

Abstract
In this work, highly infiltrative brain tumors with a stem-like phenotype were established by xenotransplantation of human brain tumors in immunodeficient nude rats. These tumors coopted the host vasculature and presented as an aggressive disease without signs of angiogenesis. The malignant cells expressed neural stem cell markers, showed a migratory behavior similar to normal human neural stem cells, and gave rise to tumors in vivo after regrafting. Serial passages in animals gradually transformed the tumors into an angiogenesis-dependent phenotype. This process was characterized by a reduction in stem cells markers. Gene expression profiling combined with high throughput immunoblotting analyses of the angiogenic and nonangiogenic tumors identified distinct signaling networks in the two phenotypes. Furthermore, proinvasive genes were up-regulated and angiogenesis signaling genes were down-regulated in the stem-like tumors. In contrast, proinvasive genes were down-regulated in the angiogenesis-dependent tumors derived from the stem-like tumors. The described angiogenesis-independent tumor growth and the uncoupling of invasion and angiogenesis, represented by the stem-like cancer cells and the cells derived from them, respectively, point at two completely independent mechanisms that drive tumor progression. This article underlines the need for developing therapies that specifically target the stem-like cell pools in tumors.
Keywords: glioma, invasiveness, vessel cooption
A basic principle in tumor progression is the requirement for angiogenesis, yet several clinical studies have reported limited efficacy of angiogenesis inhibitors to control tumor growth (1–7). This finding has been explained by pharmacokinetic parameters such as the mode of delivery, inadequate biodistribution, and misfolding of the therapeutic proteins (8). Still, some studies suggest that the nature of this problem may not be inherent in the therapeutic compound, but rather underlies the concept of angiogenesis-dependency itself (9–11). An alternative mechanism for obtaining essential nutrients may be that the malignant cells are sustained by the preexisting vasculature of the host tissue, as they invade the surrounding parenchyma.
Stem cells and tumor cells share the ability of cell division. Moreover, EGF and FGF, which maintain neural stem cells in a proliferative state in vitro, also increase proliferation of glioma cells (12–14). Similar to migrating neural stem cells grafted in adult rat brain, invading glioma cells may be supported by the vascular network in the normal brain (15–19). However, studies suggest that although tumor cells initially coopt surrounding vessels, subsequent growth requires angiogenesis (20, 21). Thus, the prevailing view is that solid tumor growth is angiogenesis-dependent (22–24).
Glioblastomas (GBMs) are highly vascular brain tumors that are considered to be attractive candidates for antiangiogenic therapy (25). GBMs are classified as high-grade gliomas because of the presence of necrosis and microvascular proliferations, and most often arise de novo in patients not previously diagnosed with a low-grade glioma. They are then referred to as primary GBMs and display a characteristic set of genetic changes (26, 27). However, these tumors may also arise from the malignant progression of invasive, low-grade gliomas without microvascular proliferations (26, 28). Apart from the onset of angiogenesis, this transition is characterized by progressive genetic changes different from those observed in primary GBMs (29). In this work, we xenografted 10 biopsies from primary glioblastomas into nude rat brains. Surprisingly, the resulting tumors recapitulated the infiltrative growth pattern of low-grade gliomas, coopting the host vasculature without any signs of angiogenesis or necrosis. Upon passaging in vivo, they progressed toward a highly malignant phenotype displaying tumor angiogenesis and large necrotic regions. This progression was not paralleled by progressive genetic derangements because the angiogenic and nonangiogenic phenotypes had almost identical array comparative genomic hybridization (CGH) profiles. However, they displayed distinct gene-expression profiles, suggesting that transcriptional modulation mediated the phenotypic shift. Our findings demonstrate that even highly vascular and aggressive tumors, with no definable precursor lesions, contain tumor cells that can revert and adapt the growth characteristics of low-grade tumors. Subsequently, these tumors can again progress to become vascular and necrotic. Our results show that the cellular heterogeneity and adaptive behavior demonstrated by these tumor cells bears a resemblance to the plasticity of stem cells and implies that anti-angiogenic cancer therapy should be combined with a therapy that targets the invasive stem-like cell populations.
Results
Patient Characteristics, Immunohistochemistry, and Engraftment Rate of Tumor Biopsies and Glioma Spheroids.
Spheroids derived from biopsy tissue of 10 patients with GBM all developed tumors (hereafter termed first-generation tumors) when transplanted into the CNS of nude rats (30, 31), although at varying rates (Table 1). All tumors were previously untreated, primary glioblastomas, with histological features defined by nuclear pleomorphism, mitosis, necrosis, and endothelial cell proliferation (Fig. 6a, which is published as supporting information on the PNAS web site). The tissue specimens were minced and cultured in vitro in serum containing medium to form glioma spheroids before implantation (Fig. 6 Right). Immunohistochemical staining displayed a strong expression of glial fibrillary acidic protein (GFAP) both in the tumor biopsies and the biopsy spheroids (Fig. 6b), whereas nestin was up-regulated in the spheroids (Fig. 6c). The tumor biopsies showed some staining for the cancer stem cell marker CD133, in contrast to the spheroids, which were CD133 negative (Fig. 6d).
Table 1.
Tumor take, engraftment rate, and passaging data on 10 primary GBM biopsies
| Case | Tumor take (%) | Survival, days, mean ± SEM* | Passaged in vivo |
|---|---|---|---|
| 1 | 12 of 13 (92) | 117.5 ± 8.6 | No |
| 2 | 7 of 7 (100) | 97 ± 1.7 | Yes |
| 3 | 4 of 5 (80) | 169.5 ± 22.1 | No |
| 4 | 3 of 5 (60) | 252 ± 1.6 | No |
| 5 | 7 of 8 (88) | 64 ± 1.5 | No |
| 6 | 2 of 10 (20) | 93.5 ± 10.6 | No |
| 7 | 6 of 6 (100) | 104.5 ± 1.4 | Yes |
| 8 | 8 of 8 (100) | 119.5 ± 3.5 | Yes |
| 9 | 12 of 14 (86) | 137.5 ± 5 | No |
| 10 | 7 of 7 (100) | 126.5 ± 2.9 | Yes |
*Survival data were recorded only from animals where tumor take was confirmed after histological examination.
Highly Vascular Brain Tumors Contain Cancer Cells with the Capacity to Generate New Tumors Without Angiogenesis.
To study tumor progression, we used longitudinal MRI over three time points (Fig. 1a). The T2 scans displayed diffuse lesions that occupied most of the hemispheres in the terminal stage, causing a shift of midline structures. Although engraftment took place from all of the biopsies, the xenografts from seven patients developed without signs of contrast enhancement (Fig. 1b). For two biopsies, only minor enhancement was visible, and only one biopsy developed into a tumor with contrast enhancement (data not shown). Animals displaying no contrast enhancements were subsequently infused with 18F-3′-deoxy-3′-fluorothymidine ([18F]FLT) and examined by positron emission tomography (32). The scans showed a diffuse intracranial uptake of [18F]FLT, indicating a disseminated spread of dividing tumor cells throughout the brain (Fig. 1c). Similarly, brain sections from rats pulsed with BrdU before killing, showed BrdU-positive cells spreading over the corpus callosum to the contralateral hemisphere (Fig. 1d). Moreover, we performed triple staining for the basement membrane marker collagen IV and BrdU in rats systemically injected with Hoechst 33342 (Fig. 1e). BrdU-positive cells were observed between blood vessels with no Hoechst leakage into the surrounding parenchyma, suggesting a normal vascular morphology and a functionally intact blood–brain barrier. Immunostaining and morphometric quantification for the vascular marker CD31 revealed that the area fraction representing vascular elements and vascular counts per field was slightly lower in the tumors compared with the normal brain (Fig. 1f, g, and j). This result may be attributed to cells infiltrating the vascular bed, thereby increasing the distance between neighboring vessels. Double staining for Ki67 and von Willebrand factor showed dividing tumor cells among quiescent normally sized blood vessels (Fig. 1h). No dividing endothelial cells were observed in the tumors. Double staining for collagen IV and the hypoxia marker pimonidazol revealed no sign of hypoxia in the first-generation tumor (Fig. 1i). Sections of rat brains perfused with India ink (Fig. 7a, which is published as supporting information on the PNAS web site) and transmission electron microscopy (Fig. 7b) also revealed a normal endothelial morphology and tight junctions between the endothelial cells. Immunohistochemical detection of reactive endothelial cells by staining for angiopoietin-2 was negative (Fig. 8a, which is published as supporting information on the PNAS web site).
Fig. 1.

Tumor growth without angiogenesis. (a) MRI scans (T2 sequence) at three different time points. The midline structure at 18 weeks, as indicated by arrowheads. (b) T1 sequence after gadodiamid administration. (c) A [18F]FLT positron emission tomography scan of a rat brain with a tumor. (d) Coronary rat brain section costained with BrdU (green) and collagen IV (red). (e) Triple staining of the tumor bed for BrdU (green), collagen IV (red), and Hoechst (blue). (f) CD31 staining of vessels in the normal brain. (g) CD31 staining of vessels in the tumor. (h) Costaining for von Willebrand factor (red) and Ki67 (brown). Ki67-positive tumor cell nucleus (arrow), and Ki67-negative endothelial nucleus (arrowhead) are shown. (i) Double staining for collagen IV (red) and pimonidazol (green). (j) Morphometric quantification of vascular parameters in the first-generation tumors and in the normal brain. Error bars show SEM. [Scale bars: 1 cm (c and d); 100 μm (e–g); 40 μm (h); and 5 mm (i).]
Angiogenesis-Independent Growth Is Mediated by Tumor Cells That Exhibit Stem-Like Characteristics.
Rat brains harvested at the time of MRI (Fig. 1a) allowed comparison with histological sections from corresponding regions (Fig. 2a). In all regions of the brain, we identified Ki67-positive tumor cells, which also stained positive for human-specific vimentin, a marker present in neuroepithelial progenitors and stem cells (33) (Fig. 2 b–d%). Tumor cells migrating along the corpus callosum, entering the cortex, also expressed the neural stem cell marker nestin (Fig. 2e). For comparison, transplanted glioma spheroids and human neural stem cells (HNSC 100) were stained for vimentin and showed a striking similarity in their migratory pattern (Fig. 2 f and g). The tumor cells also expressed the neural stem cell marker musashi-1 (Fig. 2h), an RNA-binding protein involved in asymmetric cell division during Drosophila neural development (34).
Fig. 2.

Nonangiogenic tumors contain cells with stem-like features. (a) Brain sections at different time points corresponding to the MRI scans. The main tumor mass has a purple color because of immunostaining with a human-specific antibody against vimentin. Costaining with anti-human vimentin (red) and Ki67 (brown) show dividing and nondividing tumor cells in different regions of the brain: corpus callosum (b), tumor bulk (c), and contralateral hemisphere (d). (e) Nestin-positive cancer cells (brown) invading the parenchyma in the contralateral hemisphere. (f and g) Migration along corpus callosum of vimentin-positive cancer cells (brown) from a tumor spheroid (f) and of human neural stem cells (g). (h) Musashi-1-positive cells (green) migrating from a tumor spheroid (red). (Scale bars: 50 μm.)
Serial Passaging in Vivo Changes the Nonangiogenic Tumor to a Highly Vascular Phenotype.
To further investigate tumor progression, first-generation tumors from four patients (Table 1) were removed and serially passaged in rats for 4–5 generations (hereafter termed high-generation tumors). In the subsequent generations, the tumors became more vascular and circumscribed (Fig. 3a) with emerging necrotic regions (Fig. 3 b and c). Moreover, MRI scans showed less invasive (Fig. 3d), strongly contrast-enhancing tumors (Fig. 3e) in the high generation. The less-invasive nature of the high-generation tumors was also confirmed by positron emission tomography scans, where they appeared sharply demarcated (Fig. 3f). Immunohistochemistry revealed tumors with a disordered vasculature, dilated vessels, and endothelial cell proliferations (Fig. 3 g and h). Triple staining for collagen IV and the hypoxia marker pimonidazole in rats infused with Hoechst 33342 revealed numerous hypoxic areas surrounded by dilated vessels with Hoechst leakage into the surrounding parenchyma (Fig. 3i). This leakage was also confirmed in rats that had received systemic injections of India ink (data not shown). A morphometric quantification of the vascular parameters (Fig. 3j) revealed lower vascular counts per visual field in the high-generation tumors compared with normal rat brain, whereas the area fraction representing endothelial cells per visual field was increased. Finally, the proliferative capillary index was 6% in the tumors compared with 0% in the normal brain. The onset of angiogenesis coincided with a significant decrease in survival from 113 ± 2.6 SEM to 43 ± 2.1 SEM days (Fig. 3k).
Fig. 3.

Angiogenesis-independent stem-like tumors progress to become vascular and necrotic tumors. (a) H&E staining of a high-generation tumor. Dashed lines indicate the tumor periphery. (b) Picture of the same tumor exhibiting macroscopic necrosis (arrowhead). (c) H&E staining of a high-generation tumor at high magnification with enlarged vessels and arrowheads indicating necrotic areas. (d and e) T2-weighted (d) and gadodiamid-enhanced T1-weighted (e) MRI scans of a high-generation tumor. White area in e represents contrast enhancement. (f) Positron emission tomography scan of the rat brain tumor. (Scale bar: 1 cm.) (g) CD31 staining (brown) of the tumor bed. (h) Costaining for von Willebrand (red) and Ki67 (brown). (Inset) Proliferating endothelial cells (arrowheads). (i) Triple staining of a tumor section against pimonidazol (green), collagen IV (red), and Hoechst (blue). (j) Quantification of vascular parameters and comparison with normal brain. (k) Kaplan–Meyer curves presenting survival data for animals grafted with four patient biopsies that were passaged from first- to high-generation. (Scale bars: 100 μm, unless otherwise indicated.)
Angiogenesis-Independent and -Dependent Phenotypes Are Genetically Similar, but Display Different Gene and Protein Expression Profiles and Distinct Patterns of Intracellular Signaling.
Array CGH showed that the human biopsy and the first- and high-generation tumors displayed nearly identical chromosomal profiles (Fig. 4a), where they showed loss on chromosome 5p, gain on 7 with EGFR amplification, INK4A/ARF homozygous deletion, loss of chromosome 10, and interstitial loss of 15q. The striking similarities in the array CGH profiles between the tumors suggested that transcriptional regulation is an important component in the phenotypic shift observed. Therefore, a comprehensive gene-expression analysis comparing first- and high-generation tumors was performed. In total, we found 77 genes whose differential expression was 2-fold or more between the two tumor phenotypes, using three different microarray platforms [16,000-oligonucleotide cDNA; Agilent Technologies (Palo Alto, CA), 44,000-oligonucleotide; Agilent Technologies, and 37,000-oligonucleotide microarrays; Applied Biosystems (Foster City, CA)] (Fig. 4b, and Table 2, which is published as supporting information on the PNAS web site). Furthermore, two of the array platforms contained vimentin and nestin, which where up-regulated 200% and 70% in the first-generation, respectively. To ensure that this up-regulation was human-specific and not caused by reactive host-derived cells, we designed primers specific for rat vimentin. Quantitative real-time PCR (RT-qPCR) from low- and high-generation confirmed that the expression was from the tumor cells (Fig. 9, which is published as supporting information on the PNAS web site). Moreover, a comprehensive Kinetworks multiimmunoblotting screen was performed, which represents a systems-biology approach providing simultaneous expression and phosphorylation states of hundreds of target proteins. The Kinetworks screen revealed numerous proteins to be differentially expressed, including main components of intracellular signaling pathways (Fig. 4c). Based on the gene-expression profiles and the Kinetworks screen, we found that components of the Wnt, PI3K, and NF-kβ signaling pathways were overexpressed in the invasive first-generation tumors compared with the high-generation tumors. In addition, although components of the Ras signaling pathway were expressed in both first- and high-generation tumors, they were significantly up-regulated in the high generation (Fig. 4d). Moreover, the first-generation tumors displayed up-regulation of genes involved in fetal development and cell motility (Table 3, which is published as supporting information on the PNAS web site).
Fig. 4.

Comparison of chromosomal DNA, gene expression, and protein profiles between first- and high-generation tumors. (a) Array CGH showing the relative chromosome copy numbers of the parent biopsy, first- and high-generation tumors. (b) Bar graph presenting the genes with the biggest difference in expression levels between the first- and high-generation tumors. (c) Immunoblot analysis of protein extracts from first- and high-generation tumor tissue. VEGF was analyzed from cerebrospinal fluid. (d) Signaling pathways differentially activated in the two tumor phenotypes.
Invasion and Angiogenesis, Two Independent Strategies for Tumor Progression.
The tumor cell invasion marker SPARC (35–37) was up-regulated in first-generation tumors, whereas the high-generation tumors displayed weak or no staining (Fig. 5a). Furthermore, spheroids from first-generation tumors were highly invasive when tested in a collagen-invasion-gel assay, whereas the high-generation tumor spheroids only displayed a modest invasion in the gel (Fig. 5b).
Fig. 5.

Inverse relationship between angiogenesis and invasion. (a) SPARC immunostaining (brown) at the tumor periphery in first- and high-generation tumors. (b) Invasion of tumor cells in a collagen gel from first- and high-generation glioma spheroids. (c and d) Hif-1α and VEGF expression (brown), respectively, in first- and high-generation tumors. (e) Aortic ring explants incubated with conditioned medium from first- and high-generation tumor spheroids. Pictures from aortic ring and collagen-invasion assays were all taken on day 5. (Scale bars: 100 μm.)
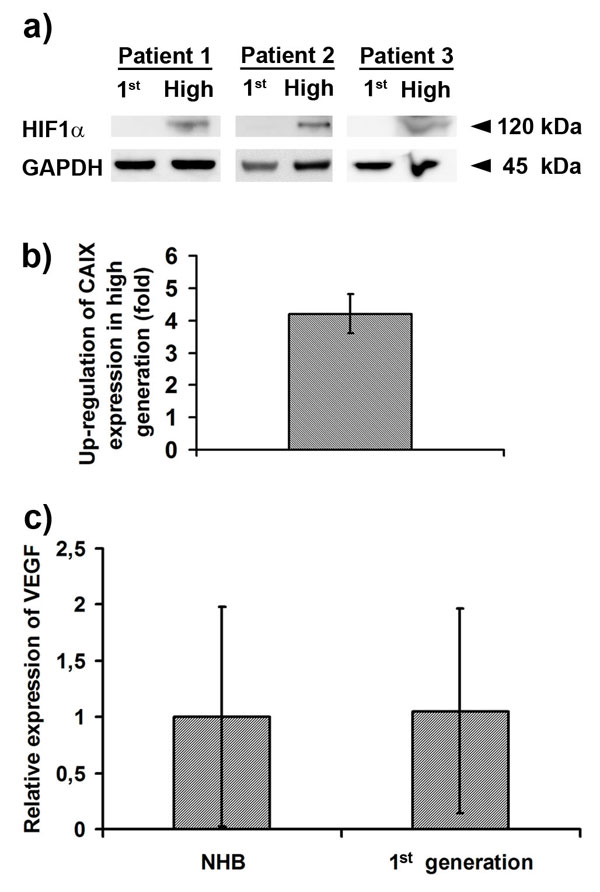
Conversely, immunostaining for HIF-1α and VEGF were negative in sections from first-generation tumors, whereas staining for both markers were positive in the high-generation tumors (Fig. 5 c and d). The same staining pattern was seen for angiopoitin-2 (Fig. 8). Furthermore, Western blotting for HIF-1α and quantitative real-time PCR (RT-qPCR) for its target gene carbon anhydrase IX (CAIX) showed up-regulation in the high-generation tumors, whereas RT-qPCR for VEGF in the first-generation tumors showed levels comparable with normal brain (Fig. 10 a–c%, which is published as supporting information on the PNAS web site). In addition, HIF-1α was not detected in tumor spheroids in normoxic conditions but was up-regulated in hypoxia, followed by an increase of CAIX expression (Fig. 11 a and b, which is published as supporting information on the PNAS web site). Moreover, we functionally assessed the angiogenic potential of first- and high-generation tumors in a rat aortic ring assay (Fig. 5e). Endothelial cell sprouting was evident only from aortic rings that received conditioned medium from high-generation tumor spheroids. Conditioned media from first-generation tumor spheroids induced no outgrowth of endothelial cells during the observation period of 11 days, suggesting that first-generation tumors do not secrete the necessary amounts of angiogenic factors to trigger angiogenesis.
Discussion
Malignant gliomas are the most common cancers in the brain and remain difficult to cure despite advances in surgery and adjuvant therapy. Recent studies have identified tumor cell subpopulations that might be responsible for tumor initiation and progression. Cancer stem cells have been identified in leukemias and breast, prostate, and brain cancer (38–44). In some cases, these tumor-initiating cells can be distinguished from the non-tumor-initiating cancer cells based on cell surface marker expression. For instance, it has been found that only CD44+/CD24−/lineage− breast cancer cells form new tumors in animals (45). Similarly, CD133 has been proposed as a cancer stem cell marker in brain cancers (46). However, we established tumors in vivo from GBM-derived spheroids that contained nestin+/GFAP+/CD133− cells. This discrepancy may be due to different culture conditions because we cultured our biopsy material in serum-containing medium. The implanted tumor spheroids developed tumors with a stem-like, nonangiogenic and highly invasive phenotype. The first-generation tumors mediated a fulminant fatal disease course, and 7 of 10 specimens produced this phenotype. Two other specimens developed into highly invasive tumors with a predominantly normal vasculature, and only one biopsy produced contrast enhancement. Although the cellular program mediating the nonangiogenic phenotype is possibly a remnant of fetal development that lies dormant during normal tumorigenesis, the program may be reactivated to drive tumor progression in a clinical setting when patients are treated with angiogenic inhibitors. In contrast to the dormant tumors that become malignant only after the onset of angiogenesis (21), our results challenge the current view of malignant tumor growth as an angiogenesis-dependent process.
Despite the fact that the nonangiogenic phenotype recapitulates developmental signaling pathways and expresses stem cell markers, it is not clear whether these cells are derived from transformed neural stem cells, from stem cell fusion events (47), or from otherwise restricted subpopulations within the tumor. The genetic similarities between the different tumor phenotypes, as demonstrated by almost identical array CGH profiles, do not support a major involvement of clonal selection, but suggest that transcriptional regulation mediates the phenotypes observed. Furthermore, it has been shown that an astrocytoma cell line became more invasive after knocking out the HIF-1α gene (48).
In later generations, transition to a vascular tumor phenotype is mediated by cells where the Ras-signaling pathway is activated. Thus, the capacity for tumor growth is neither limited to a genetic subclone nor to a certain cell phenotype, but is shared between groups of phenotypically diverse cells, where some are characterized by a diffuse growth pattern and others by angiogenesis. Accordingly, the uncoupling of invasion and angiogenesis, represented by the stem-like cancer cells and the cells derived from them respectively, points at two different mechanisms that drive tumor progression. Although the mechanism behind the phenotypic shift is not fully understood, HIF-1α expression seems to be triggered by hypoxia, because it was not constitutively expressed by high-generation tumor spheroids cultured under normoxic conditions. The results showing that both phenotypes can mediate a fulminant disease course suggest that even a 100%-effective therapy directed toward one of the biological entities (either invasion or angiogenesis) will not cure the cancer. Cancer treatment strategies need to pursue both the invasive stem-like cancer cells and angiogenic targets. A major challenge will be to design therapies that target the stem-like cancer cells without destroying the normal stem cell pools that are needed to maintain normal tissue function.
Materials and Methods
Cell Culture and in Vitro Assays.
Biopsy spheroids were prepared as described (49). After 1–2 weeks in culture, spheroids with diameters between 200 and 300 μm were selected for intracerebral implantation.
In Vivo Experiments.
Nude immunodeficient rats (Han: rnu/rnu Rowett) were fed a standard pellet diet and were provided with water ad libitum. All procedures were approved by The National Animal Research Authority. Biopsy spheroids were stereotactically implanted into the right brain hemisphere, and the rats were killed when symptoms developed.
Immunohistochemistry.
After deparaffinization, all sections were boiled in citrate buffer, pH 6.2, for 20 min, except for the von Willebrand staining, where the sections were treated with proteinase K (DAKO, Glostrup, Denmark) for 10 min. Sections were then treated with protein-blocking solution (DAKO) for 10 min, and the primary antibody was incubated for 45 min at room temperature, washed four times, incubated for 35 min with En Vision+ Systems polymer-conjugated secondary antibody (DAKO), washed four times, and finally incubated with DAB for 5 min.
Transmission Electron Microscopy.
The rats were perfusion fixed, and the brains were removed and embedded in Epon 812, followed by ultrathin sectioning in preparation for electron microscopy.
Hypoxia Experiment.
Spheroids were cultured at 37°C with 5% CO2, 94% N2, and 1%O2 for 16 h in a Mini Galaxy incubator (RS Biotech, Ayrshire, Scotland, U.K.).
Western Blotting.
Cerebrospinal fluid was run on SDS/PAGE by using NuPage precast gels (Invitrogen, Carlsbad, CA). After blotting, the nitrocellulose membrane was blocked for 30 min at room temperature and incubated overnight at 4°C in buffer (TBS with 0.1% Tween 20, 5% milk powder) containing anti-VEGF-A diluted 1:100 (Abcam, Cambridge, U.K.), anti-HIF1α diluted 1:100 (BD Biosciences, San Diego, CA), or anti-GAPDH diluted 1:2,000 (Abcam). The primary antibody was detected by using an HRP-conjugated goat anti-rabbit/mouse secondary antibody (Immunotech, Fullerton, CA) diluted 1:2,500. Extraction of protein from cultured spheroids was done by washing in PBS two times and homogenizing in lysis buffer by sonication twice for 15 sec by using Sonics Vibra Cell (Cole–Parmer Instruments, Vernon Hills, IL). Whole lysate was used for subsequent analysis. Twenty micrograms of protein was applied in each well.
Protein Kinase and Phosphosite Screening.
The procedure is described in refs. 50 and 51). The following screens were performed: KPKS-1.2A, KPKS-1.2B, KPSS-2.1, KPSS-4.1, and KPSS-1.3. For details, see the Kinexus (Advent Software, San Francisco, CA) home page www.kinexus.com.
Quantitative RT-PCR.
cDNA was generated by using the iScript cDNA synthesis kit according to the manufacturers instructions (Bio-Rad, Hercules, CA). Each reaction is in triplicate on the plate, and a similar plate was repeated three times. The reactions were performed by using iQ SYBR green Supermix reagents kit (Bio-Rad), and the PCR was run on a BioRad iCycle detection system (Bio-Rad).
Gene-Expression Analysis.
Single-stranded cDNA was reverse transcribed from 2 μg of total RNA and T7 RNA polymerase promoter-containing double-stranded cDNAs, and T7 RNA polymerase-amplified RNAs (cRNAs) were generated according to the T7 Megakit protocol (Ambion, Austin, TX) as described (52).
Agilent DNA Microarrays.
The Agilent 16,000-oligonucleotide cDNA microarrays were processed as described (53).
ABI1700 DNA Oligonucleotide Microarrays.
The Human Genome Survey Microarray, Chemiluminescence Detection kit, Applied Biosystems Chemiluminescent RT-IVT Labeling kit, and Applied Biosystems 1700 Chemiluminescent Microarray Analyzer was used as recommended.
Bioinformatic Analysis of DNA Microarray Data.
In total, six hybridizations were performed, two for each platform. The result files from the three different image-processing software programs were all imported into the analysis software J-Express (54). Controls and flagged spots were removed. J-Express is available at www.molmine.com.
Array CGH.
To determine the copy number across all chromosomes, we did comparative genomic hybridizations on whole-genome arrays of 2,400 chromosomally mapped BAC clones (Hum.Array1.14) following methods described in ref. 55.
Supporting Information.
For more information, see Supporting Materials and Methods, which is published as supporting information on the PNAS web site.
Supplementary Material
Acknowledgments
We thank Aina Johannessen, Linda Vabø, and Tore-Jacob Raa for technical assistance. This work was supported by the Norwegian Cancer Society, the Norwegian Research Council, Innovest AS, Helse-Vest, Haukeland University Hospital, the Bergen Translational Research Program, the Centre Recherche de Public Santé Luxembourg, and the European Commission 6th Framework Program Contract 504743.
Abbreviations
- CGH
comparative genomic hybridization
- GBM
glioblastoma.
Footnotes
The authors declare no conflict of interest.
References
- 1.Eisterer W, Jiang X, Bachelot T, Pawliuk R, Abramovich C, Leboulch P, Hogge D, Eaves C. Mol Ther. 2002;5:352–359. doi: 10.1006/mthe.2002.0573. [DOI] [PubMed] [Google Scholar]
- 2.Garber K. Nat Biotechnol. 2002;20:1067–1068. doi: 10.1038/nbt1102-1067. [DOI] [PubMed] [Google Scholar]
- 3.Brem S, Grossman SA, Carson KA, New P, Phuphanich S, Alavi JB, Mikkelsen T, Fisher JD. Neuro-oncol. 2005;7:246–253. doi: 10.1215/S1152851704000869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Akella NS, Twieg DB, Mikkelsen T, Hochberg FH, Grossman S, Cloud GA, Nabors LB. J Magn Reson Imaging. 2004;20:913–922. doi: 10.1002/jmri.20202. [DOI] [PubMed] [Google Scholar]
- 5.Gagner JP, Law M, Fischer I, Newcomb EW, Zagzag D. Brain Pathol. 2005;15:342–363. doi: 10.1111/j.1750-3639.2005.tb00119.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hansma AH, Broxterman HJ, van der Horst I, Yuana Y, Boven E, Giaccone G, Pinedo HM, Hoekman K. Ann Oncol. 2005;16:1695–1701. doi: 10.1093/annonc/mdi318. [DOI] [PubMed] [Google Scholar]
- 7.Wedam SB, Low JA, Yang SX, Chow CK, Choyke P, Danforth D, Hewitt SM, Berman A, Steinberg SM, Liewehr DJ, et al. J Clin Oncol. 2006;24:769–777. doi: 10.1200/JCO.2005.03.4645. [DOI] [PubMed] [Google Scholar]
- 8.Marshall E. Science. 2002;295:2198–2199. doi: 10.1126/science.295.5563.2198. [DOI] [PubMed] [Google Scholar]
- 9.Kunkel P, Ulbricht U, Bohlen P, Brockmann MA, Fillbrandt R, Stavrou D, Westphal M, Lamszus K. Cancer Res. 2001;61:6624–6628. [PubMed] [Google Scholar]
- 10.O'Donnell A, Padhani A, Hayes C, Kakkar AJ, Leach M, Trigo JM, Scurr M, Raynaud F, Phillips S, Aherne W, et al. Br J Cancer. 2005;93:876–883. doi: 10.1038/sj.bjc.6602797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bouscary D, Legros L, Tulliez M, Dubois S, Mahe B, Beyne-Rauzy O, Quarre MC, Vassilief D, Varet B, Aouba A, et al. Br J Haematol. 2005;131:609–618. doi: 10.1111/j.1365-2141.2005.05817.x. [DOI] [PubMed] [Google Scholar]
- 12.Engebraaten O, Bjerkvig R, Pedersen PH, Laerum OD. Int J Cancer. 1993;53:209–214. doi: 10.1002/ijc.2910530206. [DOI] [PubMed] [Google Scholar]
- 13.Ignatova TN, Kukekov VG, Laywell ED, Suslov ON, Vrionis FD, Steindler DA. Glia. 2002;39:193–206. doi: 10.1002/glia.10094. [DOI] [PubMed] [Google Scholar]
- 14.Dvorak P, Dvorakova D, Hampl A. FEBS Lett. 2006;580:2869–2874. doi: 10.1016/j.febslet.2006.01.095. [DOI] [PubMed] [Google Scholar]
- 15.Englund U, Fricker-Gates RA, Lundberg C, Bjorklund A, Wictorin K. Exp Neurol. 2002;173:1–21. doi: 10.1006/exnr.2001.7750. [DOI] [PubMed] [Google Scholar]
- 16.Hurelbrink CB, Armstrong RJ, Dunnett SB, Rosser AE, Barker RA. Eur J Neurosci. 2002;15:1255–1266. doi: 10.1046/j.1460-9568.2002.01959.x. [DOI] [PubMed] [Google Scholar]
- 17.Aboody KS, Brown A, Rainov NG, Bower KA, Liu S, Yang W, Small JE, Herrlinger U, Ourednik V, Black PM, et al. Proc Natl Acad Sci USA. 2000;97:12846–12851. doi: 10.1073/pnas.97.23.12846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Visted T, Enger PO, Lund-Johansen M, Bjerkvig R. Front Biosci. 2003;8:e289–304. doi: 10.2741/1026. [DOI] [PubMed] [Google Scholar]
- 19.Brabletz T, Jung A, Spaderna S, Hlubek F, Kirchner T. Nat Rev Cancer. 2005;5:744–749. doi: 10.1038/nrc1694. [DOI] [PubMed] [Google Scholar]
- 20.Holash J, Maisonpierre PC, Compton D, Boland P, Alexander CR, Zagzag D, Yancopoulos GD, Wiegand SJ. Science. 1999;284:1994–1998. doi: 10.1126/science.284.5422.1994. [DOI] [PubMed] [Google Scholar]
- 21.Naumov GN, Bender E, Zurakowski D, Kang SY, Sampson D, Flynn E, Watnick RS, Straume O, Akslen LA, Folkman J, Almog N. J Natl Cancer Inst. 2006;98:316–325. doi: 10.1093/jnci/djj068. [DOI] [PubMed] [Google Scholar]
- 22.Carmeliet P. Oncology 69 (Suppl) 2005;3:4–10. doi: 10.1159/000088478. [DOI] [PubMed] [Google Scholar]
- 23.Folkman J. N Engl J Med. 1971;285:1182–1186. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 24.Ribatti D. Br J Haematol. 2005;128:303–309. doi: 10.1111/j.1365-2141.2004.05291.x. [DOI] [PubMed] [Google Scholar]
- 25.Ribatti D, Vacca A. Curr Cancer Drug Targets. 2005;5:573–578. doi: 10.2174/156800905774932806. [DOI] [PubMed] [Google Scholar]
- 26.Collins VP. J Neurol Neurosurg Psychiatry. 2004;75(Suppl 2):ii2–ii11. doi: 10.1136/jnnp.2004.040337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu L, Backlund LM, Nilsson BR, Grander D, Ichimura K, Goike HM, Collins VP. J Mol Med. 2005;83:917–926. doi: 10.1007/s00109-005-0700-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karcher S, Steiner HH, Ahmadi R, Zoubaa S, Vasvari G, Bauer H, Unterberg A, Herold-Mende C. Int J Cancer. 2006;118:2182–2189. doi: 10.1002/ijc.21648. [DOI] [PubMed] [Google Scholar]
- 29.Kleihues P, Ohgaki H. Neuro-oncol. 1999;1:44–51. doi: 10.1093/neuonc/1.1.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Engebraaten O, Hjortland GO, Hirschberg H, Fodstad O. J Neurosurg. 1999;90:125–132. doi: 10.3171/jns.1999.90.1.0125. [DOI] [PubMed] [Google Scholar]
- 31.Mahesparan R, Read TA, Lund-Johansen M, Skaftnesmo KO, Bjerkvig R, Engebraaten O. Acta Neuropathol (Berlin) 2003;105:49–57. doi: 10.1007/s00401-002-0610-0. [DOI] [PubMed] [Google Scholar]
- 32.Shields AF, Grierson JR, Dohmen BM, Machulla HJ, Stayanoff JC, Lawhorn-Crews JM, Obradovich JE, Muzik O, Mangner TJ. Nat Med. 1998;4:1334–1336. doi: 10.1038/3337. [DOI] [PubMed] [Google Scholar]
- 33.Villa A, Snyder EY, Vescovi A, Martinez-Serrano A. Exp Neurol. 2000;161:67–84. doi: 10.1006/exnr.1999.7237. [DOI] [PubMed] [Google Scholar]
- 34.Okabe M, Imai T, Kurusu M, Hiromi Y, Okano H. Nature. 2001;411:94–98. doi: 10.1038/35075094. [DOI] [PubMed] [Google Scholar]
- 35.Schultz C, Lemke N, Ge S, Golembieski WA, Rempel SA. Cancer Res. 2002;62:6270–6277. [PubMed] [Google Scholar]
- 36.Rich JN, Hans C, Jones B, Iversen ES, McLendon RE, Rasheed BK, Dobra A, Dressman HK, Bigner DD, Nevins JR, West M. Cancer Res. 2005;65:4051–4058. doi: 10.1158/0008-5472.CAN-04-3936. [DOI] [PubMed] [Google Scholar]
- 37.Shi Q, Bao S, Maxwell JA, Reese ED, Friedman HS, Bigner DD, Wang XF, Rich JN. J Biol Chem. 2004;279:52200–52209. doi: 10.1074/jbc.M409630200. [DOI] [PubMed] [Google Scholar]
- 38.Yuan X, Curtin J, Xiong Y, Liu G, Waschsmann-Hogiu S, Farkas DL, Black KL, Yu JS. Oncogene. 2004;23:9392–9400. doi: 10.1038/sj.onc.1208311. [DOI] [PubMed] [Google Scholar]
- 39.Reya T, Morrison SJ, Clarke MF, Weissman IL. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 40.Marx J. Science. 2003;301:1308–1310. doi: 10.1126/science.301.5638.1308. [DOI] [PubMed] [Google Scholar]
- 41.Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB. Cancer Res. 2003;63:5821–5828. [PubMed] [Google Scholar]
- 42.Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Cancer Res. 2005;65:10946–10951. doi: 10.1158/0008-5472.CAN-05-2018. [DOI] [PubMed] [Google Scholar]
- 43.Nakano I, Kornblum HI. Pediatr Res. 2006;59:54R–58R. doi: 10.1203/01.pdr.0000203568.63482.f9. [DOI] [PubMed] [Google Scholar]
- 44.Bonnet D. Cell Prolif. 2005;38:357–361. doi: 10.1111/j.1365-2184.2005.00353.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Proc Natl Acad Sci USA. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 47.Bjerkvig R, Tysnes BB, Aboody KS, Najbauer J, Terzis AJ. Nat Rev Cancer. 2005;5:899–904. doi: 10.1038/nrc1740. [DOI] [PubMed] [Google Scholar]
- 48.Blouw B, Song H, Tihan T, Bosze J, Ferrara N, Gerber HP, Johnson RS, Bergers G. Cancer Cell. 2003;4:133–146. doi: 10.1016/s1535-6108(03)00194-6. [DOI] [PubMed] [Google Scholar]
- 49.Bjerkvig R, Tonnesen A, Laerum OD, Backlund EO. J Neurosurg. 1990;72:463–475. doi: 10.3171/jns.1990.72.3.0463. [DOI] [PubMed] [Google Scholar]
- 50.Wang J, Laschinger C, Zhao XH, Mak B, Seth A, McCulloch CA. Biochem Biophys Res Commun. 2005;330:123–130. doi: 10.1016/j.bbrc.2005.02.140. [DOI] [PubMed] [Google Scholar]
- 51.Lin HJ, Hsieh FC, Song H, Lin J. Br J Cancer. 2005;93:1372–1381. doi: 10.1038/sj.bjc.6602862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Halvorsen OJ, Oyan AM, Bo TH, Olsen S, Rostad K, Haukaas SA, Bakke AM, Marzolf B, Dimitrov K, Stordrange L, et al. Int J Oncol. 2005;26:329–336. [PubMed] [Google Scholar]
- 53.Gjertsen BT, Oyan AM, Marzolf B, Hovland R, Gausdal G, Doskeland SO, Dimitrov K, Golden A, Kalland KH, Hood L, Bruserud O. J Hematother Stem Cell Res. 2002;11:469–481. doi: 10.1089/15258160260090933. [DOI] [PubMed] [Google Scholar]
- 54.Dysvik B, Jonassen I. Bioinformatics. 2001;17:369–370. doi: 10.1093/bioinformatics/17.4.369. [DOI] [PubMed] [Google Scholar]
- 55.Snijders AM, Nowak N, Segraves R, Blackwood S, Brown N, Conroy J, Hamilton G, Hindle AK, Huey B, Kimura K, et al. Nat Genet. 2001;29:263–264. doi: 10.1038/ng754. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}