

Abstract
Diabetes mellitus is a major risk factor for the development of vascular complications. We hypothesized that hyperglycemia decreases endothelial cell (EC) proliferation and survival via phosphatidylinositol 3-kinase (PI3k) and Akt signaling pathways. We cultured human umbilical vein ECs (HUVEC) in 5, 20, or 40 mM d-glucose. Cells grown in 5, 20, and 40 mM mannitol served as a control for osmotic effects. We measured EC proliferation for up to 15 days. We assessed apoptosis by annexin V and propidium iodide staining and flow cytometry, analyzed cell lysates obtained on culture day 8 for total and phosphorylated PI3k and Akt by Western blot analysis, and measured Akt kinase activity using a GSK fusion protein. HUVEC proliferation was also tested in the presence of pharmacological inhibitors of PI3k-Akt (wortmannin and LY294002) and after transfection with a constitutively active Akt mutant. ECs in media containing 5 mM d-glucose (control) exhibited log-phase growth on days 7–10. d-Glucose at 20 and 40 mM significantly decreased proliferation versus control (P < 0.05 for both), whereas mannitol did not impair EC proliferation. Apoptosis increased significantly in HUVEC exposed to 40 mM d-glucose. dGlucose at 40 mM significantly decreased tyrosine-phosphorylated PI3k, threonine 308-phosphorylated-Akt, and Akt activity relative to control 5 mM d-glucose. Pharmacological inhibition of PI3k-Akt resulted in a dose-dependent decrease in EC proliferation. Transfection with a constitutively active Akt mutant protected ECs by enhancing proliferation when grown in 20 and 40 mM d-glucose. We conclude that d-glucose regulates Akt signaling through threonine phosphorylation of Akt and that hyperglycemia-impaired PI3k-Akt signaling may promote EC proliferative dysfunction in diabetes.
Keywords: endothelial cell proliferation, diabetes mellitus, phosphatidylinositol 3-kinase, protein kinase B, Akt, endothelial cell apoptosis
diabetes mellitus is a major risk factor for the development of vascular complications leading to a threefold increase in death relative to nondiabetic patients (21). Complications characteristically involve both macrovascular and microvascular circulations in several organs (8a, 43a). Microvascular complications include proliferative retinopathy, autonomic neuropathy, and nephropathy (9, 24, 35). However, macrovascular complications such as ischemic heart disease (45), peripheral vascular disease (36), and thromboembolic stroke (26) are the major contributors to morbidity and mortality in diabetes (4, 15). Endothelial cell (EC) injury and proliferative dysfunction are considered to be the initial events in the development of atherosclerosis (2), postangioplasty restenosis (23, 25), plaque erosion, and thromboembolism (2), which are contributors to macrovascular complications. However, the mechanisms by which diabetes effects EC dysfunction remain poorly identified. The central importance of hyperglycemia in this process is becoming increasingly evident (8a, 43a). Population studies show that higher levels of blood glucose are associated with an incremental risk of cardiovascular disease (4). Because poor glycemic control is associated with increased vascular complications, it is possible that deficient EC proliferation and survival may be secondary to high glucose concentrations.
Insulin sequentially activates the insulin receptor, phosphatidylinositol 3-kinase (PI3k), and Akt (12, 37, 47) through phosphorylation at threonine 308 (Thr308) and serine 473 (Ser473) (10). High glucose concentrations may modulate post-receptor insulin signaling in myocytes, lipocytes, and hepatocytes by reducing Akt activity resulting in the inhibition of GLUT4-mediated glucose transportation (16, 42). Uncoupling of downstream insulin signaling at PI3k-Akt in myocytes, lipocytes, and hepatocytes has been implicated in the development of insulin resistance and Type II diabetes (44). However, in ECs, PI3k-Akt signaling plays a crucial role in survival, proliferation, microvascular permeability, and angiogenesis (11, 18). It is plausible that hyperglycemia-induced uncoupling of postreceptor insulin signaling at PI3k-Akt in ECs may be responsible for the development of vascular complications in diabetes. High glucose concentration has been reported to increase EC apoptosis (30) and permeability (8). Conversely, EC proliferation has been variously reported to increase (33) or decrease (39) as a result of high glucose exposure in different studies. However, the role of PI3k-Akt signaling in the context of hyperglycemia and EC proliferation has not been fully explored. Therefore, we hypothesized that prolonged exposure of ECs to high glucose concentrations would result in reduced proliferation and survival through altered PI3k-Akt signaling.
MATERIALS AND METHODS
ECs and chemicals
Human umbilical vein ECs (HUVEC) were obtained from Clonetics (Cambrex, Walkersville, MD) and cultured in endothelial growth medium (EGM, Clonetics), containing fetal bovine serum, bovine brain extract, human epidermal growth factor, hydrocortisone, gentamicin, and amphotericin B, at 37°C in humidified 5% CO2 in air. Cells from passages 2–5 were used in this study. The d-glucose concentration in baseline EGM was 5 mM. d-Glucose, mannitol, anti-Akt polyclonal antibody, anti-PI3k polyclonal antibody, monoclonal phospho-tyrosine antibody, and anti-rabbit and anti-mouse horseradish peroxidase (HRP) antibody were purchased from Sigma Chemicals (St. Louis, MO). Anti-phospho-specific (Ser473 and Thr308) Akt polyclonal antibodies were obtained from Upstate Biotechnology (Lake Placid, NY). Annexin V-FITC and propidium iodide were obtained from Molecular Probes (Eugene, OR). Akt activity kit, including immobilized Akt IG-1 monoclonal antibody, phospho-GSK antibody, GSK-3 fusion protein, ATP, and kinase buffer was purchased from Cell Signaling Technology (Beverley, MA). Constitutively, active and inactive Akt1 mutants in pUSEamp vector were purchased from Upstate Biotechnology. LY294002 and wortmannin were obtained from Sigma Chemicals.
EC proliferation assay
The effect of elevated d-glucose on HUVEC proliferation was studied by growing cells in EGM containing d-glucose concentrations of 5 (baseline control), 20, or 40 mM. The effect of hyperosmolarity was assessed by using 5, 20, and 40 mM mannitol in the growth medium. HUVEC were plated on polystyrene 24-well plates at 103 cells/cm2. After 24 h, the medium was changed to 20 and 40 mM d-glucose or to 5, 20, and 40 mM mannitol. Thereafter, media was changed every 48 h. HUVEC proliferation was assessed on days 2, 5, 7, 8, 10, 14, and 15 to obtain complete cellular proliferation kinetics. Cell number was determined by manual Coulter counting of a trypsinized aliquot of cells as described previously (28). For subsequent experiments, cell numbers were counted on day 8 (midlog-phase of growth). Cell viability was assessed by using Trypan blue staining.
ECs apoptosis assay
HUVEC were grown in EGM containing 5 (baseline control), 20, or 40 mM d-glucose for 8 days. Five microliters of annexin V-FITC and ten microliters of propidium iodide (50 μg/ml in 1× PBS) were added to a trypsinized aliquot of HUVEC (106 cells/ml) in binding buffer (Cell Signaling). Cell necrosis and apoptosis were measured by using flow cytometry within 30 min after fluorescent labeling by using standard quantitative methods (fluorescense activated cell sorter caliber by Becton-Dickinson).
Preparation of cellular lysates and immunoblot analysis
HUVEC were cultured in media containing 5, 20, and 40 mM d-glucose or 5, 20, and 40 mM mannitol for 8 days. Cells were then washed with ice-cold PBS, and whole cell extracts were prepared by exposing the cells to modified Tris-NaCl-EDTA buffer [20 mM HEPES (pH 7.4), 1% Triton X-100, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM sodium fluoride, 1 mM sodium orthovanadate, 1 mM PMSF, 10 μg/ml leupeptin, 10 μg/ml aprotinin, and 1.5 μM pepstatin]. Protein quantification of the samples was carried out by using the bicinchoninic acid assay (Pierce, Rockford, IL). All immunoblots were standardized to the same amount of protein per well. Equal amounts of protein were applied for SDS-PAGE under reducing conditions and transferred to polyvinylidene difluoride (PVDF) membranes. Western blot analysis was carried out by incubating the membranes with antibodies to PI3k, Akt, phosphospecific (Ser473 and Thr308) Akt, and myc. The signal was visualized by using horseradish peroxidase-conjugated secondary antibodies and the enhanced chemiluminescence assay (Amersham; Piscataway, NJ). Band intensities were determined by using an IS-1000 imaging system (Alpha Innotech; San Leandro, CA).
Immunoprecipitation
HUVEC were cultured in media containing 5, 20, and 40 mM d-glucose for 8 days. Cells were then washed with ice-cold PBS, and whole cell extracts were prepared (see Preparation of cellular lysates and immunoblot analysis). The samples were diluted to a final concentration of 500 μg/ml and precleared by incubating with 20 μl of protein A-Sepharose bead slurry for 30 min at 4°C. The samples were then incubated with 0.5 μl of polyclonal PI3k antibody. The immunocomplex was captured with protein ASepharose beads slurry and resuspended by boiling in 2× sample buffer. SDS-PAGE was performed with the supernatant and transferred to the PVDF membranes. The membranes were probed for phospho-tyrosine using monoclonal phospho-tyrosine antibody.
Akt kinase activity assay
HUVEC were cultured in media containing 5, 20, and 40 mM d-glucose for 8 days. Cells were then washed with ice-cold PBS, and whole cell extracts were prepared (see Preparation of cellular lysates and immunoblot analysis). Akt was selectively immunoprecipitated from HUVEC lysates using resuspended immobilized Akt antibody slurry. The resulting immunoprecipitate was incubated with GSK-3 fusion protein in the presence of ATP and kinase buffer. This allowed Akt to phosphorylate GSK-3. Phosphorylation of GSK-3 was measured by Western blot analysis using a phospho-GSK-3α/β (Ser 21/9) antibody.
Pharmacological inhibition of PI3k-Akt
In separate experiments, HUVEC were grown in 5 mM d-glucose with increasing doses of wortmannin (1, 10, and 100 nM) or LY294002 (0.1, 1, 10, and 100 μM). Cells were counted after 8 days to determine the effect of pharmacological inhibition of PI3k-Akt on EC proliferation.
Akt transfection
HUVEC were transiently transfected with myc-tagged constitutively active (myr-Akt1) and dominant negative [Akt1 (K179M)] mutants of Akt according to the manufacturer's protocol. Successful transfection was tested with immunoblotting with anti-myc and anti-Akt antibodies. Transfected cells were allowed to grow in media containing 5, 20, and 40 mM d-glucose, and cells were counted on day 8 to determine whether transfection with constitutively active Akt could protect ECs from the effects of high glucose concentrations.
Statistical analysis
Proliferation data are means ± SE. Immunoblot data are means ± SD of band intensity relative to control. All experiments were repeated for n = 6. Statistical analysis was performed using GraphPad Prism 3.0 (San Diego, CA). Groups were analyzed for differences by one-way ANOVA followed by Tukey's test. Significance was accepted at P < 0.05.
RESULTS
High glucose concentrations and EC proliferation and survival
Baseline HUVEC proliferation in EGM containing 5 mM d-glucose followed standard growth kinetics with a clearly defined lag (0–6 days), log (7–10 days), and plateau (11–15 days) phase (Fig. 1A). Because day 8 was well within the log phase of cell proliferation, further comparisons were made on that day. The number of proliferating HUVEC on day 8 were significantly lower when cultured in 20 and 40 mM d-glucose than when cultured in 5 mM d-glucose (P < 0.01 each; Fig. 1A). HUVEC grown in EGM containing equimolar concentrations of mannitol (3, 20, and 40 mM) did not demonstrate a decrease in proliferation (Fig. 1B). Therefore, the effect of d-glucose on proliferation was not secondary to osmotic load. To ascertain whether the decreased cell counts in 20 and 40 mM d-glucose were due to reduced proliferation or increased cell death, we performed flow cytometry with propidium iodide staining. There was no significant difference in the percentage of necrotic cells when cultured in 20 or 40 mM d-glucose, relative to 5 mM d-glucose (P > 0.05 for both, Fig. 1C). These results indicate that HUVEC necrosis was not enhanced by the increasing d-glucose concentrations. Because apoptosis may occur in the absence of necrosis, we performed flow cytometry using annexin V-FITC staining to detect apoptosis (Fig. 1C). Cells cultured in 5 mM d-glucose had significantly less apoptotic cells than necrotic cells (P < 0.05). Cells cultured in 20 mM d-glucose demonstrated an increasing trend (P < 0.05), but those grown in 40 mM d-glucose presented a significantly higher percentage of apoptotic cells than those cultured in 5 mM d-glucose (2.35% apoptotic cells in 5 mM vs. 4.81% in 40 mM; P < 0.01).
Fig. 1.
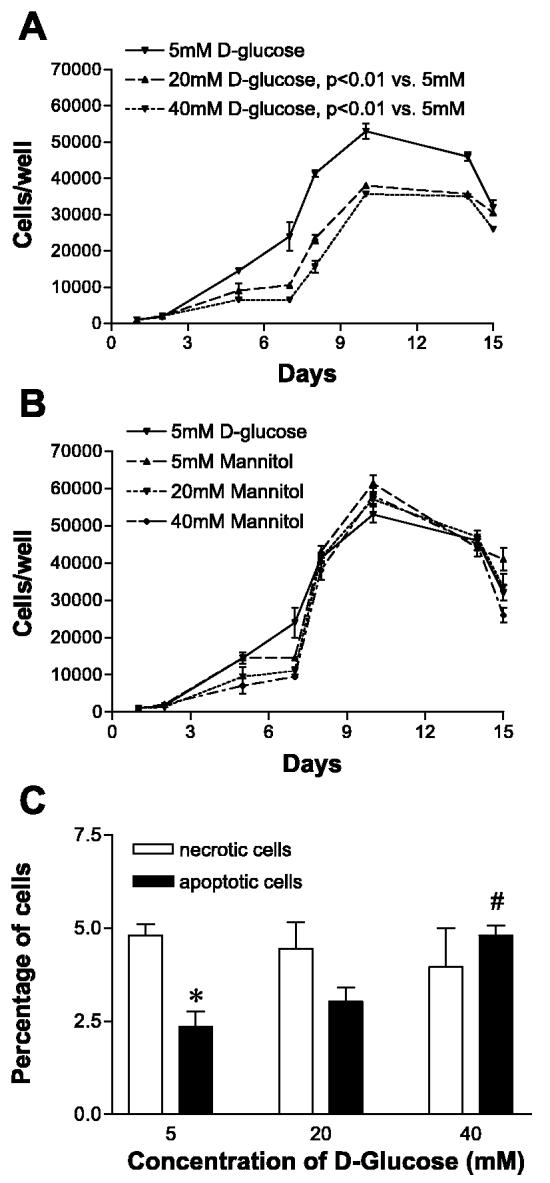
Increasing d-glucose concentrations decrease endothelial cell (EC) proliferation independent of osmotic changes or cell death. A: human umbilical vein ECs (HUVEC) were seeded at 1,000 cells/well and grown in endothelial growth medium containing baseline control (5 mM) and high (20 and 40 mM) concentrations of d-glucose. Cell counts were performed through 15 days. Log phase of growth was between days 7 and 10. Number of proliferating HUVEC on day 8 were significantly lower when cultured in 20 (P < 0.01) and 40 mM (P < 0.01) d-glucose compared with control (5 mM d-glucose). B: mannitol at 5, 20, and 40 mM did not change HUVEC proliferation. C: percentage of cells undergoing necrosis was not significantly different among groups. Relative percentage of apoptotic cells was lower than necrotic cells in 5 mM d-glucose (*P < 0.05). There was an increase in percentage of apoptotic cells in 40 mM d-glucose vs. 5 mM d-glucose (#P < 0.01).
High glucose concentrations and PI3k expression in ECs
Total PI3k was not significantly altered in HUVEC cultured in 20 or 40 mM d-glucose compared with 5 mM d-glucose (Fig. 2). The p85 subunit of PI3k undergoes tyrosine phosphorylation to become activated. To ascertain whether phosphorylated PI3k was decreased in HUVEC when cultured in 20 and 40 mM d-glucose, we performed immunoblots for tyrosine-phosphorylated proteins after immunoprecipitation of PI3k. There was a significant reduction in the levels of phosphorylated p85 subunit of PI3k in cells cultured in 40 mM d-glucose compared with 5 mM d-glucose (Fig. 2; P < 0.05). To determine whether this difference was because of a reduction in the total immunoprecipitated PI3k or a true decrease in phosphorylated PI3k, we stripped these membranes and reprobed them for total PI3k. Total PI3k was not different between groups (Fig. 2).
Fig. 2.
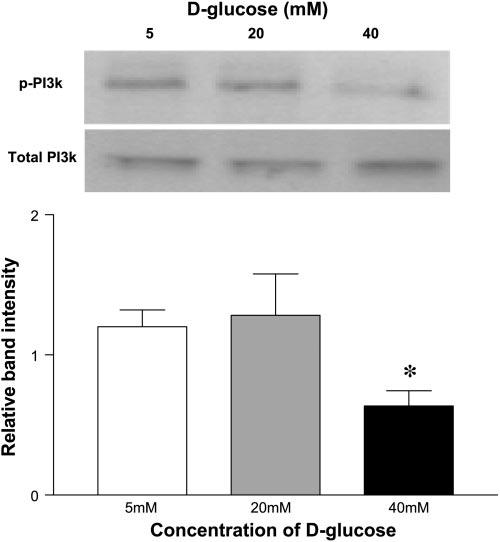
Concentration of 40 mM d-glucose decreases phospho-phosphatidylinositol 3-kinase (PI3k) expression in ECs. Immunoblots were performed for tyrosine-phosphorylated proteins after immunoprecipitation of PI3k from lysates obtained from HUVEC cultured in 5, 20, and 40 mM d-glucose. Total PI3k was evaluated by reprobing membranes. Bar graph shows the mean relative band intensity ± SE of tyrosine-phosphorylated PI3k in lysates. *P < 0.05 compared with control (5 mM d-glucose).
High glucose concentrations and Akt expression in ECs
Total Akt in HUVEC lysates remained unchanged when cultured in 5, 20, or 40 mM d-glucose and in 5, 20, or 40 mM mannitol (Fig. 3A). Akt is primarily activated by PI3k-mediated phosphorylation of its Thr308 and Ser473 residues. These phosphorylations additively activate Akt serine/threonine kinase activity. We therefore measured the levels of phosphorylated Akt at Thr308 and Ser473 by immunoblotting with their respective phospho-antibodies. Whereas the level of phosphoSer473 Akt was not significantly altered among groups (Fig. 3B), there was a significant decrease in phospho-Thr308 Akt in HUVEC cultured in 20 and 40 mM d-glucose compared with control (5 mM; P < 0.05 and P < 0.003, respectively, Fig. 3C). Exposure to 5, 20, or 40 mM mannitol did not alter the expression of phospho-Ser473 Akt or phospho-Thr308 Akt relative to 5 mM d-glucose control (Fig. 3, B and C).
Fig. 3.
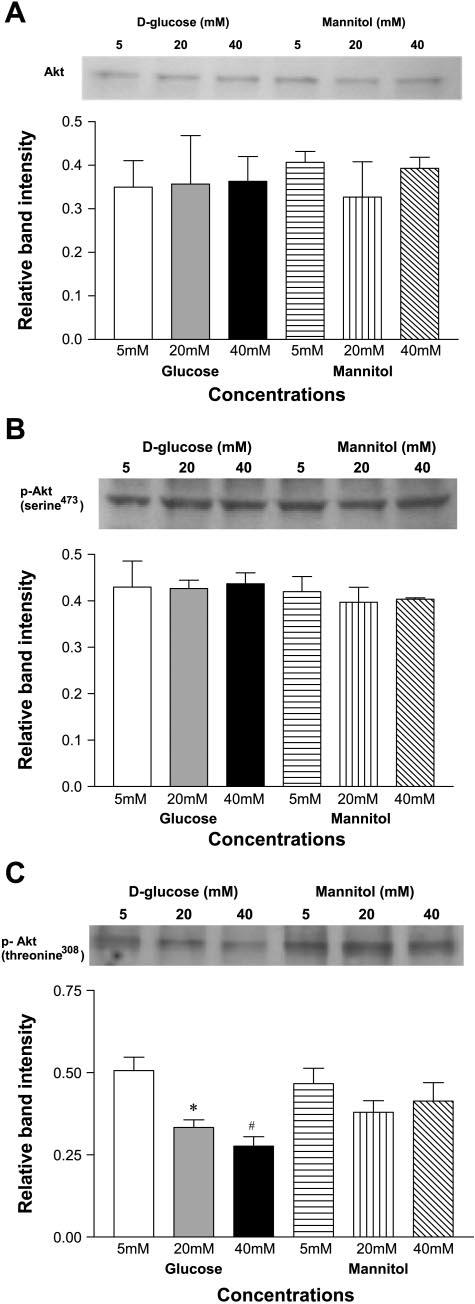
A: high d-glucose concentration does not alter total Akt expression in ECs. HUVEC were cultured in 5, 20, or 40 mM d-glucose and in 5, 20, and 40 mM mannitol. Bar graphs show the means relative band intensity ± SE of Akt in lysates. B: high d-glucose concentration does not alter expression of phosphorylated Akt at Serine 473 in ECs. C: high d-glucose concentration decreases expression of phosphorylated Akt at Threonine 308 in ECs. *P < 0.05, #P < 0.01.
High glucose concentrations and Akt activity in ECs
We performed an Akt activity assay to determine whether the quantitative differences in phosphorylated PI3k and phosphoThr308 Akt affected the activity of Akt in HUVEC grown in increasing concentrations of d-glucose. HUVEC were cultured in 5, 20, and 40 mM d-glucose, and Akt was selectively immunoprecipitated from HUVEC lysates and incubated with GSK-3 fusion protein in the presence of ATP and kinase buffer. Akt kinase activity was assessed by measuring the phosphorylation of GSK-3 by Western blot analysis using a phospho-GSK-3α/β (Ser 21/9) antibody. We found that Akt activity was markedly reduced in lysates from HUVEC cultured in 20 mM d-glucose (P < 0.05) and 40 mM d-glucose (P < 0.001; Fig. 4).
Fig. 4.
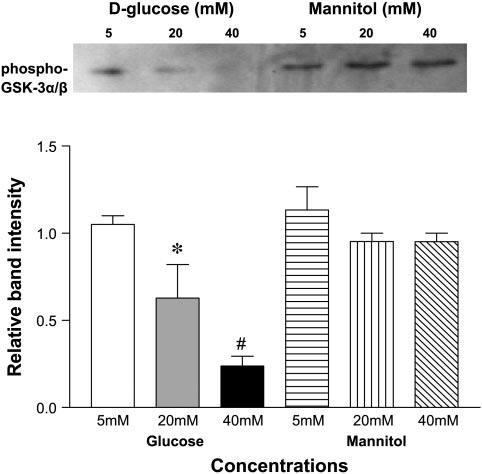
High d-glucose concentration decreases Akt activity in ECs. Akt was immunoprecipitated from HUVEC lysates using immobilized Akt antibody slurry. Immunoprecipitate was incubated with GSK-3 fusion protein in presence of ATP and kinase buffer. Phosphorylation of GSK-3 was used as a measure of Akt activity. Bar graphs show the means relative band intensity ± SE of phospho-GSK-3α/β (Ser 21/9) in lysates. *P < 0.05 and #P < 0.001 compared with control (5 mM d-glucose).
Pharmacological inhibition of PI3k-Akt and EC proliferation
We tested EC proliferation in various concentrations of pharmacological inhibitors selective for PI3k-Akt to determine whether the decrease in proliferation seen in ECs grown in high glucose concentrations could be reproduced. HUVEC were grown in normal glucose concentrations (5 mM) with increasing concentrations of wortmannin (1, 10, and 100 nM) or LY294002 (0.1, 1, 10, and 100 μM). EC proliferation was attenuated in the presence of all three concentrations of wortmannin (P < 0.05 for each dose vs. 5 mM d-glucose; Fig. 5A). Additionally, there was a dose-dependent reduction in EC proliferation when a more specific PI3k-Akt blocker LY294002 was used (P < 0.05 for 1 μM, and P < 0.001 for 10 μM vs. 5 mM d-glucose; Fig. 5B).
Fig. 5.
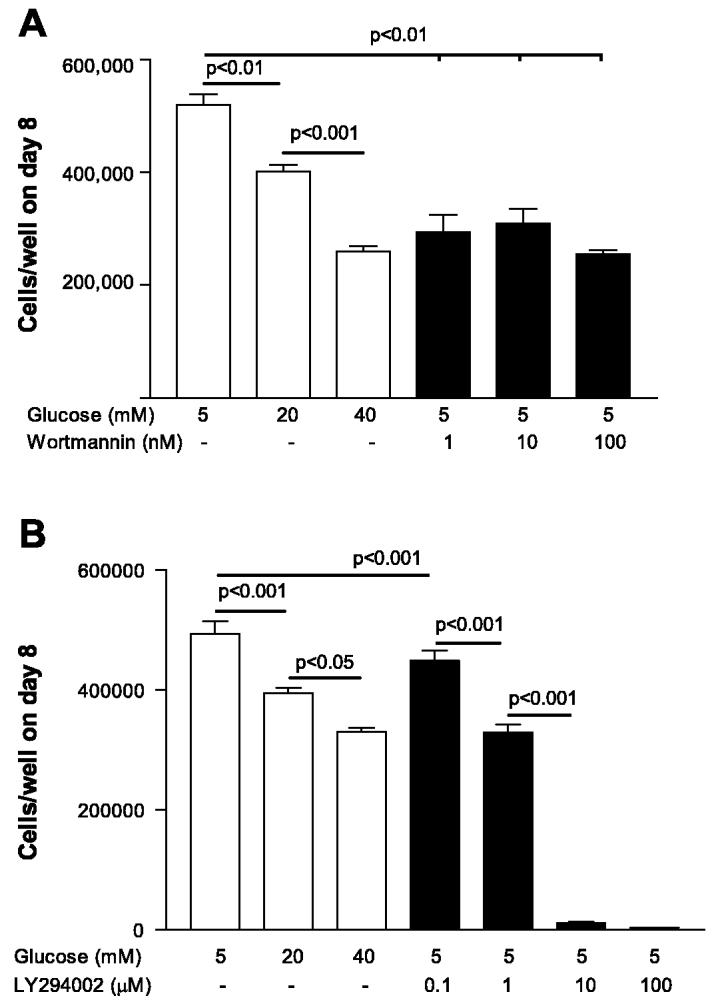
Pharmacological inhibition of PI3k-Akt inhibits EC proliferation. A: EC proliferation was attenuated by wortmannin at all concentrations (P < 0.05 vs. 5 mM d-glucose). B: LY294002 produced dose-dependent reduction in EC proliferation (P < 0.05 for 1 μM and P < 0.001 for 10 μM vs. 5 mM d-glucose).
High glucose concentrations and Akt transfection
We performed transient transfections of Akt mutants to test whether overexpression of Akt could protect ECs from the proliferation inhibition induced by exposure to high glucose concentrations. HUVEC were transiently transfected with myc-tagged constitutively active (myr-Akt1) and dominant negative [Akt1 (K179M)] mutants of Akt, and cells were counted on day 8. Transfection with an empty pUSEamp vector served as a control. Successful transfection was confirmed by immunoblotting with anti-myc and anti-Akt antibodies (Fig. 6A). Notably, endogenous Akt was not depleted in the presence of dominant negative Akt. When grown in 5 mM d-glucose, HUVEC with constitutively active Akt demonstrated significantly higher proliferation compared with nontransfected ECs and ECs transfected with empty vector or Akt−/− mutant (Fig. 6B). When grown in 20 mM d-glucose, HUVEC with constitutively active Akt demonstrated significantly higher proliferation compared with nontransfected ECs and ECs transfected with empty vector or Akt−/− mutant. Notably, the protective effect allowed Akt+/+ ECs grown in 20 mM d-glucose to proliferate, as well as nontransfected ECs grown in 5 mM d-glucose (Fig. 6C). When grown in 40 mM d-glucose, HUVEC with constitutively active Akt demonstrated significantly higher proliferation compared with nontransfected ECs and ECs transfected with empty vector or Akt−/− mutant. Notably, protection was only partial, allowing Akt+/+ ECs grown in 40 mM d-glucose to proliferate more than nontransfected ECs grown in 40 mM d-glucose but not as much as nontransfected ECs grown in 5 mM d-glucose (Fig. 6D).
Fig. 6.
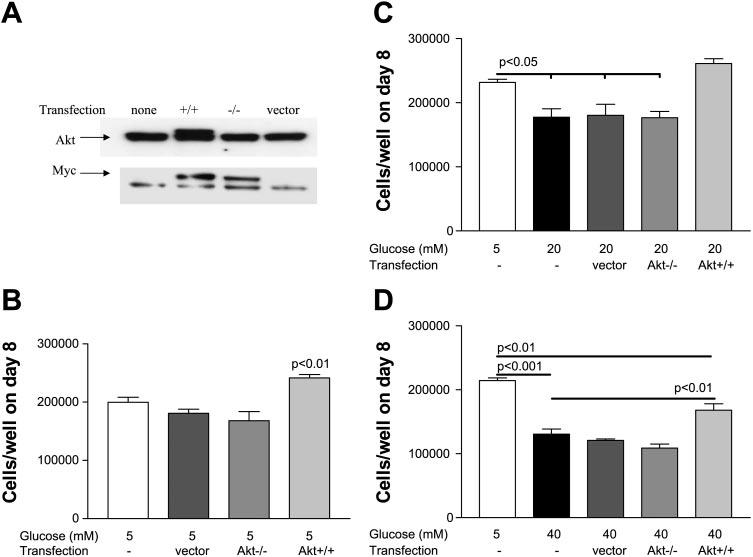
Overexpression of active Akt increases proliferation and protects EC from deleterious effects of elevated d-glucose. A: successful transfection was confirmed by immunoblotting with anti-myc and anti-Akt antibodies. B: constitutively active Akt+/+ significantly increased proliferation of HUVEC grown in 5 mM d-glucose compared with nontransfected EC, and EC transfected with empty vector or Akt−/− mutant. C: Akt+/+ protected against proliferative impairment induced by 20 mM d-glucose and restored proliferation to control levels (5 mM d-glucose). D: Akt+/+ protected against proliferative impairment induced by 40 mM d-glucose but did not restore proliferation to control levels (5 mM d-glucose).
DISCUSSION
PI3k and Akt are downstream effectors of insulin signaling (12), as well as important signaling molecules in the regulation of glycogen metabolism in myocytes, lipocytes, and hepatocytes (16, 42). Uncoupling of insulin signaling at PI3k-Akt in these cell types in response to high glucose concentrations has been implicated in the pathogenesis of insulin resistance and Type II diabetes (44). However, PI3k-Akt also plays an important role in ECs by regulating angiogenesis (18), proliferation (13), microvascular permeability (29), survival (10), cellular transformation, and embryonic differentiation (41). Our results are the first to show unequivocally the role of PI3k and Akt in hyperglycemia-induced effects in ECs. Our novel findings suggest a unified mechanism through which hyperglycemia causes insulin resistance in nonvascular cells and causes proliferative dysfunction in ECs. The need for research into the role of Akt signaling in the pathophysiology of complications of diabetes mellitus has been recently emphasized (46).
Diabetes is associated with a two-to-fourfold increase in myocardial infarction (45) and stroke (26), a 15- to 40-fold increase in limb loss from lower extremity arterial occlusive disease (36), and an increase in restenosis rates after intravascular interventions (23, 25). Although diabetes is associated with several microvascular complications (9, 24, 35), the macrovascular complications are the major cause of mortality and account for 77% of hospitalizations in these patients (15). The pathogenesis of macrovascular complications in diabetes mellitus is not fully understood. Decreased EC proliferation may play a central role in the development of atherosclerosis, decreased collateral arteriogenesis around occlusive lesions (1, 17, 40), and decreased reendothelialization of injured blood vessels (23, 25). Tight control of hyperglycemia is the only currently effective method of limiting diabetic cardiovascular complications, thereby indicating that hyperglycemia contributes significantly to their pathogenesis. The results of our study are consistent with these clinical observations. We demonstrate that exposure to increasing concentrations of glucose results in diminished EC proliferation and survival. Our results are in agreement with previous reports that high glucose concentrations reduced proliferation in HUVEC (7), human pulmonary artery ECs (31), human dermal microvascular ECs (20), porcine aortic ECs (14), and bovine retinal ECs (27). Conversely, other investigators report that hyperglycemia enhances proliferation of retinal microvascular ECs (34, 38) and of HUVEC (39). These conflicting reports may be related to differences in species or experimental conditions (passage number, extent of growth arrest before glucose exposure, concentration and duration of glucose exposure, and whether the experiments were controlled for insulin in the media). The observation that retinal ECs have increased proliferation with high glucose is consistent with the clinical finding that diabetic patients have high rates of proliferative retinopathy (24). However, the effect of diabetes on other vascular beds (coronary and lower extremity vascular disease) may be related to deficient EC proliferation and survival.
In our experiments, we used 5 mM d-glucose as a control because this is the normal concentration in EGM and is equivalent to the serum glucose concentration in normal individuals (90 mg/dl). Both 20 and 40 mM d-glucose were used as challenge doses because they reflected increasing serum glucose concentrations in diabetic individuals (360 and 720 mg/dl, respectively). Cells were exposed to prolonged hyperglycemia (8 days), beyond what has been usually reported in the literature, to better mimic the actual disease process and to act according to the proliferation kinetics demonstrating log-phase growth at this time. There is evidence that short-term (72 h) exposure to high glucose concentrations resulted in reduced EC proliferation and concomitantly increased p38-mitogen activated protein kinase (p38 MAPK) expression (33). Another study noted increased transforming growth factor-β expression under similar circumstances (32). If confirmed with additional transfection studies, the above data would emphasize that several pathways may contribute to the observed proliferation changes. Our data indicate an important role of PI3k-Akt because pharmacological inhibition resulted in inhibition of EC proliferation. We provide additional evidence that overexpression of Akt increases proliferation and protects ECs from the deleterious effect of hyperglycemia. Akt overexpression restored proliferation of ECs grown in 20 mM d-glucose to normal levels and significantly improved proliferation ECs grown in 40 mM d-glucose but not to normal levels.
EC apoptosis contributes to the pathogenesis of atherosclerosis and mediates plaque cap erosion and thromboembolization (2). We demonstrate that a significantly higher percentage of cells cultured in 40 mM d-glucose underwent early apoptosis, whereas cell necrosis was not enhanced. Decreased Akt activity in cells cultured in 40 mM d-glucose could be responsible for the increased percentage of apoptotic cells in this group, relative to control (5 mM d-glucose). However, the overall low levels of apoptosis even at 40 mM d-glucose exposure (4.81%) indicate that apoptosis was not contributing significantly to the reduced cell numbers observed on day 8. Therefore, we conclude that hyperglycemia affects cellular proliferation more than apoptosis under these conditions.
Increasing concentrations of mannitol did not alter proliferation in ECs. This indicates that high glucose concentrations do not act through an osmotic challenge but more likely through altered cellular signaling. Because PI3k and Akt play a crucial role in insulin signaling as well as in microvascular permeability, angiogenesis, EC proliferation, and EC survival, we hypothesized that defects in PI3k and Akt signaling were responsible for the altered proliferation and apoptosis in our experiments. Our experiments demonstrate an inverse relationship between phosphorylation of Akt and d-glucose concentration. Additionally, Akt kinase activity was significantly diminished in ECs cultured in 20 and 40 mM d-glucose relative to control (5 mM). Because phospho-Thr308 Akt was significantly lower in HUVEC cultured in 20 and 40 mM d-glucose, this may be the mechanism by which high glucose causes a decrease in Akt kinase activity. Furthermore, Ser473 Akt phosphorylation did not vary significantly among the treatment groups. These findings suggest that the decreased Akt kinase activity in HUVEC cultured in 20 and 40 mM d-glucose is due to a selective and specific change in threonine phosphorylation at site 308 and not serine phosphorylation at site 473 on the Akt molecule. We interpret our data to indicate that Thr308 phosphorylation of Akt is the most glucose-sensitive element of the signaling cascade. Our interpretation considers the experimental results that PI3k and serine-phosphorylation of Akt were not affected by 20 mM d-glucose, whereas Thr308 was affected. It is important to note that Akt activity was reduced at 20 and 40 mM d-glucose, supporting an important role for Thr308 phosphorylation in the enzymatic activity.
It is becoming increasingly clear that hyperglycemia is responsible for diabetic microvascular complications. Two multicenter trials have demonstrated that intensive control of hyperglycemia can reduce the incidence or progression of microvascular complications of diabetes (8a, 43a). However, there is limited data with respect to the mechanisms of the macrovascular complications of diabetes. Our study suggests that hyperglycemic conditions would cause reduced proliferation and survival in vascular ECs. It also provides novel insights into an endothelial signaling dysfunction (reduced PI3k and Akt activity) that may explain these changes in diabetic patients. These observations linking PI3k-Akt activity to hyperglycemia-induced EC-proliferation defects have great clinical relevance because restoration of insulin signaling is a major therapeutic intervention being tested in current clinical trials (19). Downstream insulin signaling through PI3k-Akt may be blocked in diabetics through hyperglycemia-induced activation of PKC-βII. Ruboxistaurin (LY333531), an inhibitor of PKC-βII (19), has been shown to prevent and/or reverse diabetic microvascular complications such as retinopathy (3), nephropathy (43), and neuropathy (22). If PI3k-Akt signaling defects contribute to diabetic macrovascular complications, then therapeutic restoration of activity may have clinical significance. We suggest that upregulation of PI3k and Akt signaling pathways within the endothelium should be considered as a target for future therapeutic modalities in protecting the diabetic patient from macrovascular complications.
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
This work was supported by a grant from the American College of Surgeons and National Heart, Lung, and Blood Institute Grant 5RO1-HL-70634.
REFERENCES
- 1.Abaci A, Oguzhan A, Kahraman S, Eryol NK, Unal S, Arinc H, Ergin A. Effect of diabetes mellitus on formation of coronary collateral vessels. Circulation. 1999;99:2239–2242. doi: 10.1161/01.cir.99.17.2239. [DOI] [PubMed] [Google Scholar]
- 2.Berk BC, Abe JI, Min W, Surapisitchat J, Yan C. Endothelial atheroprotective and anti-inflammatory mechanisms. Ann NY Acad Sci. 2001;947:93–109. doi: 10.1111/j.1749-6632.2001.tb03932.x. discussion 109–111. [DOI] [PubMed] [Google Scholar]
- 3.Comer GM, Ciulla TA. Pharmacotherapy for diabetic retinopathy. Curr Opin Ophthalmol. 2004;15:508–518. doi: 10.1097/01.icu.0000143685.60479.3b. [DOI] [PubMed] [Google Scholar]
- 4.Coutinho M, Gerstein HC, Wang Y, Yusuf S. The relationship between glucose and incident cardiovascular events. A metaregression analysis of published data from 20 studies of 95,783 individuals followed for 12.4 years. Diabetes Care. 1999;22:233–240. doi: 10.2337/diacare.22.2.233. [DOI] [PubMed] [Google Scholar]
- 7.Curcio F, Ceriello A. Decreased cultured endothelial cell proliferation in high glucose medium is reversed by antioxidants: new insights on the pathophysiological mechanisms of diabetic vascular complications. In Vitro Cell Dev Biol. 1992;28A:787–790. doi: 10.1007/BF02631069. [DOI] [PubMed] [Google Scholar]
- 8.Dang L, Seale JP, Qu X. Reduction of high glucose and phorbolmyristate-acetate-induced endothelial cell permeability by protein kinase C inhibitors LY379196 and hypocrellin A. Biochem Pharmacol. 2004;67:855–864. doi: 10.1016/j.bcp.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 8a.The Diabetes Control and Complications Trial Research Group The effect of intensive treatment of diabetes on the development, and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med. 1993;329:977–986. doi: 10.1056/NEJM199309303291401. [DOI] [PubMed] [Google Scholar]
- 9.Eastman R. Neuropathy in diabetes. In: Harris MI, Cowie CC, Stern MP, Boyo EJ, Reiber GE, Bennett PH, editors. Diabetes in America. 2nd ed. National Institutes of Health; Bethesda, MD: 1995. pp. 339–348. chapt. 15. [Google Scholar]
- 10.Franke TF, Kaplan DR, Cantley LC. PI3K: downstream AKTion blocks apoptosis. Cell. 1997;88:435–437. doi: 10.1016/s0092-8674(00)81883-8. [DOI] [PubMed] [Google Scholar]
- 11.Franke TF, Kaplan DR, Cantley LC, Toker A. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science. 1997;275:665–668. doi: 10.1126/science.275.5300.665. [DOI] [PubMed] [Google Scholar]
- 12.Galetic I, Andjelkovic M, Meier R, Brodbeck D, Park J, Hemmings BA. Mechanism of protein kinase B activation by insulin/insulinlike growth factor-1 revealed by specific inhibitors of phosphoinositide 3-kinase–significance for diabetes and cancer. Pharmacol Ther. 1999;82:409–425. doi: 10.1016/s0163-7258(98)00071-0. [DOI] [PubMed] [Google Scholar]
- 13.Gousseva N, Kugathasan K, Chesterman CN, Khachigian LM. Early growth response factor-1 mediates insulin-inducible vascular endothelial cell proliferation and regrowth after injury. J Cell Biochem. 2001;81:523–534. doi: 10.1002/1097-4644(20010601)81:3<523::aid-jcb1066>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 14.Graier WF, Grubenthal I, Dittrich P, Wascher TC, Kostner GM. Intracellular mechanism of high d-glucose-induced modulation of vascular cell proliferation. Eur J Pharmacol. 1995;294:221–229. doi: 10.1016/0014-2999(95)00534-x. [DOI] [PubMed] [Google Scholar]
- 15.Harris MI. Summary: descriptive epidemiology. In: Harris MI, Cowie CC, Stern MP, Boyo EJ, Reiber GE, Bennett PH, editors. Diabetes in America. 2nd ed. National Institutes of Health; Bethesda, MD: 1995. pp. 1–14. chapt. 1. [Google Scholar]
- 16.Hernandez R, Teruel T, Lorenzo M. Akt mediates insulin induction of glucose uptake and up-regulation of GLUT4 gene expression in brown adipocytes. FEBS Lett. 2001;494:225–231. doi: 10.1016/s0014-5793(01)02353-5. [DOI] [PubMed] [Google Scholar]
- 17.Ito WD, Arras M, Scholz D, Winkler B, Htun P, Schaper W. Angiogenesis but not collateral growth is associated with ischemia after femoral artery occlusion. Am J Physiol Heart Circ Physiol. 1997;273:H1255–H1265. doi: 10.1152/ajpheart.1997.273.3.H1255. [DOI] [PubMed] [Google Scholar]
- 18.Jiang BH, Zheng JZ, Aoki M, Vogt PK. Phosphatidylinositol 3-kinase signaling mediates angiogenesis and expression of vascular endothelial growth factor in endothelial cells. Proc Natl Acad Sci USA. 2000;97:1749–1753. doi: 10.1073/pnas.040560897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jirousek MR, Gillig JR, Gonzalez CM, Heath WF, McDonald JH, III, Neel DA, Rito CJ, Singh U, Stramm LE, Melikian-Badalian A, Baevsky M, Ballas LM, Hall SE, Winneroski LL, Faul MM. (S)-13-[(dimethylamino)methyl]-10,11,14,15-tetrahydro-4,9:16, 21-dimetheno-1H, 13H-dibenzo[e,k]pyrrolo[3,4-h][1,4,13]oxadiazacyclohexadecene-1,3(2H)-d ione ( LY333531) and related analogues: isozyme selective inhibitors of protein kinase C beta. J Med Chem. 1996;39:2664–2671. doi: 10.1021/jm950588y. [DOI] [PubMed] [Google Scholar]
- 20.Kamal K, Du W, Mills I, Sumpio BE. Antiproliferative effect of elevated glucose in human microvascular endothelial cells. J Cell Biochem. 1998;71:491–501. doi: 10.1002/(sici)1097-4644(19981215)71:4<491::aid-jcb4>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 21.Kannel WB, McGee DL. Diabetes and cardiovascular disease. The Framingham study. JAMA. 1979;241:2035–2038. doi: 10.1001/jama.241.19.2035. [DOI] [PubMed] [Google Scholar]
- 22.Kim H, Sasaki T, Maeda K, Koya D, Kashiwagi A, Yasuda H. Protein kinase Cbeta selective inhibitor LY333531 attenuates diabetic hyperalgesia through ameliorating cGMP level of dorsal root ganglion neurons. Diabetes. 2003;52:2102–2109. doi: 10.2337/diabetes.52.8.2102. [DOI] [PubMed] [Google Scholar]
- 23.Kip KE, Faxon DP, Detre KM, Yeh W, Kelsey SF, Currier JW. Coronary angioplasty in diabetic patients. The National Heart, Lung, and Blood Institute Percutaneous Transluminal Coronary Angioplasty Registry. Circulation. 1996;94:1818–1825. doi: 10.1161/01.cir.94.8.1818. [DOI] [PubMed] [Google Scholar]
- 24.Klein R, Klein BEK. Vision disorders in diabetes. In: Harris MI, Cowie CC, Stern MP, Boyo EJ, Reiber GE, Bennett PH, editors. Diabetes in America. 2nd ed. National Institutes of Health; Bethesda, MD: 1995. pp. 293–338. chapt. 14. [Google Scholar]
- 25.Kornowski R, Lansky AJ. Current perspectives on interventional treatment strategies in diabetic patients with coronary artery disease. Catheter Cardiovasc Interv. 2000;50:245–254. doi: 10.1002/(sici)1522-726x(200006)50:2<245::aid-ccd22>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 26.Kuller LH. Stroke and diabetes. In: Harris MI, Cowie CC, Stern MP, Boyo EJ, Reiber GE, Bennett PH, editors. Diabetes in America. 2nd ed. National Institutes of Health; Bethesda, MD: 1995. pp. 449–456. chapt. 20. [Google Scholar]
- 27.La Selva M, Beltramo E, Pagnozzi F, Bena E, Molinatti PA, Molinatti GM, Porta M. Thiamine corrects delayed replication and decreases production of lactate and advanced glycation end-products in bovine retinal and human umbilical vein endothelial cells cultured under high glucose conditions. Diabetologia. 1996;39:1263–1268. doi: 10.1007/s001250050568. [DOI] [PubMed] [Google Scholar]
- 28.Lal BK, Saito S, Pappas PJ, Padberg FT, Jr, Cerveira JJ, Hobson RW, II, Durán WN. Altered proliferative responses of dermal fibroblasts to TGF-beta1 may contribute to chronic venous stasis ulcer. J Vasc Surg. 2003;37:1285–1293. doi: 10.1016/s0741-5214(02)75295-6. [DOI] [PubMed] [Google Scholar]
- 29.Lal BK, Varma S, Pappas PJ, Hobson RW, II, Durán WN. VEGF increases permeability of the endothelial cell monolayer by activation of PKB/akt, endothelial nitric-oxide synthase, and MAP kinase pathways. Microvasc Res. 2001;62:252–262. doi: 10.1006/mvre.2001.2338. [DOI] [PubMed] [Google Scholar]
- 30.Liu B, Bhat M, Nagaraj RH. AlphaB-crystallin inhibits glucose-induced apoptosis in vascular endothelial cells. Biochem Biophys Res Commun. 2004;321:254–258. doi: 10.1016/j.bbrc.2004.06.151. [DOI] [PubMed] [Google Scholar]
- 31.Liu W, Schoenkerman A, Lowe WL., Jr. Activation of members of the mitogen-activated protein kinase family by glucose in endothelial cells. Am J Physiol Endocrinol Metab. 2000;279:E782–E790. doi: 10.1152/ajpendo.2000.279.4.E782. [DOI] [PubMed] [Google Scholar]
- 32.McGinn S, Poronnik P, King M, Gallery ED, Pollock CA. High glucose and endothelial cell growth: novel effects independent of auto-crine TGF-β1 and hyperosmolarity. Am J Physiol Cell Physiol. 2003;284:C1374–C1386. doi: 10.1152/ajpcell.00466.2002. [DOI] [PubMed] [Google Scholar]
- 33.McGinn S, Saad S, Poronnik P, Pollock CA. High glucose-mediated effects on endothelial cell proliferation occur via p38 MAP kinase. Am J Physiol Endocrinol Metab. 2003;285:E708–E717. doi: 10.1152/ajpendo.00572.2002. [DOI] [PubMed] [Google Scholar]
- 34.Morisaki N, Watanabe S, Fukuda K, Saito Y. Angiogenic interaction between retinal endothelial cells and pericytes from normal and diabetic rabbits, and phenotypic changes of diabetic cells. Cell Mol Biol (Noisy-le-grand) 1999;45:67–77. [PubMed] [Google Scholar]
- 35.Nelson RG, Knowler WC, Pettitt DJ, Bennett PH. Kidney diseases in diabetes. In: Harris MI, Cowie CC, Stern MP, Boyo EJ, Reiber GE, Bennett PH, editors. Diabetes in America. 2nd ed. National Institutes of Health; Bethesda, MD: 1995. pp. 349–400. chapt. 16. [Google Scholar]
- 36.Palumbo PJ, Melton LJ., III . Peripheral vascular disease and diabetes. In: Harris MI, Cowie CC, Stern MP, Boyo EJ, Reiber GE, Bennett PH, editors. Diabetes in America. 2nd ed. National Institutes of Health; Bethesda, MD: 1995. pp. 401–408. chapt. 17. [Google Scholar]
- 37.Ravichandran LV, Esposito DL, Chen J, Quon MJ. Protein kinase C-zeta phosphorylates insulin receptor substrate-1 and impairs its ability to activate phosphatidylinositol 3-kinase in response to insulin. J Biol Chem. 2001;276:3543–3549. doi: 10.1074/jbc.M007231200. [DOI] [PubMed] [Google Scholar]
- 38.Rymaszewski Z, Szymanski PT, Abplanalp WA, Myatt L, Di Salvo J, Cohen RM. Human retinal vascular cells differ from umbilical cells in synthetic functions and their response to glucose. Proc Soc Exp Biol Med. 1992;199:183–191. doi: 10.3181/00379727-199-43345. [DOI] [PubMed] [Google Scholar]
- 39.Sank A, Wei D, Reid J, Ertl D, Nimni M, Weaver F, Yellin A, Tuan TL. Human endothelial cells are defective in diabetic vascular disease. J Surg Res. 1994;57:647–653. doi: 10.1006/jsre.1994.1195. [DOI] [PubMed] [Google Scholar]
- 40.Schaper W, Buschmann I. Collateral circulation and diabetes. Circulation. 1999;99:2224–2226. doi: 10.1161/01.cir.99.17.2224. [DOI] [PubMed] [Google Scholar]
- 41.Shioi T, McMullen JR, Kang PM, Douglas PS, Obata T, Franke TF, Cantley LC, Izumo S. Akt/protein kinase B promotes organ growth in transgenic mice. Mol Cell Biol. 2002;22:2799–2809. doi: 10.1128/MCB.22.8.2799-2809.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tremblay F, Lavigne C, Jacques H, Marette A. Defective insulin-induced GLUT4 translocation in skeletal muscle of high fat-fed rats is associated with alterations in both Akt/protein kinase B and atypical protein kinase C (zeta/lambda) activities. Diabetes. 2001;50:1901–1910. doi: 10.2337/diabetes.50.8.1901. [DOI] [PubMed] [Google Scholar]
- 43.Tuttle KR, Anderson PW. A novel potential therapy for diabetic nephropathy and vascular complications: protein kinase C beta inhibition. Am J Kidney Dis. 2003;42:456–465. doi: 10.1016/s0272-6386(03)00741-8. [DOI] [PubMed] [Google Scholar]
- 43a.UK Prospective Diabetes Study Group Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment, and risk of complications in patients with type 2 diabetes (U.KPDS 33) Lancet. 1998;352:837–853. [PubMed] [Google Scholar]
- 44.Vosseller K, Wells L, Lane MD, Hart GW. Elevated nucleocytoplasmic glycosylation by O-GlcNAc results in insulin resistance associated with defects in Akt activation in 3T3-L1 adipocytes. Proc Natl Acad Sci USA. 2002;99:5313–5318. doi: 10.1073/pnas.072072399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wingard DL, Barrett-Conner E. Heart disease and diabetes. In: Harris MI, Cowie CC, Stern MP, Boyo EJ, Reiber GE, Bennett PH, editors. Diabetes in America. 2nd ed. National Institutes of Health; Bethesda, MD: 1995. pp. 429–448. chapt. 19. [Google Scholar]
- 46.Zdychova J, Komers R. Emerging role of Akt kinase/protein kinase B signaling in pathophysiology of diabetes and its complications. Physiol Res. 2005;54:1–16. doi: 10.33549/physiolres.930582. [DOI] [PubMed] [Google Scholar]
- 47.Zeng G, Nystrom FH, Ravichandran LV, Cong LN, Kirby M, Mostowski H, Quon MJ. Roles for insulin receptor, PI3-kinase, and Akt in insulin-signaling pathways related to production of nitric oxide in human vascular endothelial cells. Circulation. 2000;101:1539–1545. doi: 10.1161/01.cir.101.13.1539. [DOI] [PubMed] [Google Scholar]