

Abstract
The early genetic pathway(s) triggering the pathogenesis of coronary artery disease (CAD) and myocardial infarction (MI) remain largely unknown. Here, we describe an autosomal dominant form of CAD/MI (adCAD1) that is caused by the deletion of seven amino acids in transcription factor MEF2A. The deletion disrupts nuclear localization of MEF2A, reduces MEF2A-mediated transcription activation, and abolishes synergistic activation by MEF2A and by the transcription factor GATA-1 through a dominant-negative mechanism. The MEF2A protein demonstrates strong expression in the endothelium of coronary arteries. These results identify a pathogenic gene for a familial vascular disease with features of CAD and implicate the MEF2A signaling pathway in the pathogenesis of CAD/MI.
Coronary artery disease (CAD) and its most important complication, acute myocardial infarction (MI), are leading causes of disability and death in the developed world (1). Although relatively little is known about the genetic basis of CAD/MI, multiple risk factors have been identified, including family history, hypertension, hypercholesterolemia, obesity, smoking, and diabetes (2–5). Several genome-wide linkage scans of affected sibling pairs have identified four susceptibility loci for CAD and MI (6–9), although the specific genes remain to be identified.
We studied a large family with 13 patients who displayed an autosomal dominant pattern of CAD (kindred QW1576 in Fig. 1A and Table 1). Multiple risk factors, including dyslipidemia, hypertension, and cigarette smoking, were present in some family members (table S1). Nine of the 13 patients developed acute MI (Table 1 and table S1). We carried out a genome-wide linkage scan with 382 markers that span chromosomes 1 to 22, with an average interval of 10 cM. The positive linkage was identified for marker D15S120 with a lod score (logarithm of the odds ratio for linkage) of 4.19 at a recombination fraction of 0. Further haplotype analysis with markers D15S1014, D15S212, and D15S87 verified the observed linkage (Fig. 1). These data identified a significant linkage to autosomal dominant CAD/MI on chromosome 15q26; this locus is designated as adCAD1 for the first autosomal dominant CAD and MI locus.
Fig. 1.
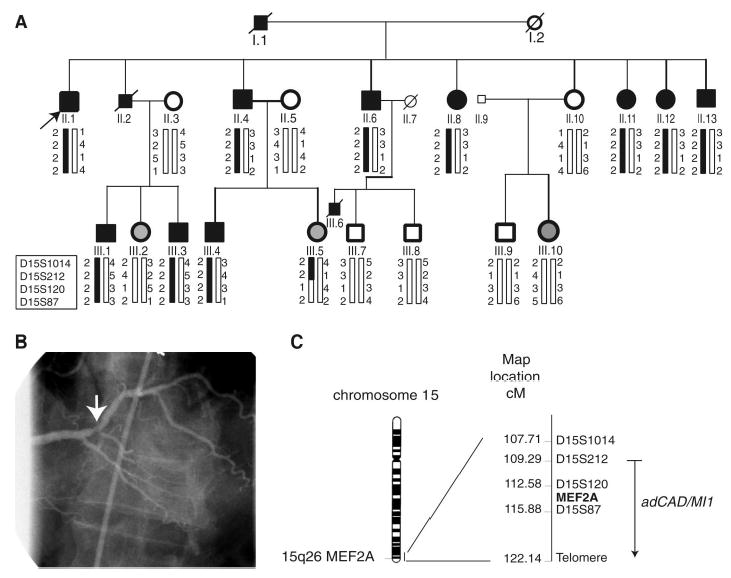
Genetic linkage of CAD/MI to chromosome 15q26 (adCAD1). (A) Pedigree structure and genotypic analysis of kindred QW1576. Individuals with CAD ( Table 1) are indicated by solid squares (males) or solid circles (females). Unaffected individuals are indicated by open symbols. Normal males under the age of 50 years or normal females under 55 years are shown in light-gray as uncertain phenotype. Deceased individuals are indicated by a slash (/). The proband is indicated by an arrow. Genotypes for markers D15S1014, D15S212, D15S120, and D15S87 are shown below each symbol. Initial linkage was identified with D15S120, which yielded a lod score of 4.19 at a recombination fraction of 0. Haplotype cosegregating with the disease is indicated by the black vertical bars. (B) Coronary angiogram from the proband, who experienced an inferior MI attributed to a plaque rupture lesion (arrow) with a 70% narrowing in the distal right coronary artery. This lesion is at a bifurcation site typical of the pattern of coronary atherosclerosis. The lesion was stented, and follow-up angiography of the site demonstrated wide patency, without any renarrowing. (C) Ideogram of chromosome 15 with the Geimsa banding pattern and localization of the adCAD1 locus. The genetic map with chromosome 15q26 markers and the location of the MEF2A gene is shown on the right.
Table 1.
Clinical characteristics of the family members in kindred QW1576, a family with CAD and MI. PTCA, percutaneous coronary angioplasty; CABG, coronary artery bypass surgery; CATH, angiogram.
| Individual ID no. | Age (years)* | Clinical diagnosis and treatment (age in years)† |
|---|---|---|
| I.1 | – | MI (45) |
| II.1 | 63 | MI (63), PTCA (63) |
| II.2 | – | MI (65), CABG (65) |
| II.4 | 81 | MI (80), CABG (65), CATH (>70% stenosis; 65) |
| II.5 | 81 | Normal |
| II.6 | 61 | MI (61), CABG (61) |
| II.8 | 77 | MI (61), CATH (>70% stenosis; 61) |
| II.10 | 72 | Normal |
| II.11 | 68 | PTCA (68) |
| II.12 | 63 | MI (63) |
| II.13 | 65 | PTCA (64) |
| III.1 | 51 | PTCA (35) |
| III.2 | 49 | Uncertain (no symptoms, female, but ≤55 years of age) |
| III.3 | 47 | PTCA (46) |
| III.4 | 49 | MI (42), CABG (42) |
| III.5 | 50 | Uncertain (no symptoms, female, but ≤55 years of age) |
| III.6 | – | MI (40) |
| III.7 | 54 | Normal (no symptoms, male, ≥50 years of age) |
| III.8 | 50 | Normal (no symptoms, male, ≥ 50 years of age) |
| III.9 | 50 | Normal (no symptoms, male, ≥50 years of age) |
| III.10 | 46 | Uncertain (no symptoms, female, but ≤55 years of age) |
Current age.
Age at time of diagnosis or treatment.
The candidate adCAD1 region contains ~93 genes (table S2), which consist of 43 known genes and 50 hypothetical genes. Among the known genes, MEF2A, which encodes a member of the myocyte enhancer factor–2 (MEF2) family of transcription factors (10), became a strong candidate, because MEF2A mRNA has been detected in blood vessels during mouse early embryogenesis (11). Furthermore, MEF2A protein expression was detected as early as day 8.5 postcoitum in cells of the embryonic vasculature (serving as an early marker for cells of vasculature). The overall expression pattern of MEF2A is similar to those of vascular endothelial growth factor receptor 2 (VEGFR2) in endothelial cell precursors and the Von Willebrand factor (an endothelial cell marker) (12). These studies suggest that MEF2A can be an early marker for vasculogenesis and may play an important role in controlling vascular morphogenesis. MEF2A contains a MADS domain at the N terminus that mediates dimerization and DNA binding; a 29–amino acid MEF2-specific domain required for high-affinity DNA binding, dimerization, and cofactor interactions; and a C terminus for transcription activation and nuclear localization (10, 13). MEF2A can interact with a variety of other protein factors, including MyoD, GATA-binding proteins (GATA), Cabin, and histone deacetylase, to regulate the expression of the downstream target genes (10, 13).
Because MEF2A was viewed as a potential candidate for causing CAD/MI susceptibility, we undertook a systematic mutational screening of the entire MEF2A gene using direct DNA sequence analysis (table S3). A 21–base pair (bp) deletion was identified in exon 11 in all ten living affected members in the family (Fig. 2), resulting in a deletion of seven amino acids of MEF2A (ΔQ440P441P442Q443P444Q445P446 or Δ7aa). These seven amino acids are conserved among MEF2A proteins in the human, the mouse (QPPQPQP), the pig (pqPQPQa), and the Chamek spider monkey (QPqQPQP). Δ7aa is located in the conserved C-terminal region between MEF2A and MEF2C (a MEF2A homolog), a location that has been demonstrated to be important for nuclear localization of these two proteins (14). Δ7aa was not identified in the family members with normal phenotypes and was absent in 119 individuals with normal angiograms, strongly suggesting that the deletion was responsible for CAD and MI in this large family.
Fig. 2.
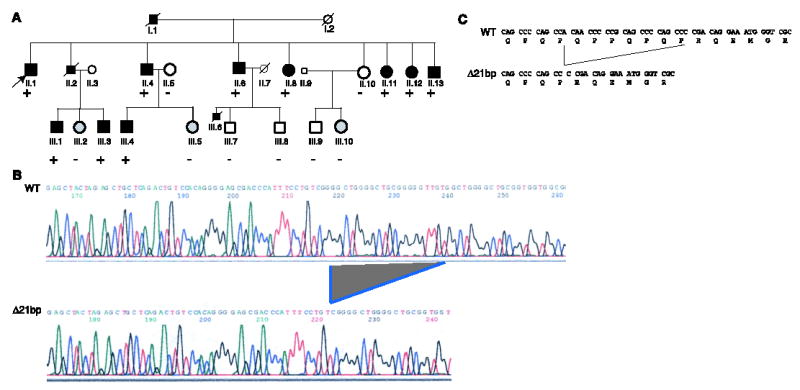
MEF2A intragenic deletion cosegregates with CAD in kindred QW1576. (A) The pedigree of kindred QW1576, showing genetic status: + indicates the presence of the 21-bp deletion of MEF2A (heterozygous); − indicates the absence of the deletion. (B) DNA sequence analysis of the wild-type ( WT ) allele and the 21-bp deletion allele (Δ21bp) of MEF2A. Sequence analysis of exon 11 of MEF2A in the proband (II.1) revealed the presence of a deletion. The wild-type and deletion alleles were separated by a 3% agarose gel and a single strand conformation polymorphism gel, purified and sequenced directly. The location of Δ21bp is indicated. (C) Δ21bp results in a deletion of seven amino acids of MEF2A (ΔQ440P441P442Q443P444Q445P446).
We hypothesized that Δ7aa may cause a conformational change of the MEF2A protein that might result in protein trafficking defects that could prevent its function as a transcription factor. To test this hypothesis, we examined the cellular localization of mutant MEF2A protein by immunofluorescence staining. As expected, wild-type MEF2A localized to the nucleus (Fig. 3, A to C). However, Δ7aa caused a marked defect in MEF2A trafficking, with a block of MEF2A entry into the nucleus (Fig. 3, A to C). The mechanism by which the MEF2A deletion mutant is retained in the cytoplasm is not clear, although the corresponding region of MEF2C has been found to play an important role in its nuclear localization (14).
Fig. 3.
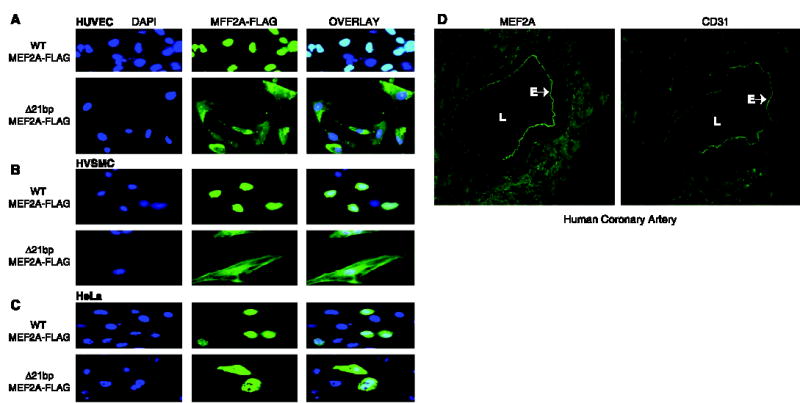
Functional characterization of wild-type and Δ7aa MEF2A proteins by immunofluorescence. (A to C) MEF2A deletion Δ7aa causes a defect in nuclear localization of the MEF2A protein in three cell types: (A) human umbilical vascular endothelial cells (HUVEC); (B) human aortic smooth muscle cells (HVSMC); and (C) HeLa cells. Cells were transfected with expression constructs for wild-type and mutant MEF2A proteins tagged with a FLAG epitope. Green, MEF2A signal; blue, nucleus. DAPI, 4′,6-diamidino-2-phenylindole. (D) Colocalization of MEF2A and CD31 (PECAM, an endothelial cell–specific marker) in the endothelium of human coronary arteries. Cryosections (6 μm thick) of human coronary arteries were immunostained with the rabbit polyclonal antiserum to MEF2A. The adjacent sections were used for immunostaining with a monoclonal antibody to CD31. L, lumen; E, endothelium.
The functional consequence of the MEF2A deletion was also explored by transcription activation assays. The atrial natriuretic factor promoter (ANF−700) can be activated by cooperation between MEF2A and GATA-1, a member of the GATA family of zinc-finger transcription factors (15). Mutant MEF2A with Δ7aa has only a third of wild-type MEF2A transcription activity (Fig. 4), indicating that the deletion is a functional mutation that reduces transcription activation by MEF2A. Coexpression of the mutant and wild-type MEF2A showed transcription activity similar to that of mutant MEF2A alone (Fig. 4). Cotransfection of MEF2A and GATA-1 showed synergistic activation of the ANF−700 promoter as reported previously (15). The synergistic activation of transcription by MEF2A and GATA-1 was abolished by coexpression of the mutant Δ7aa MEF2A with normal wild-type MEF2A (Fig. 4). Together, these data suggest that Δ7aa acts by a dominant-negative mechanism. The dominant-negative effect of the mutant Δ7aa MEF2A can be explained by the findings that MEF2A can function as a dimer (13) or in a complex with GATA factors (15).
Fig. 4.
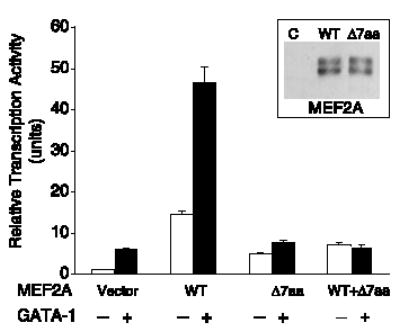
Functional characterization of wild-type and Δ7aa MEF2A proteins by transcriptional activation assays. The effect of Δ7aa on the transcription activation activity of MEF2A was analyzed in the presence or absence of GATA-1 with the ANF−700 promoter. Transcriptional activity is shown as relative luciferase activity on the y axis. The transcriptional activity of the reporter gene only (vector) was set arbitrarily to 1. WT/Δ7aa data represents coexpression of both wild-type and mutant MEF2As. Inset: Western blot analysis with rabbit polyclonal antiserum to MEF2A showed that both wild-type and mutant MEF2A were successfully expressed in transfected HeLa cells. Vector only (C) was used as a negative control. The MEF2A antibody detected two bands as previously reported (18). The data shown are from two independent experiments in triplicate and are expressed as mean ± SEM.
Immunostaining for MEF2A revealed strong MEF2A protein signal within the endothelial cell layer of coronary arteries (Fig. 3D). This pattern of expression is similar to that of CD31 [also known as platelet endothelial cell adhesion molecule (PECAM)], an endothelial cell–specific marker (Fig. 3D). Immunostaining and reverse-transcription polymerase chain reaction also detected MEF2A expression in human umbilical vascular endothelial cells (fig. S1). Collectively, our data and previous work (11, 12) implicate an important role for MEF2A in endothelial cell development and function. A genetic defect in MEF2A may lead to a defective or abnormal vascular endothelium, which may promote the diapedesis of monocytes and expose the subendothelial matrix to the genesis of atherosclerotic plaque or thrombosis.
MEF2A mRNA was also detected in cultured proliferating rat smooth muscle cells (SMCs) (16). MEF2A protein expression was detected in proliferating SMCs but not in differentiated SMCs in the rat model of arterial injury and clinical restenosis. We have also detected expression of MEF2A protein in the nuclei of proliferating human SMCs (fig. S1). Increased SMC proliferation was found to be associated with accelerated atherosclerosis (2). Therefore, the Δ7aa deletion in MEF2A may also affect the activity of proliferating SMCs, influencing the process of atherogenesis.
Mice deficient in MEF2A have been created (17). Homozygous MEF2a− − mice in the 129sv genetic background die suddenly within the first week of life; however, mice on a mixed genetic background survive (17). No structural heart defects have been detected before death, but dilation of the right ventricle was detected for the homozygous MEF2a−/− mice with sudden death at necropsy (17). The cause of the sudden death phenotype in homozygous MEF2a−/− mice remains largely unknown. Further phenotypic characterization of MEF2a−/− mice will determine whether these mice show any phenotype related to human CAD and MI. Heterozygous MEF2a+/− mice exhibit a normal phenotype. The phenotypic difference between heterozygous MEF2a+/− mice and the human patients with heterozygous MEF2A 21-bp deletion may reflect the inherent differences between the two species and/or specific effects of the seven–amino acid deletion in MEF2A.
Like other cardiovascular diseases such as long QT syndrome and hypertrophic cardiomyopathy, familial CAD/MI is likely to be genetically heterogeneous. Our linkage analysis suggests that three other large families with CAD and MI are not linked to the chromosome 15q26 adCAD1 locus. Mutational analysis failed to detect any MEF2A mutations in 50 sporadic patients with CAD/MI. MEF2A mutations may, therefore, be a rare cause of CAD and MI; however, the true prevalence rate of MEF2A mutations in the CAD/MI patient population may be revealed by future studies with large sample sizes. Future studies may also reveal whether single nucleotide polymorphisms (SNPs) with less profound changes in MEF2A may be associated with common complex CAD and MI. Because we do not have histopathologic data for our patients, it is possible that the CAD and MI manifested in this family represent a distinct pathophysiologic entity, clinically homologous but not representative of typical atherosclerosis. Finally, although unlikely, we cannot exclude the possibility that another mutation in a yet-unidentified gene, which is in disequilibrium with the MEF2A Δ7aa deletion, also contributes to the development of CAD/MI.
Our results define a genetic pathway and provide a molecular mechanism for the pathogenesis of familial CAD and MI. These findings open avenues for understanding the complex pathogenic mechanisms of CAD and MI and imply that other genes in the MEF2A signaling pathway, as well as genes regulating endothelial development and function, may be pathophysiologically relevant to this complex disease process.
Footnotes
Supporting Online Material
www.sciencemag.org/cgi/content/full/302/5650/1578/DC1
Materials and Methods
Fig. S1
Tables S1 to S3
References and Notes
References
- 1.Murray CJ, Lopez AD. Lancet. 1997;349:1498. doi: 10.1016/S0140-6736(96)07492-2. [DOI] [PubMed] [Google Scholar]
- 2.Lusis AJ. Nature. 2000;407:233. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nora JJ, et al. Circulation. 1980;61:503. doi: 10.1161/01.cir.61.3.503. [DOI] [PubMed] [Google Scholar]
- 4.Schildkraut JM, Myers RH, Cupples LA, Kiely DK, Kannel WB. Am J Cardiol. 1989;64:555. doi: 10.1016/0002-9149(89)90477-3. [DOI] [PubMed] [Google Scholar]
- 5.Wang Q, Pyeritz RE. In: Textbook of Cardiovascular Medicine. EJ Topol., editor. Vol. 3. Lippincott, Williams & Wilkins; New York: 2000. pp. 1–13. [Google Scholar]
- 6.Broeckel U, et al. Nature Genet. 2002;30:210. doi: 10.1038/ng827. [DOI] [PubMed] [Google Scholar]
- 7.Francke S, et al. Hum Mol Genet. 2001;10:2751. doi: 10.1093/hmg/10.24.2751. [DOI] [PubMed] [Google Scholar]
- 8.Harrap SB, et al. Arterioscler Thromb Vasc Biol. 2002;22:874. doi: 10.1161/01.atv.0000016258.40568.f1. [DOI] [PubMed] [Google Scholar]
- 9.Pajukanta P, et al. Am J Hum Genet. 2000;67:1481. doi: 10.1086/316902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Black BL, Molkentin JD, Olson EN. Mol Cell Biol. 1998;18:69. doi: 10.1128/mcb.18.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Edmondson DG, Lyons GE, Martin JF, Olson EN. Development. 1994;120:1251. doi: 10.1242/dev.120.5.1251. [DOI] [PubMed] [Google Scholar]
- 12.Subramanian SV, Nadal-Ginard B. Mech Dev. 1996;57:103. doi: 10.1016/0925-4773(96)00542-4. [DOI] [PubMed] [Google Scholar]
- 13.McKinsey TA, Zhang CL, Olson EN. Trends Biochem Sci. 2002;27:40. doi: 10.1016/s0968-0004(01)02031-x. [DOI] [PubMed] [Google Scholar]
- 14.Borghi S, et al. J Cell Sci. 2001;114:4477. doi: 10.1242/jcs.114.24.4477. [DOI] [PubMed] [Google Scholar]
- 15.Morin S, Charron F, Robitaille L, Nemer M. EMBO J. 2000;19:2046. doi: 10.1093/emboj/19.9.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Firulli AB, et al. Circ Res. 1996;78:196. doi: 10.1161/01.res.78.2.196. [DOI] [PubMed] [Google Scholar]
- 17.Naya FJ, et al. Nature Med. 2002;8:1303. doi: 10.1038/nm789. [DOI] [PubMed] [Google Scholar]
- 18.Mao Z, Nadal-Ginard B. J Biol Chem. 1996;271:14371. doi: 10.1074/jbc.271.24.14371. [DOI] [PubMed] [Google Scholar]
- 19.We thank E. F. Plow and J. D. Smith for critical review of the manuscript; E. N. Olson for the MEF2A expression plasmids; C. Fersky, R. Cannata, R. Kadaba, L. Li, S. Archacki, G. Shen, S. Chen, X. Tian, A. Bakos, E. Zirzow, S. Rao, S. You, and other members of Wang laboratory for help and discussion; and the study participants for their enthusiasm and support for this study. Supported by the Cleveland Clinic Foundation Center for Cardiovascular Genetics funds, a Doris Duke Innovation in Clinical Research award (Q.W. and E.J.T.), and NIH grant nos. R01 HL65630 (Q.W.) and R01 HL66251 (Q.W.).