doi: 10.1056/NEJMoa013128. [DOI] [PubMed] [Google Scholar]
- 43.Rogawski MA, Wenk GL. The neuropharmacological basis for the use of memantine in the treatment of Alzheimer's disease. CNS Drug Rev. 2003;9(3):275–308. doi: 10.1111/j.1527-3458.2003.tb00254.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sadot E, Gurwitz D, Barg J, Behar L, Ginzburg I, Fisher A. Activation of m1 muscarinic acetylcholine receptor regulates tau phosphorylation in transfected PC12 cells. J Neurochem. 1996;66(2):877–80. doi: 10.1046/j.1471-4159.1996.66020877.x. [DOI] [PubMed] [Google Scholar]
- 45.Scarpini E, Scheltens P, Feldman H. Treatment of Alzheimer's disease: current status and new perspectives. Lancet Neurol. 2003;2(9):539–47. doi: 10.1016/s1474-4422(03)00502-7. [DOI] [PubMed] [Google Scholar]
- 46.Song L, De Sarno P, Jope RS. Central role of glycogen synthase kinase-3β in endoplasmic reticulum stress-induced caspase-3 activation. J Biol Chem. 2002;277(47):44701–8. doi: 10.1074/jbc.M206047200. [DOI] [PubMed] [Google Scholar]
- 47.Su Y, Ryder J, Li B, Wu X, Fox N, Solenberg P, et al. Lithium a common drug for bipolar disorder treatment, regulates amyloid-β precursor protein processing. Biochemistry. 2004;43(22):6899–908. doi: 10.1021/bi035627j. [DOI] [PubMed] [Google Scholar]
- 48.Svenningsson P, Tzavara ET, Carruthers R, Rachleff I, Wattler S, Nehls M, et al. Diverse psychotomimetics act through a common signaling pathway. Science. 2003;302(5649):1412–5. doi: 10.1126/science.1089681. [DOI] [PubMed] [Google Scholar]
- 49.Takashima A, Murayama M, Murayama O, Kohno T, Honda T, Yasutake K, et al. Presenilin 1 associates with glycogen synthase kinase-3β and its substrate tau. Proc Natl Acad Sci USA. 1998;95(16):9637–41. doi: 10.1073/pnas.95.16.9637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Takashima A, Noguchi K, Sato K, Hoshino T, Imahori K. Tau protein kinase I is essential for amyloid β-protein-induced neurotoxicity. Proc Natl Acad Sci USA. 1993;90(16):7789–93. doi: 10.1073/pnas.90.16.7789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Takashima A, Yamaguchi H, Noguchi K, Michel G, Ishiguro K, Sato K, et al. Amyloid β peptide induces cytoplasmic accumulation of amyloid protein precursor via tau protein kinase I/glycogen synthase kinase-3β in rat hippocampal neurons. Neurosci Lett. 1995;198(2):83–6. doi: 10.1016/0304-3940(95)11964-x. [DOI] [PubMed] [Google Scholar]
- 52.Tang X, Batty IH, Downes CP. Muscarinic receptors mediate phospholipase C-dependent activation of protein kinase B via Ca2+, ErbB3, and phosphoinositide 3-kinase in 1321N1 astrocytoma cells. J Biol Chem. 2002;277(1):338–44. doi: 10.1074/jbc.M108927200. [DOI] [PubMed] [Google Scholar]
- 53.Tariot PN, Farlow MR, Grossberg GT, Graham SM, McDonald S, Gergel I. Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: a randomized controlled trial. JAMA. 2004;291(3):317–24. doi: 10.1001/jama.291.3.317. [DOI] [PubMed] [Google Scholar]
- 54.Terry AV, Jr, Buccafusco JJ. The cholinergic hypothesis of age and Alzheimer's disease-related cognitive deficits: recent challenges and their implications for novel drug development. J Pharmacol Exp Ther. 2003;306(3):821–7. doi: 10.1124/jpet.102.041616. [DOI] [PubMed] [Google Scholar]
- 55.Wei H, Leeds PR, Qian Y, Wei W, Chen R, Chuang D. β-Amyloid peptide-induced death of PC 12 cells and cerebellar granule cell neurons is inhibited by long-term lithium treatment. Eur J Pharmacol. 2000;392(3):117–23. doi: 10.1016/s0014-2999(00)00127-8. [DOI] [PubMed] [Google Scholar]
- 56.Zhang Y, Hong Y, Bounhar Y, Blacker M, Roucou X, Tounekti O, et al. p75 neurotrophin receptor protects primary cultures of human neurons against extracellular amyloid β peptide cytotoxicity. J Neurosci. 2003;23(19):7385–94. doi: 10.1523/JNEUROSCI.23-19-07385.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]