

1. Introduction
1.1. Background
Reaction and separation are the two important aspects of organic synthesis. Traditional solution-phase synthesis emphasizes reaction, while resin-based solid-phase combinatorial synthesis and polymer-assisted solution-phase parallel synthesis emphasize separation. Fluorous synthesis, which successfully integrates solution-phase reaction conditions with the phase-tag separation, has been recently introduced as a “beadless” high-speed synthetic technology.1,2 Perfluoroalkyl chains instead of resins are used as the phase tags to facilitate the separation process. Compared to traditional solution-phase and solid-phase synthesis, fluorous synthesis has the following features:
Fluorous reactions have homogeneous solution-phase reaction kinetics;
Fluorous molecules can be separated by the fluorous separations as well as conventional methods such as chromatography, distillation, and recrystallization;
Fluorous reactions can be monitored by conventional analytical methods such as TLC, HPLC, IR and NMR;
Fluorous tags are chemically stable and have minor effect on the reactivity of the attached molecules;
The solubility of fluorous compounds in organic solvents can be fine-tuned by the fluorine content as well as temperature;
Fluorous synthesis does not need large excess of fluorous reagents to complete the reaction;
More than one fluorous reagent can be used in a single reaction;
Fluorous methods are less problematic in adaptation of literature reaction conditions than that of solid-phase synthesis;
Fluorous synthesis can be combined with other methods such as microwave reactions, supercritical CO2 reactions, and solid-phase synthesis;
Fluorous materials can be recovered after fluorous separation.
The development of fluorous technologies started in the early 1990’s. Among the pioneers, Vogt and Zhu explored the synthesis utility of temperature-dependent miscibility of fluorous solvents with organic solvents.3–4 Horvath and Rabai invented the fluorous biphasic catalysis for the recovery of catalysts.5 The Curran group and Fluorous Technologies, Inc. (FTI) developed the “light fluorous” synthesis to eliminate the use of fluorous solvents.6,7 Other major advances in fluorous technologies include mixture synthesis for making individually pure compound libraries,8 triphasic reactions which integrates the fluorous reaction and separation processes,9 and thermomorphic catalysts for fluorous solvent-free biphasic catalysis.10
Since Horvath’s seminal paper on fluorous in 1994,5 many review articles have covered different aspects of fluorous technologies at different development stages.11–27 A comprehensive and up-to-date monograph entitled “The Handbook on Fluorous Chemistry” will soon be published.28 Described in this Review are synthesis of heterocyclic systems using fluorous reagents, catalysts, scavengers, protecting groups, and tags. Application of fluorous technologies in multicomponent reactions, microwave reactions, solid-phase reactions, triphasic reactions, and mixture synthesis are also described.
1.2. Fluorous Molecules
Fluorous molecules contain a perfluorinated domain (Rf) for fluorous separation, as illustrated by the representative examples in Scheme 1. The fluorous groups are usually attached to parent molecules through a (CH2)m segment to insulate the reactive site from the electron withdrawing fluorines. A fluorous alkyl chain CnF2n+1CmH2m can be presented as Rfnhm. There are two broad classes of fluorous molecules in fluorous synthesis. The first class of fluorous molecules including reagents, scavengers, and catalysts is employed for single reaction steps. They have similar utilities as the functionalized polymeric reagents in solution-phase synthesis. The second class of fluorous molecules including reactants, protecting groups, and related tags are used to attach to the substrate and used for multistep reactions. They are similar to the polymeric linkers in solid-phase synthesis. Fluorous molecules can also be classified into two categories based on their fluorine content. Heavy fluorous molecules usually have greater than 60% of fluorine content by molecular weight. They contain two, three, or even more Rf groups to ensure good partition coefficient between fluorous and organic solvents. Light fluorous molecules contain only one or two Rf groups and the fluorine content is significantly lower than heavy fluorous molecules. The separation of light fluorous molecules can be achieved by fluorous silica gel-based solid-phase extraction or HPLC.
1.3. Fluorous Separations
The success of fluorous synthesis largely depends on the efficiency of fluorous separations. Fluorous separations are based on fluorine-fluorine interactions between the fluorous molecules and the fluorous separation media. The separation media can be fluorous solvents used for fluorous liquid/liquid extraction or fluorous silica gel used for solid-phase extraction, flash chromatography, and HPLC.22
1.3.1. Fluorous Liquid-Liquid Extractions (F-LLE)
Fluorous media are orthogonal to organic and aqueous phases. Many fluorous solvents exhibit temperature-dependent miscibility with organic solvents. This characteristic has been utilized in designing fluorous biphasic catalysis. The separation of the reaction mixture is conducted with an organic/fluorous biphasic extraction or an organic/aqueous/fluorous triphasic extraction if water-soluble materials are involved (Figure 1). Fluorous liquid-liquid extractions are targeted towards heavier fluorous molecules which have good partition coefficient between fluorous and organic solvents. Commonly used fluorous solvents are perfluoroalkanes, perfluoroethers, and perfluoroamines such as perfluorohexane (FC-72), perfluoroheptane, perfluoromethylcyclohexane (PFMC). Benzotrifluoride (BTF) is not fluorous because of relatively fluorine content. It has been widely used in fluorous synthesis as a hybrid solvent.
Figure 1.
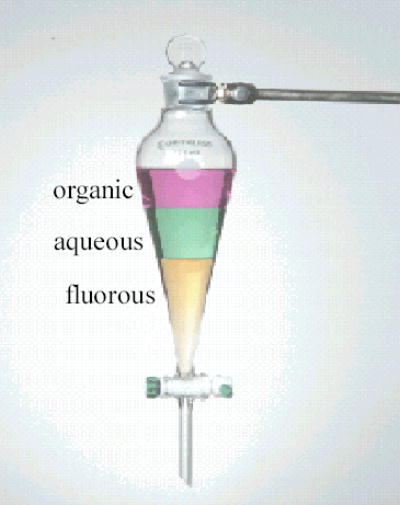
Fluorous liquid-liquid extraction
1.3.2. Fluorous Solid-Phase Extractions (F-SPE)
FluoroFlash silica gel has a bonded phase of Si(CH3)2CH2CH2C8F17.29 A reaction mixture containing fluorous molecules bearing tags such as C6F13 or C8F17 can be easily separated from the non-fluorous molecules by F-SPE. In a typical F-SPE separation, a crude reaction mixture is loaded onto the SPE cartridge with a minimum amount of organic solvent. The cartridge is then eluted with a fluorophobic solvent such as 80:20 MeOH/H2O for the nonfluorous compounds followed by a more fluorophilic solvent such as MeOH, acetone, acetonitrile, or THF for fluorous compounds. The suggested loading (amount of crude product vs silica gel) for F-SPE separation is between 5–10%. The cartridge can be conditioned for reuse. Figure 2 shows F-SPE separation of a mixture containing an organic dye (blue) and a fluorous dye (orange). Since two dyes have similar polarities, they can be separated by chromatography but not by SPE with normal or reverse-phase silica gels. Using a fluorous silica gel cartridge, however, the separation can be done by SPE. The blue dye quickly elutes with 80:20 MeOH/H2O, whereas the orange dye is retained on the cartridge until elution with 100% MeOH. Depending on the chemistry, the desired products can be the non-fluorous compounds collected in the MeOH/H2O fraction or the fluorous compounds in the MeOH fraction.30
Figure 2.

A dye demonstration of F-SPE
1.3.3. Fluorous HPLC (F-HPLC)
A fluorous mixture can be separated in a high performance mode by F-HPLC. A F-HPLC column packed with FluoroFlash silica gel (5 μm, bonded with Si(CH3)2CH2CH2C8F17) behaves much differently from a standard reverse-phase C8 or C18 column. On a fluorous HPLC column, the non-fluorous compounds have very weak retention, whereas fluorous compounds can be retained and separated in an order of increasing fluorine content.31,32 The mobile phase is usually a gradient of MeOH/H2O. The MeOH can be replaced by other solvents such as MeCN or THF. In F-HPLC separation, fluorous tags dominate the separation. The organic part of the molecules only has minor separation effect. An F-HPLC trace shown in Figure 3 demonstrates separation of a mixture of seven fluorous tagged mappicine analogs bearing different R1 and Rf groups.33
Figure 3.
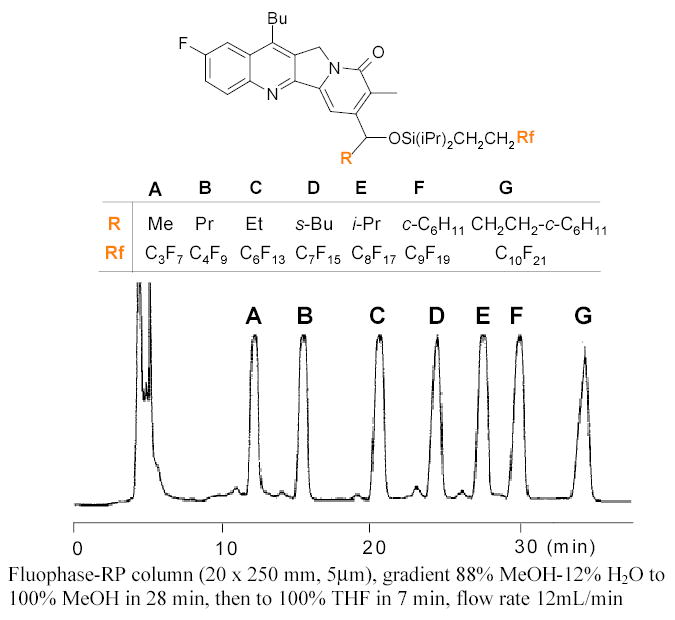
Semipreparative F-HPLC demixing of a 7-component mixture
In addition to the FluoroFlash HPLC column, FluoFix (bonded with branched C6F13 groups), FluoPhase-RP (bonded with liner C6F13 groups), and FluoPhase-PFP (bonded with perfluorophenyl groups) columns are also commercially available.34 FluoFix column developed in late 1970’s by de Galan and coworkers was originally used as a modified reverse phase column.35–38 Its special utility for fluorous compounds separation has not been fully recognized until Curran’s work almost two decades later.22 A comparison of different fluorous HPLC columns in fluorous separation has been reported.33
Recently, the Mikami and Takeuchi groups used β-cyclodextrin (β-CD) columns to separate analogs bearing different length of fluorous tags and also to separate enantiomers bearing the same fluorous tags.39–41 The longer fluorous chain (C7F15) tagged enantiomers have better resolution then those tagged with shorter ones (C3F7 or CF3) (Figure 4).
Figure 4.
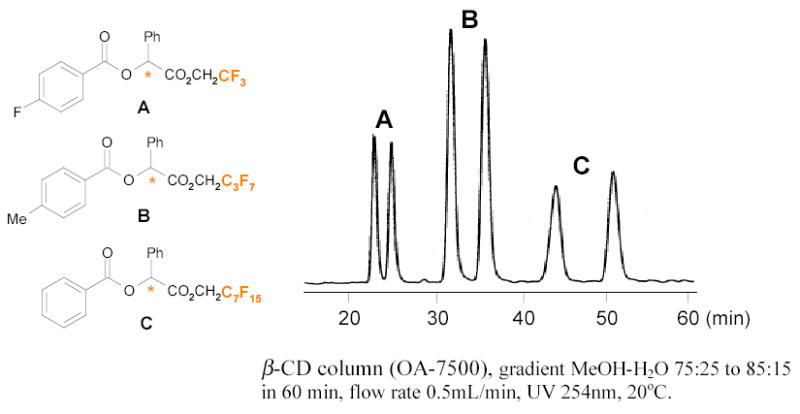
Resolution of fluorous tagged O-benzoylmadelate derivatives with a β-CD column
1.3.4. Fluorous Flash Chromatography (F-FC)
F-SPE is employed mainly for parallel synthesis. HPLC is good for small scale final product purification, but it has limitation for large scale intermediate purification. The development of F-FC has provided a scalable separation from 10 mg up to over 10 g by using different sizes of cartridges.42 A significant gap in fluorous separation techniques is plugged. Flash chromatography has much better resolution than SPE and is more scalable than HPLC. UV-triggered fraction collection provides reliable fraction cut off that gives better control on yields and purities than F-SPE. Flash chromatography systems such as those from Biotage (Horizon) and Isco (CombiFlash) are very popular in synthetic labs (Figure 5). These systems equipped with fluorous cartridges can be used for F-FC.
Figure 5.

A Biotage Horizon system with different size cartridges and Samplets
1.4. Fluorous Reaction Systems
The fluorous synthesis, which addresses both the reaction and separation issues, can be performed in biphasic, monophasic, or triphasic reaction systems.
1.4.1. Biphasic Systems
The first generation of fluorous reactions was developed in the fluorous biphasic system.5,12 Based on the temperature-dependent miscibility of fluorous solvents with organic solvents, the reaction and separation are conducted at different temperatures: monophasic at higher temperature for reactions and biphasic at lower temperature for separations (Figure 6). In many cases, the reaction is performed using minimal amount of fluorous solvent to achieve the best monophasic effect. Additional fluorous solvent is added after the reaction for biphasic separation. The biphasic system was originally designed for fluorous catalysis. It has been extended to other reactions involving heavy fluorous molecules such as reagents and scavengers. The biphasic system has good potential for relatively large scale reactions to recover fluorous components.
Figure 6.
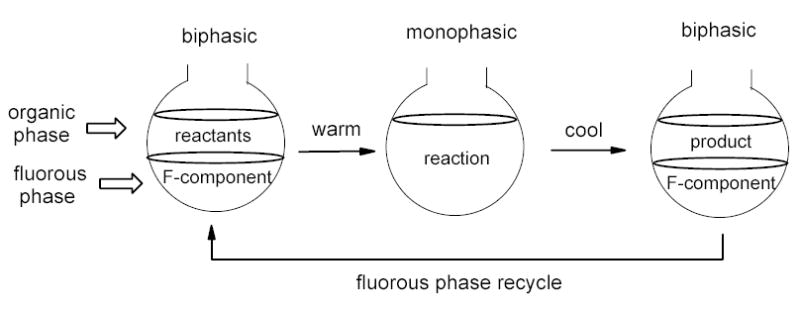
Fluorous biphasic reaction system
1.4.2. Monophasic (Fluorous Solvent-Free) Systems
Light fluorous reactions are conducted in a monophasic system with common organic solvents (Figure 7). The costly and environmentally persistent fluorous solvents can be eliminated from the reaction and separation steps. Purifications are conducted by F-SPE or F-FC for parallel synthesis and by HPLC for mixture synthesis. Since light fluorous molecules have better organic solubility and hence better reactivity than the heavy fluorous molecules, conventional solution-phase conditions can be adapted with less development effort. Fluorous solvent-free systems are commonly used for making compound libraries.6
Figure 7.
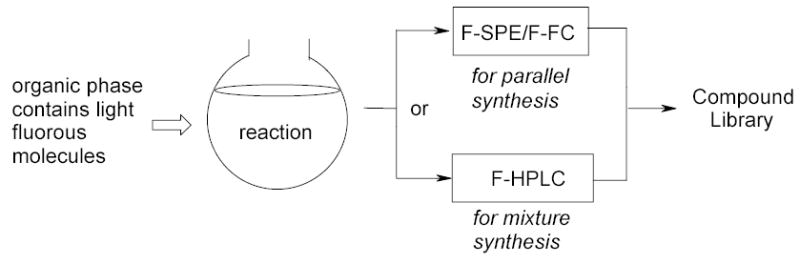
Fluorous solvent-free synthesis and separation
1.4.3. Triphasic Systems
In fluorous triphasic reactions, the reaction and separation occur simultaneously in a system with the reaction driving the separation. There are two types of applications: detagging and phase-vanishing reactions. The detagging reactions are carried out in a U-tube set up (Figure 8).9,43,44 The organic source phase and the receiving phase are separated by a fluorous phase in the middle. Fluorous molecules added to the source phase transfer to the receiving phase for detagging. At the end of a triphasic reaction, nonfluorous byproducts are retained in the source phase, the detagged product is in the receiving phase, and the fluorous tag is in the fluorous phase. In phase-vanishing reactions, fluorous solvents serve as a barrier to control the mixing hence the reaction of organic reactants and reagents.45 If one reagent (such as a halogenated one) is denser than the fluorous solvent, then the reaction can performed in a test tube set up (Figure 9). At the end of a stoichiometric reaction, the product is in the top organic phase and the bottom phase has disappeared. If all reagents and reactants are less dense than the fluorous solvent, then phase-vanishing reactions can be conducted in a U-tube set up. When the reagents are used stoichiometrically, one of the reagent/reactant phases vanishes at the end of the reaction. Fluorous triphasic reactions have good potential in chemical process and production.
Figure 8.
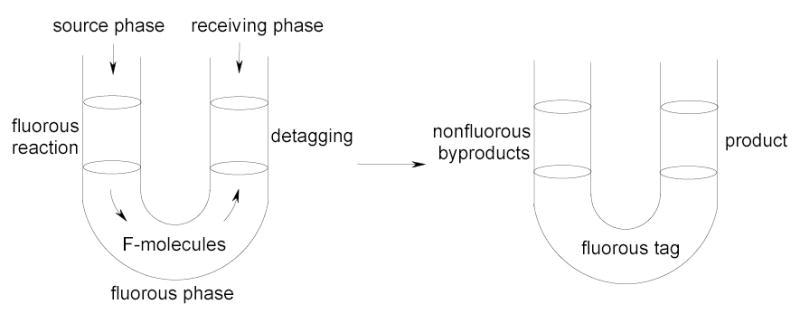
Triphasic detagging reaction
Figure 9.
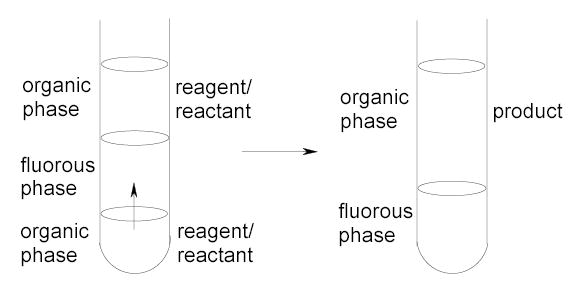
Phase vanishing reaction
1.5. Comparison of Fluorous, Solution-Phase, and Solid-Phase Syntheses
Described in this section are examples that demonstrated the value of fluorous synthesis by comparing with conventional solution-phase and solid-phase syntheses.
1.5.1. Mitsunobu Reactions
The Mitsunobu reaction efficiently forms carbon-heteroatom bonds. However, separation of byproducts derived from the azodicarboxylate and phosphine reagents usually requires chromatography. Accordingly, the Mitsunobu reaction has been the subject of much study in the strategic separation area. Initial strategies involved placing either the phosphine or the azodicarboxylate onto a solid support.46–47 These strategies still required a flash chromatography to remove the non-polymer supported byproduct, since reaction with both reagents on solid support is not possible. This limitation has led to a number of modifications to remove the non-polymer supported byproducts by scavenging or destruction.48–51 In those approaches, a second reaction is always needed before purification. The only chromatography-free Mitsunobu reaction that does not require additional reaction after the Mitsunobu transformation has been achieved by the Curran group using a fluorous DEAD reagent and a fluorous phosphine (see Section 2.2.4).52–53 The reaction was carried out in THF and the purification was conducted by a simple F-SPE.
1.5.2. Scavenging Reactions
Polymer-bound scavenging has become a popular synthetic technique. However, polymer quenching is slow and the loading levels of commercially available solid phase scavengers span a broad range. Therefore a large amount (3–5 equiv or even more) of a solid-bound scavenger is commonly employed in practice. FTI,54,55 the Lindsley group at Merck,56,57 and the Curran group58 have developed a series of light fluorous scavengers. The light fluorous scavenging occurs in a homogeneous organic solution, so it is rapid and clean. A near stoichiometric amount (or slight excess) of scavenger is commonly used. In a fluorous and polymer-supported isatoic anhydrides comparison experiment (Figure 10), reactions were conducted at room temperature using 1.0 equiv of N-phenylpiperazine and 1.5 equiv of each scavenger.59 After 60 min, 84% of the amine remained during polymer scavenging (pink line), whereas only 10% of the amine remained in fluorous scavenging (green line). Doubling the amount of polymer scavenger A to 3.0 equiv (purple line), still has 44% of the amine left after 60 min. Octyl-alkylated isatoic anhydride C was used to compare the fluorous and the non-fluorous reagents (blue line). Interestingly, the scavenging with the fluorous reagent is about 10% faster than with the non-fluorous reagent. This is most likely resulted from the electron-withdrawing effects of the fluorous tag. In another case of using thiols as electrophile scavengers,54 it was found that the fluorous scavenging is about ten times faster than the polymer scavenger despite the negative effect of the Rf group on the nucleophilicity of the thiol.
Figure 10.
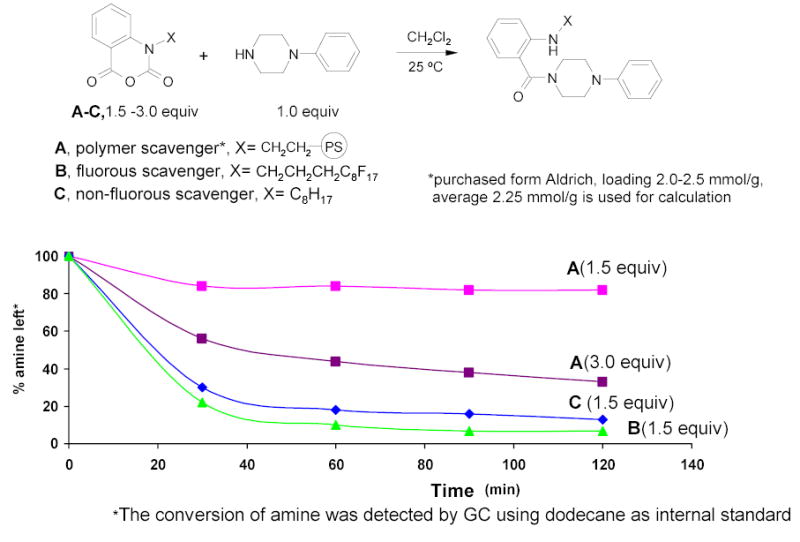
Use Different Isatoic Anhydrides as Amine Scavengers
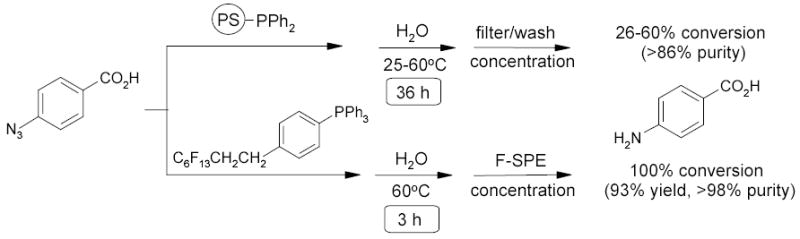
1.5.3. The Staudinger Reaction
In a comparison experiment conducted by the Lindsley group,60 polymer supported and fluorous phenyl phosphines were used to convert an azide to an amine (Scheme 2). The fluorous reaction took 3 h for 100% conversion and gave final products in greater than 98% purity after fluorous SPE, whereas the solid-supported route took 36 h for 26–60% conversion and gave final products in greater than 86% purity. The speed and purity advantages of using fluorous phenyl phosphine are clear.
In addition to three examples described above, another very promising technology to combine microwave and fluorous technologies to speed up both the reaction and separation processes is discussed in Section 2.7.2.
2. Synthesis of Heterocycles
2.1. Fluorous Ligands and Catalysts
Biphasic catalytic reactions have so far attracted the most attention of fluorous synthesis.25 A number of fluorous ligands have been synthesized but only few have been used in the synthesis of heterocycles.
2.1.1. Triphenylphosphine Ligands
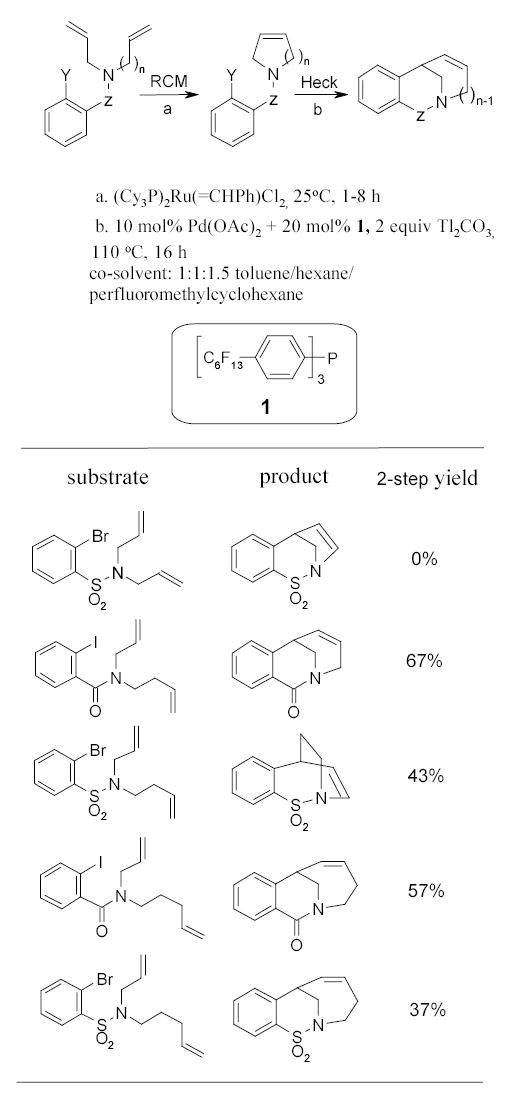
Grigg and York developed a bimetallic catalytic ring closing metathesis (RCM)/intramolecular Heck reaction sequence for making cyclic amides and sulfonamides (Scheme 3).61 Triphenylphosphine 1 was used as a ligand combined with Pd(OAc)2 for the Heck reaction. The reaction solvent was a mixture of 1:1:1.5 toluene/hexane/perfluoromethylcyclohexane. The reagents for the two step reactions were added together at the beginning of the procedure. The mixture was stirred for 1–8 h at room temp for RCM and then heated at 110 °C for 16 h for the Heck reaction.
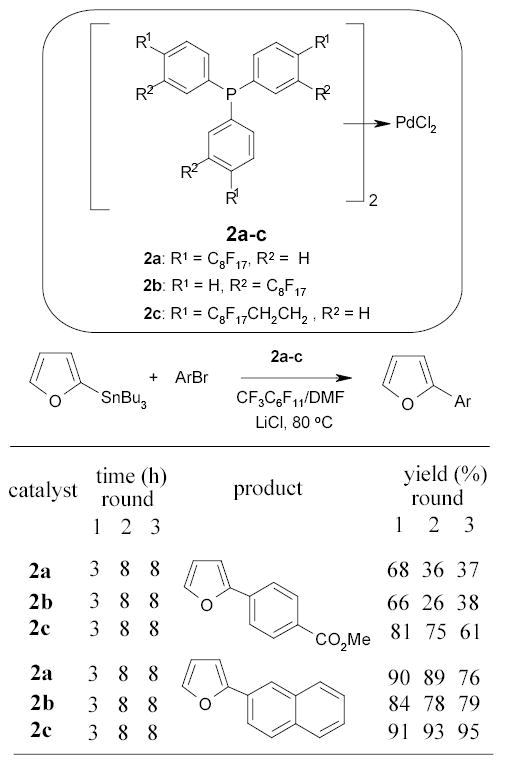
The Bannwarth group prepared several different fluorous bis-triphenylphosphane palladium complexes 2 and used them for the Stille cross-coupling reactions. Couplings of 2-bromofuran with aryltin compounds were carried out in a mixture of DMF and PFMC at 80 °C (Scheme 4).62 Catalysts were recovered and used for twice more. In another experiment, the Stille coupling was performed in supercritical CO2 instead of the biphasic solvent to improve yields.63
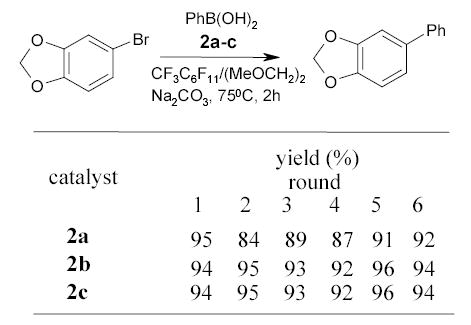
Fluorous catalysts 2a–c were also employed in the Suzuki couplings (Scheme 5).64 The catalyst recovered from the fluorous phase was reused 5 more times without significant deterioration of reaction yields.
2.1.2. Amine Ligands
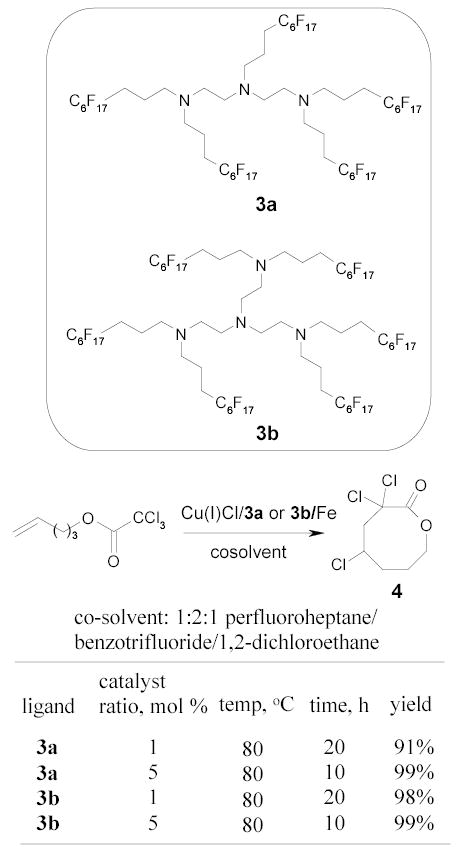
Verlhac and coworkers reported an efficient atom transfer radical reaction in the synthesis of lactones 4 (Scheme 6).65 A complex containing fluorous ligands 3a or 3b, Cu(I)Cl, and iron powder promoted the cyclization of a trichloroester. The reactions were performed in a co-solvent of 1:2:1 perfluoroheptane/BTF/1,2-dichloroethane which was almost biphasic at the reaction temperature of 80 °C. The catalyst was recovered from the fluorous layer at room temperature. The product in the organic layer was purified by flash chromatography through a silica gel plug. The turnover of the catalyst was about 100.
2.1.3. Fluorous Ruthenium Catalyst
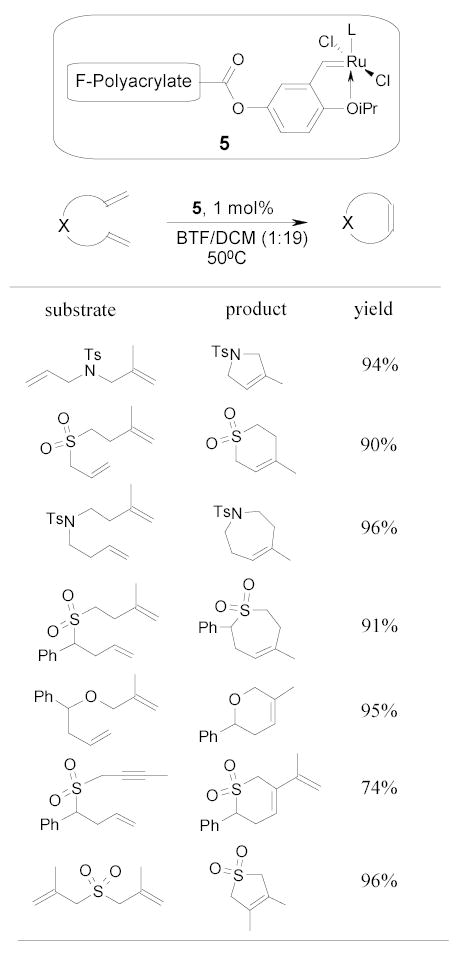
Yao and Zhang recently developed a recyclable fluorous ruthenium catalyst 5 for ring-closing metathesis.66 Reactions were conducted in a BTF/CH2Cl2 co-solvent system. The ruthenium catalyst recovered by FC-72 extraction has been reused for 6 to 20 times with very slight drop of activity (Scheme 7).
2.2. Fluorous Reagents
Fluorous reagents including organotins, organoseleniums, triphenylphosphines, coupling and oxidation agents have been synthesized and applied in heterocycle synthesis.
2.2.1. Tin Reagents and Catalysts
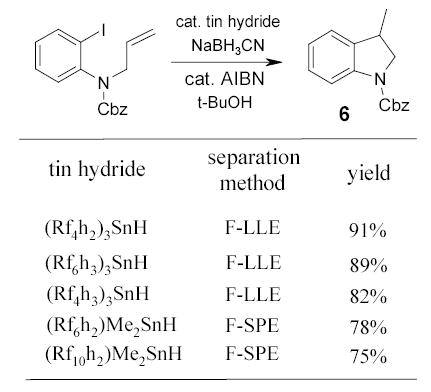
Organotin reagents have important synthetic utility. However, they are highly toxic and difficult to be removed from the reaction mixture. The Curran group introduced a number of fluorous tin reagents and evaluated their utilities in the synthesis of indolines 6 (Scheme 8).67,68 The radical cyclizations were conducted under standard catalytic procedure with NaCNBH3 in t-BuOH. No fluorous solvent was required because of acceptable organic solubility of tin reagents at the reaction temperature. Reaction mixtures were purified by either F-LLE or F-SPE. Because these tin compounds are heavily fluorinated, acetonitrile instead of 80:20 MeOH/H2O was used in F-SPE to elute the non-fluorous product. After fluorous purification, no resonances from the fluorous tin compound were detected in the 1H NMR spectra of the product 6.
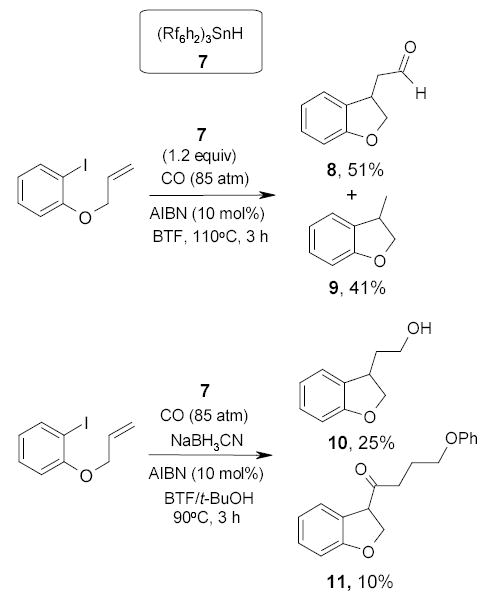
In a collaborative work by the Ryu and Curran groups, fluorous tin hydride 7 was employed to promote two kinds of radical carbonylations (Scheme 9).69 In the first reaction, a stoichiometric amount of fluorous tin hydride 7 was used and the reaction was conducted in BTF at 110 °C. Both the carbonylated benzofuran 8 and non-carbonylated benzofuran 9 were observed. The second reaction was carried out under catalytic conditions with NaCNBH3. A mixture of BTF and t-BuOH was used as a co-solvent. The desired cyclization/carbonylation product 10 was generated together with byproduct 9 (32%) and undesirable dimerization product 11. The organic compounds were separated from the tin compounds by a simple three-layer extraction with H2O/CH2Cl2/FC-72.
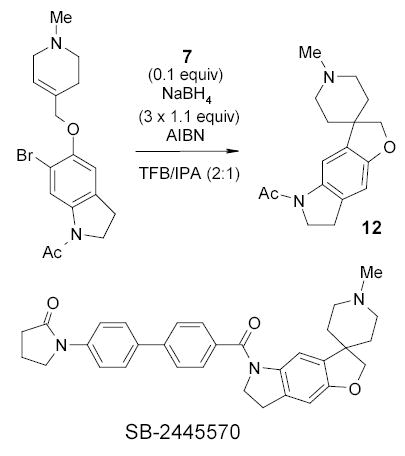
A more practical application of fluorous tin hydride was reported by Mulholland in the synthesis of SB245570 intermediate 12 (Scheme 10).70 The radical cyclization was performed in a co-solvent of 2:1 BTF/isopropanol (IPA). The F-LLE was carried out in FC-72 and CH2Cl2. Residual tin content of the spirocycle was below the detection limit of ICP/AES (7 ppm).
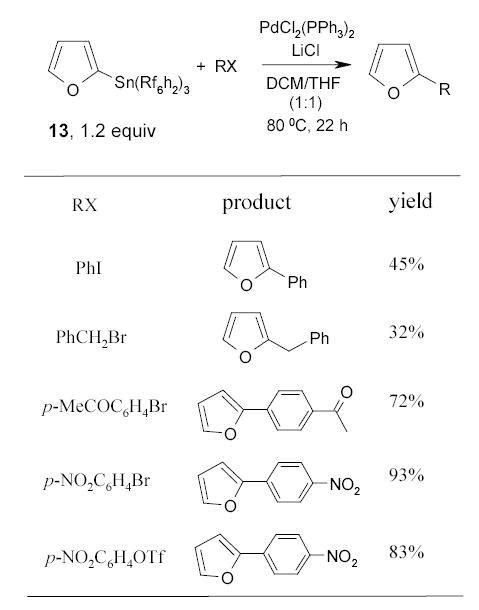
Fluorous aryl tins are another kind of reagent developed by the Curran group. Scheme 11 shows the results of the Stille coupling of 2-furyl fluorous tin 13 with different halides or triflates in the presence of 2 mol % PdCl2(PPh3)2, 3 equiv of LiCl in 1:1 DMF/THF solvent at 80 °C.71,72 The concentrated reaction mixture was partitioned in a H2O/CH2Cl2/FC-72 triphasic system. The product was collected from the organic layer, whereas the tin chloride was recovered from the FC-72 layer in 80–90% which was routinely recycled.
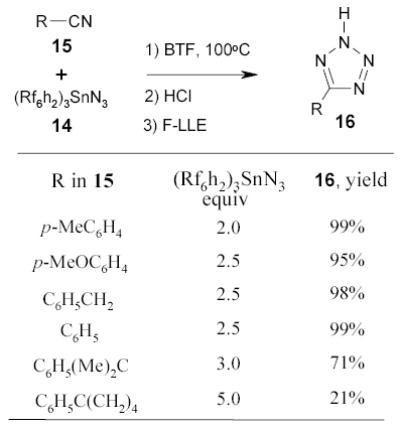
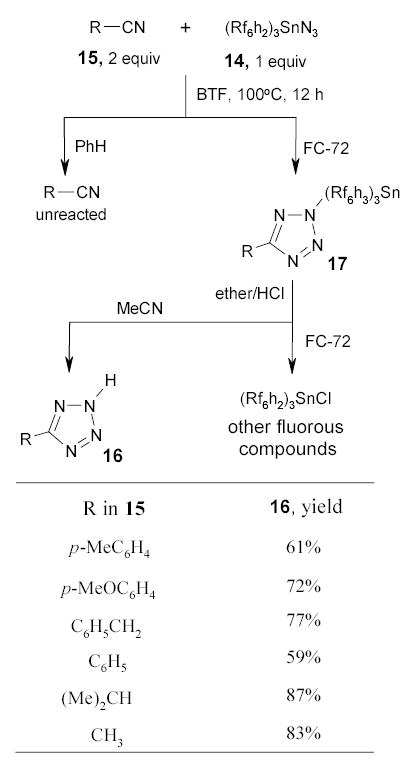
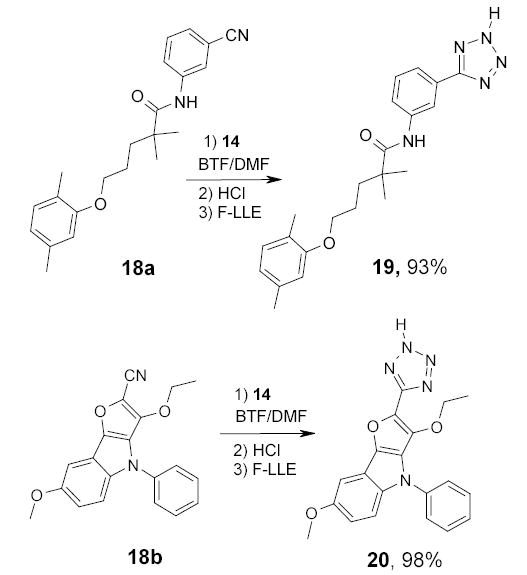
The fluorous tin azide 14 has been used as an alternative to the regular tributyltin azide in the conversion of nitriles 15 to tetrazoles 16 (Scheme 12).73 Reactions were carried out in two different ways. In a traditional one-pot mode, the intermediate tin tetrazoles not isolated, but were hydrolyzed to tetrazoles 16 by brief exposing it to ethereal HCl prior to partitioning between MeCN and FC-72. In a second so called “phase–switching” mode, the fluorous intermediates 17 were purified by F-LLE between benzene and FC-72 to remove unreacted nitriles (Scheme 13). The tin tetrazoles 17 were then treated with HCl and the crude products were purified by a second F-LLE. The double phase switching resulted in lower product yield, however, it ensured that the final product was free from both the organic and fluorous impurities.
A one-pot reaction procedure has been used to make tetrazoles from two complex nitriles 18a and 18b (Scheme 14).73 Reactions were run in a mixture of BTF and DMF because of low solubility of the nitriles in BTF. Three equivalents of azide were used to ensure completion of the reaction. After F-LLE, the acetonitrile layer yielded tetrazoles 19 and 20 in 93% and 98% yield, respectively.
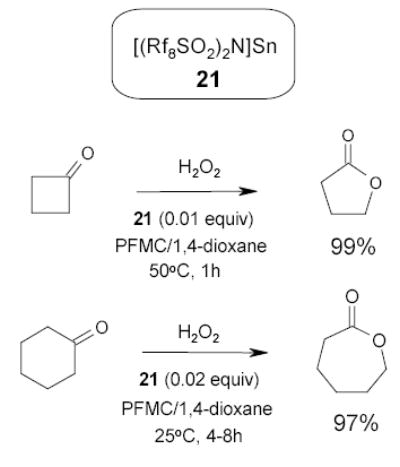
Very recently, Nishikido and coworkers reported the use of fluorous Lewis acid 21 in the fluorous biphasic system for Bayer-Villiger oxidation of cyclic ketones (Scheme 15). The catalyst gave good yield and can be recovered from the fluorous phase and reused four times without loss of activity.74–76
2.2.2. Selenium Reagents
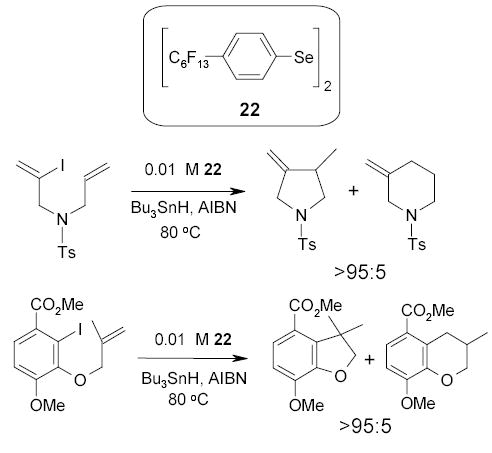
Competitive undesired radical rearrangements are general problems in the synthesis of heterocyclics by free radical cyclizations. The Crich group discovered that the use of fluorous diselenide, reduced in situ to the corresponding selenol, can significantly inhibit the formation of by products in the tributyltin hydride-promoted radical cyclizations.77,78 Scheme 16 demonstrates that diselenide 22 at 0.01 M was able to reduce the homoallyl/cyclobutyl and neophyl type rearrangements, which usually occurs during vinyl and aryl radical cyclizations. The ratio of 5-exo/6-endo cyclization was greater than 95:5. The diselenide was recovered in 90% yield from the fluorous phase by partition between toluene and PFMC.
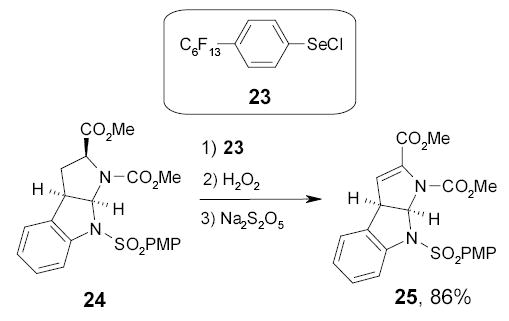
Another fluorous reagent, areneselenyl chloride 23, was found to be useful in the conversion of carbonyl compounds to their α,β-unsaturated derivatives.79 The ester 24 was α-selenated with selenyl chloride followed by oxidation and syn-elimination to give α,β-unsatuated ester 25 (Scheme 17). The fluorous reagent was recovered as the diselenide in 95% yield by continuous extraction of the organic layer with FC-72.
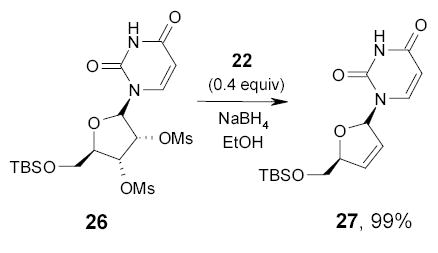
In another study conducted by Crich and coworkers, a catalytic amount of fluorous diselenide 22 was used in conjunction with stoichiometric NaBH4 to convert vicinal diol 26 to alkene 27 (Scheme 18).80 The fluorous reagent was recovered in 88% by continuous extraction with FC-72.
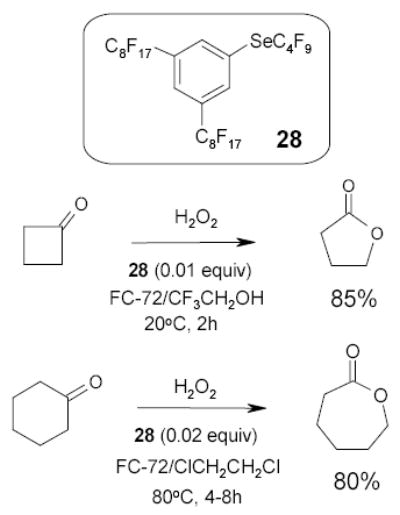
Sheldon and coworkers employed fluorous phenyl butylselenide 28 as a catalyst in the Baeyer-Villiger oxidation of cyclic ketones with hydrogen peroxide (Scheme 19).81 The reaction was carried out in a FC-72 and trifluoroethanol biphasic system. Since this co-solvent had the tendency to form emulsion, in another experiment trifluoroethanol was replaced by 1,2-dichloroethane to address the problem.
2.2.3. Triphenylphosphine Reagents
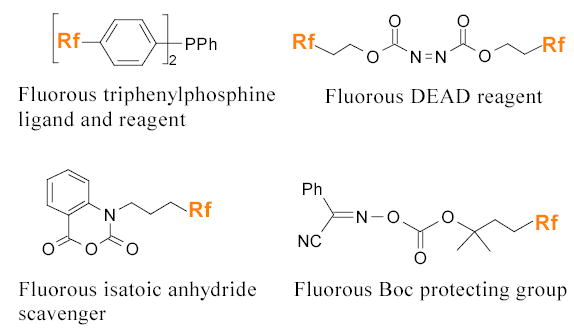
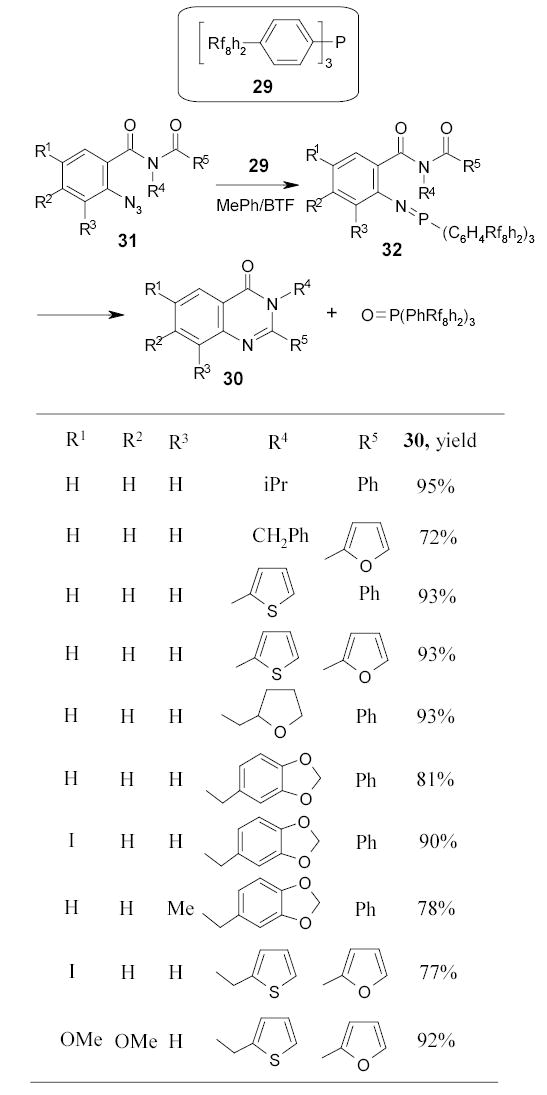
Bannwarth and coworkers employed fluorous triphenylphosphine 29 in the parallel synthesis of 3H-quinazoline-4-ones 30 (Scheme 20).82 The starting material 31 was converted to the iminophosphorane 32 by the Staudinger reaction with fluorous triphenylphosphine. The intermediate directly underwent an intramolecular Aza-Wittig reaction. The reaction solvent was a mixture of toluene and BTF. Problematic phosphine oxide species were separated easily by F-SPE in parallel.
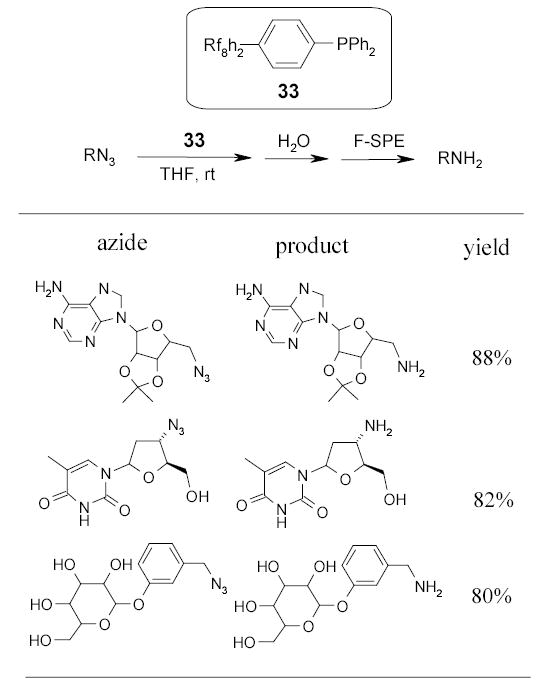
Lindsley and coworkers employed fluorous triphenylphosphine 33 for Staudinger reactions to convert more complex heterocyclic azides to the corresponding amines (Scheme 21).60
2.2.4. DEAD Reagent
Fluorous diethyl azodicarboxylate (34, DEAD) has been developed by the Curran and Dobbs groups for the Mitsunobu reaction (Scheme 22).52,53 The Dobbs group used the normal triphenylphosphine, whereas the Curran group used fluorous triphenylphosphine so that both the phosphine oxide and F-DEAD derivative were efficiently removed by F-SPE.
2.2.5. DAIB Reagent
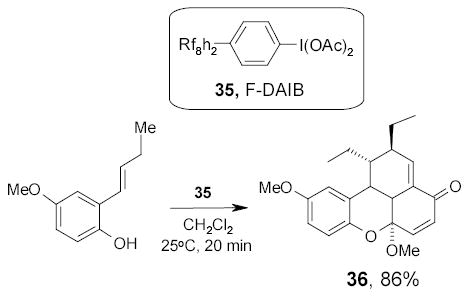
Fluorous diacetoxy iodobenzene (35, F-DAIB) has been recently synthesized by the Lindsley group and employed in the preparation of an unnatural carpanone-like molecule 36 through a homo-β,β-phenolic coupling followed by an inverse electron demand Diels-Alder reaction (Scheme 23). The excess F-DAIB and its derivatives were separated by F-SPE.83
2.3. Fluorous Scavenging
The scavenging technique has been widely used in solution-phase synthesis to improve reaction yield and facilitate the separation process by selectively removing unwanted species from the reaction mixture. The fluorous scavenging is unique because both the reaction and scavenging are conducted in a homogeneous solution phase.
2.3.1. Alkene/Alkyne Scavengers
Several heavy fluorous scavengers have been developed by the Curran and Wipf groups.67,68,84 Heavy fluorous tin hydride 7 has been employed to remove excess alkene in the nitrile oxide cycloadditions (Scheme 24).68 The reaction was conducted in BTF at room temperature. The scavenged by product was separated from the reaction mixture by F-LLE.
2.3.2. Electrophile Scavengers
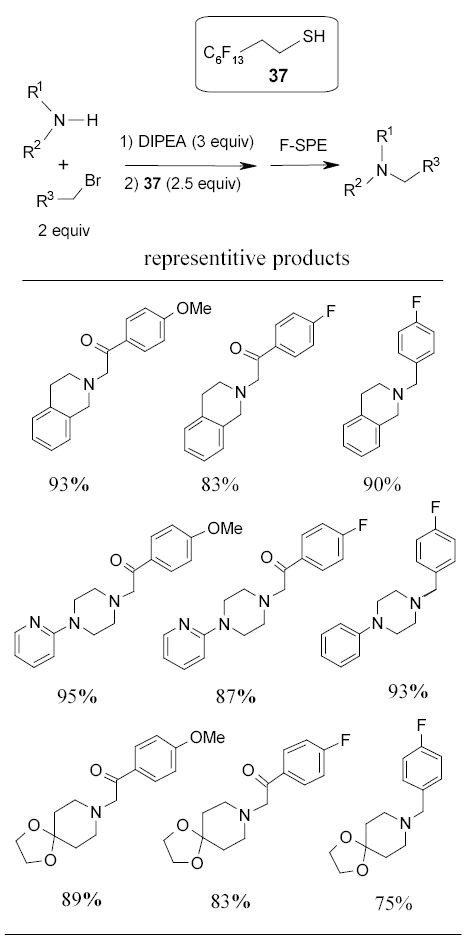
Recently the Lindsley group at Merck and researchers at FTI independently developed several light fluorous scavengers.54–57 Fluorous thiol 37 was used as a electrophile scavenger to remove α-bromoketone in the parallel synthesis of a tertiary amine library (Scheme 25).54 The quenched reaction mixture was washed with aq. NH4Cl and then purified by F-SPE. In a comparison experiment, it was found that the thio quenching with fluorous scavenger was 5–10 time faster than using a polymer-supported analog.
2.3.3. Nucleophile Scavengers
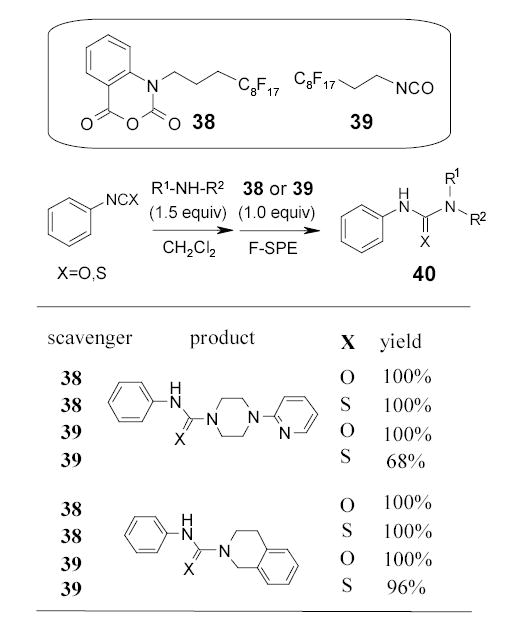
Both isatoic anhydride 38 and isocyanate 39 have been introduced by FTI as nuclephile scavengers to remove primary and secondary amines in the synthesis of urea, thiourea analogs 40 (Scheme 26), and β-hydroxyamines 41 (Scheme 27).55
2.3.4. Dienophile Scavengers
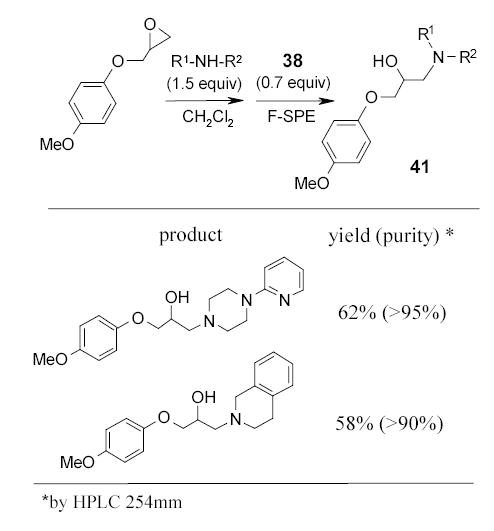
Curran and Werner recently introduced fluorous benzylmaleimide 42 and fluorous [1,2,4]triazoline-3,5-dione 43 as powerful scavengers to remove excess dienes, such as 1,2,3,4,5-pentamethylcyclopentadiene, α-terpinene, 1,4-diphenyl-1,3-butadiene, and anthracene, from the Diels-Alder reaction mixture.58 Scheme 28 shows the Diels-Alder reactions of the fluorous dienophiles 43 gave better results than that of 42 and. The Diels-Alder adducts were isolated by F-SPE.
2.4. Fluorous Tags
Fluorous tagged starting materials (reactants) can be used in multistep synthesis. At the last reaction step, the fluorous tags are usually cleaved by displacement reactions. The utility of fluorous tags is similar to the “catch and release” linkers in solid-phase synthesis. Purification of fluorous intermediates as well as the detagged product can be achieved by fluorous F-LLE or F-SPE.
2.4.1. Alcohol Tags
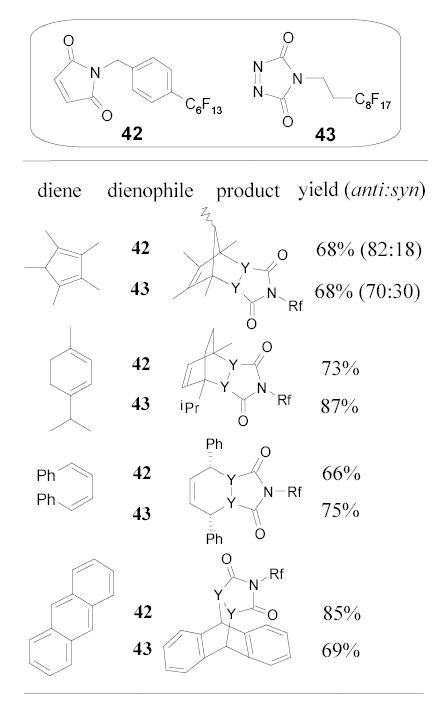
Wipf and Methot developed a new entry to dihydropyridazinone 44 using a fluorous alcohol-tagged ester 45 as a starting material (Scheme 29).85 After steps of transformations, the δ–keto ester 46 was treated with hydrazine to form the dihydropyridazinone ring. The fluorous tag was cleaved during the cyclodehydration. The product was separated from the reaction mixture by F-LLE with FC-72/MeCN.
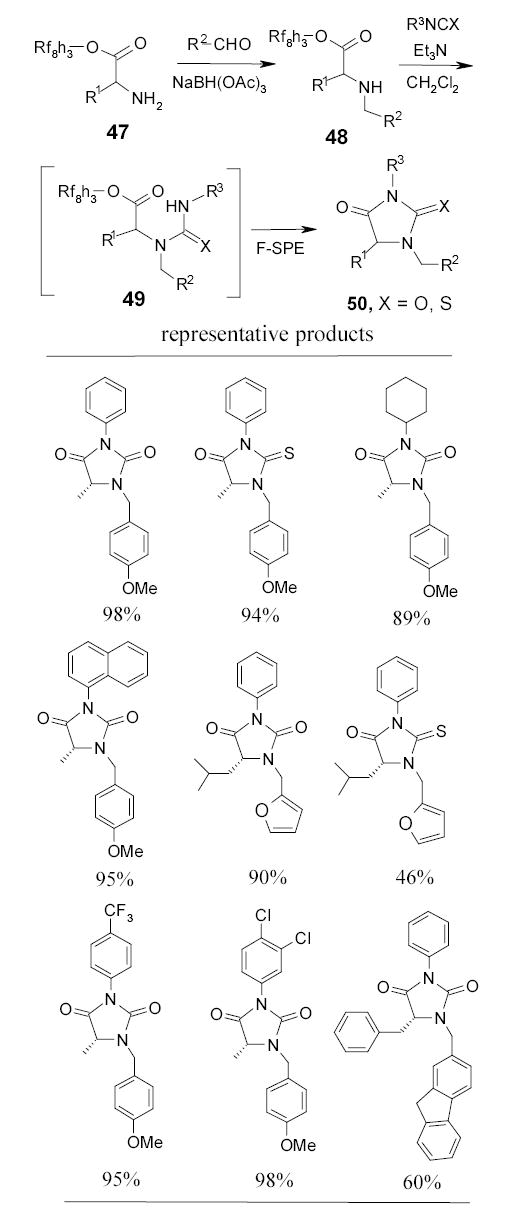
A similar cleavage strategy has been used in hydantoin synthesis.86 Using primary fluorous alcohol protected amino acids as the starting material, researchers at FTI recently prepared a 120-member hydantoin and thiohydantoin library by a parallel synthesis (Scheme 30).87 Two fluorous amino esters 47 (R1 = i-Bu and Bz) underwent reductive amination with six aldehydes. Each of the twelve intermediates 48 was further reacted with ten aryl isocyanates or aryl isothiocyanates. In situ cyclization of the resulting ureas or thioureas 49 displaced the fluorous tag and afforded heterocyclic products 50. The average yields for this two-step synthesis were around 50% and purities of the final products after F-SPE were between 85–95%.
Examples of fluorous alcohol-protected amino acids in multicomponent reaction are described in Section 2.6.2.
2.4.2. Thiol Tags
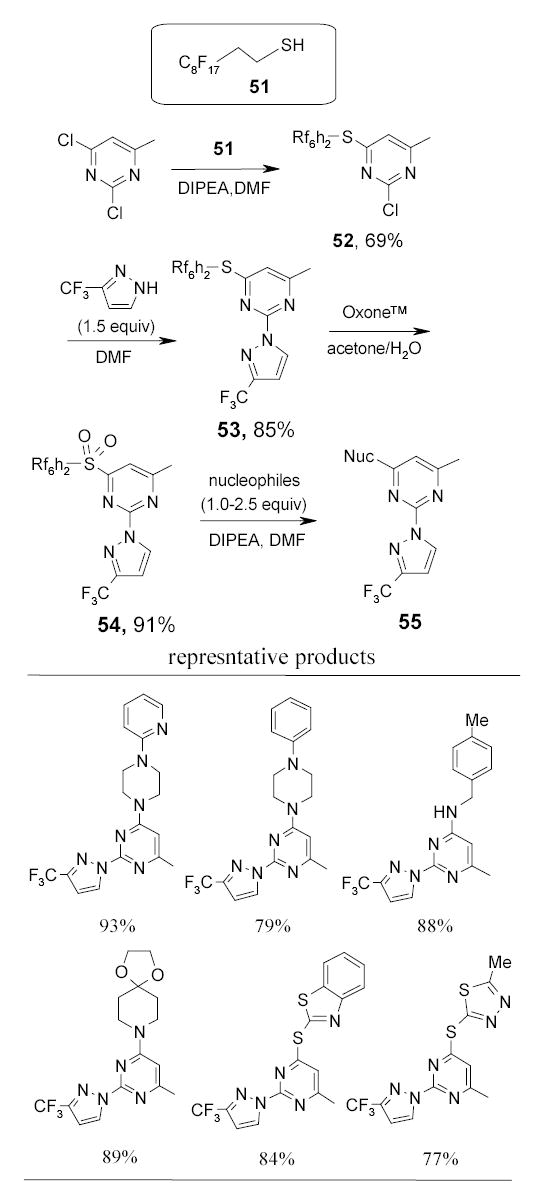
Fluorous thiol 51 has been used as a nucleophilic tag to displace the 3-chlorine of 1,2-dichloro-5-methylpyrimidine (Scheme 31).88 The tagged substrate 52 was further displaced with a 3-trifluoromethylpyrazole to give 53. The thiol tag was then activated by oxidation to a sulfone 54 and displaced by a set of nucleophiles to afford disubstituted pyrimidines 55. The purities of final products after the F-SPE were greater than 90%.
2.4.3. FluoMar
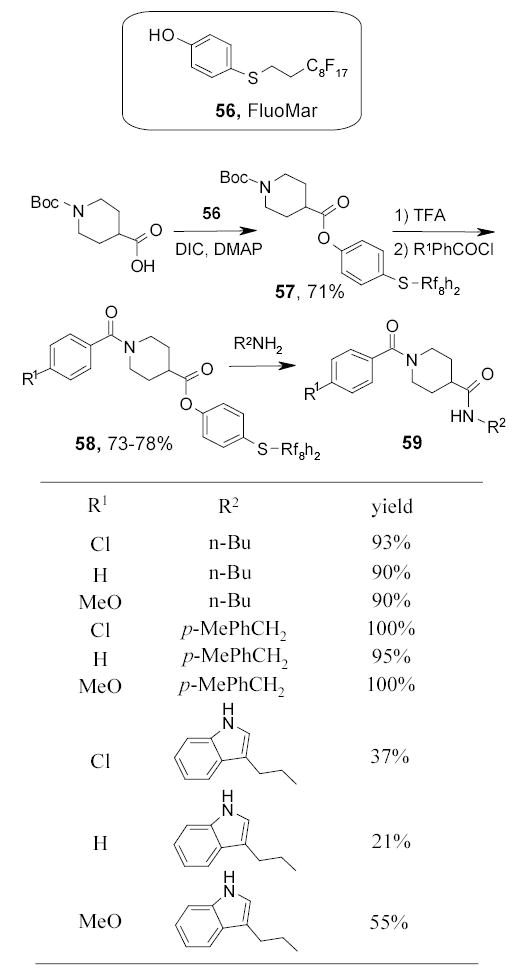
The Marshall resin is a popular linker in solid-phase organic synthesis. FluoMar has been recently introduced as a fluorous version of the Marshall resin for solution-phase synthesis (Scheme 32).89 In the preparation of a demonstration library, carboxylic acids were coupled with FluoMar 56 under standard conditions using diisopropylcarbodiimide (DIC) and dimethylaminopyridine (DMAP). The intermediate 57 was deprotected and then coupled with acid chlorides to form amides 58. The fluorous tags were finally displaced with a set of amines to give amides 59.
2.4.4. Fluorous Benzophenone Imines
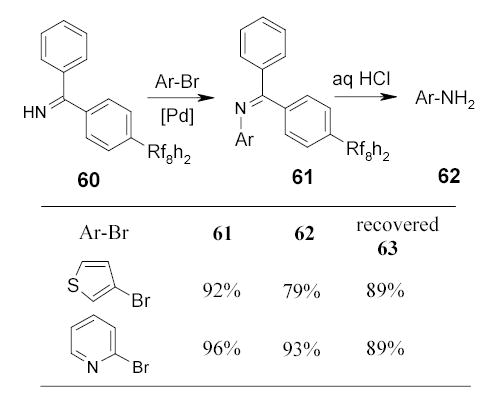
Recently, Herr and coworkers90 employed fluorous benzophenone imine 60 to react with aryl halides and triflates in a fashion analogous to Buchwald’s procedure (Scheme 33).91 Intermediate N-aryl benzophenone imines 61 were purified by F-SPE and converted to amines 62 by hydrolysis. The fluorous benzophenone 63 byproduct (not shown) was recovered by F-SPE and used to regenerate 60.
2.5. Fluorous Protecting Groups
Fluorous protecting groups have a “one stone hits two birds” effect in fluorous synthesis. The functional group protection and the fluorous tag introduction can be accomplished by a single operation. Slightly modified conventional solution-phase protections and deprotection conditions can be used for fluorous synthesis.
2.5.1. Fluorous Silyl Groups
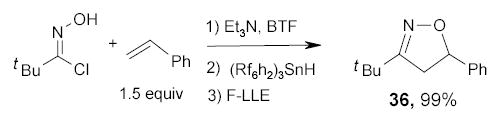
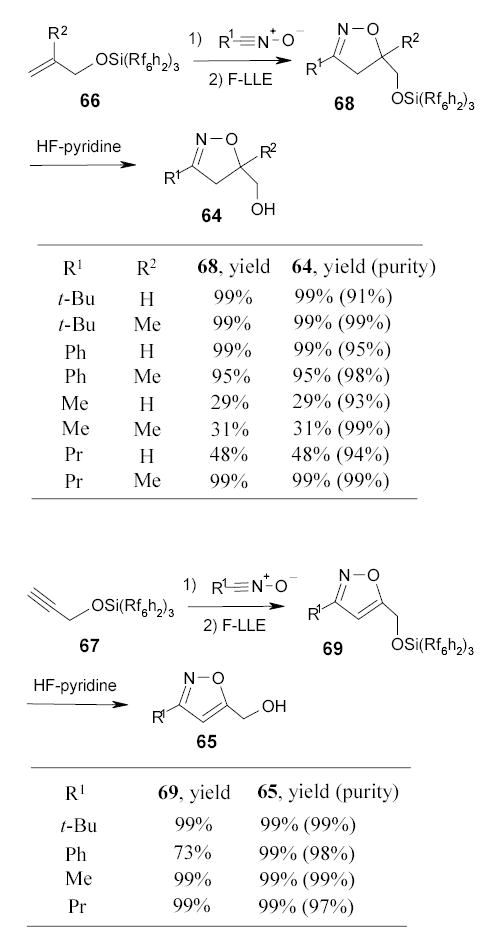
Studer and Curran developed a new approach to isoxazoline 64 and isoxazoles 65 by cycloadditions of nitrile oxides with heavy fluorous silyl-protected allyl- and propargyl alcohols 66 and 67, respectively (Scheme 34).92 Large excesses (4–8 equiv) of nitrile oxides were used to drive the cycloaddition reaction to completion. The cycloaddtion products 68 and 69 were isolated in from the unreacted nitrile oxides by the triphasic extraction with FC-72/benzene/H2O. The desilylations were performed with HF-pyridine in Et2O at room temperature. The final products 64 and 65 were isolated from the organic layer after a triphasic (FC-72, CH2Cl2, and aq. NH4Cl) extraction. More examples of fluorous silyl protections in heterocyclic synthesis are discussed in the Section 2.8.
Very recently, Manzoni reported the use of fluorous silyl reagent 70 to protect the anomeric position of sugar acceptors in the rapid synthesis of oligosaccharides by F-SPE purification (Scheme 35).93
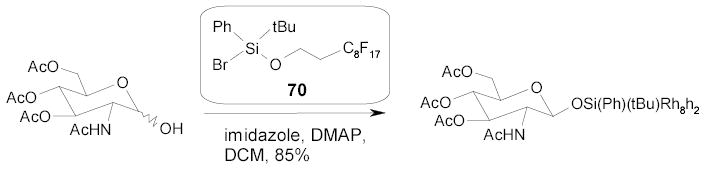
2.5.2. Fluorous Boc Groups
Curran and coworkers have prepared a series of F-BocON compounds containing different Rf chains. The Boc-ON 71 with a single C8F17 chain was used in the parallel synthesis of isonipecotic acid derivatives 72 (Scheme 36).94 The amino group of the isonipecotic acid was first protected by the F-Boc. The fluorous intermediate 73 was then coupled with eight amines (R1NHR2). After deprotection of the F-Boc with TFA, the resulting compounds were further reacted with twelve electrophiles (R3X) to give a 96-compound library.
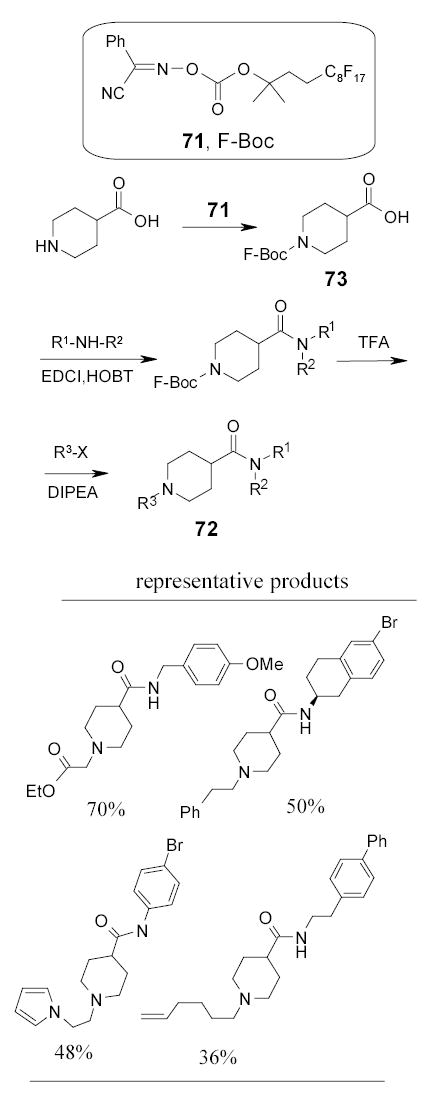
2.5.3. Fluorous Cbz Groups
F-CbzCl 74 developed by Schwinn and Bannwarth has been applied in fluorous biphasic synthesis of quinazoline-2,4-diones 75 (Scheme 37).95 Amidation of fluorous protected acid 76 followed by cyclative deprotection of 77 led to the formation of quinazoline-2,4-diones 75. This chemistry has been modified by absorption of the fluorous chains onto the fluorous silica gel via strong fluorine-fluorine interactions to eliminate the use of fluorous solvents for the reaction and separations (Section 2.8)
The Curran group and FTI recently developed a light fluorous Cbz group. This protecting group has been applied to the protection of amino acids (see Section 2.10.2).
2.5.4. Fluorous Diols
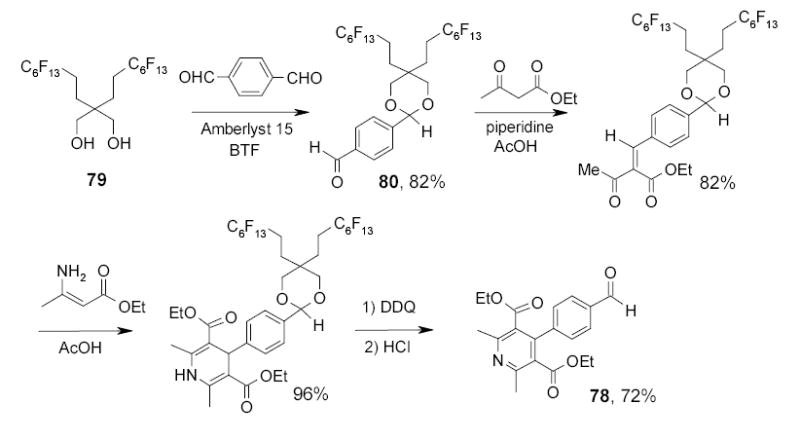
Read and Zhang recently reported the synthesis of acetals by reaction of aldehydes and ketones with fluorous 1,3-alkanediols containing mono- or difluorous chains.96 The utility of fluorous diols as the carbonyl group protecting agents has been demonstrated in the synthesis of pyridine derivative 78 (Scheme 38).97 One carbonyl group of a dialdehyde was selectively protected with fluorous diol 79. The protected compound 80 underwent condensation, cycloaddition, and oxidation reactions and finally deprotected with HCl to afford substituted pyridine 78.
2.5.5. Fluorous Benzyl Group
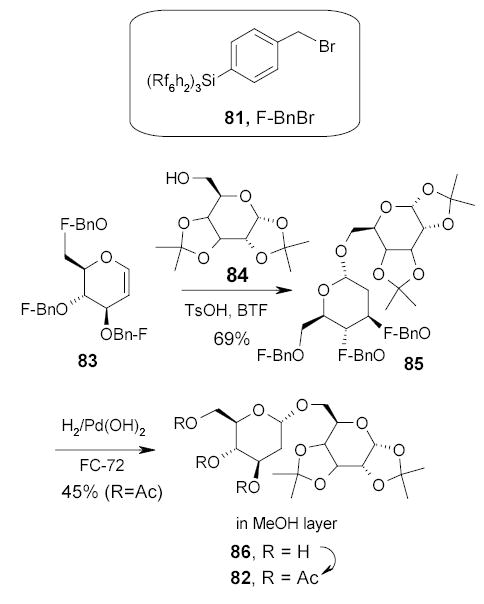
Fluorous protecting groups have also been used in oligosaccharide synthesis. The utility of F-BnBr 81 as an alcohol protecting agent was demonstrated in the synthesis of disaccharide 82 (Scheme 39).98 The hydroxyl group of d-glucal was protected with 4 equiv of F-BnBr using NaH as a base and BTF as the solvent. The crude tribenzyl glucal derivative 83 was purified by triphasic (H2O/CH2CH2/FC-72) extraction to remove organic and inorganic materials followed by flash silica gel chromatography to remove the excess benzylating agent and other impurities. Fluorous glucal 83 was then coupled with excess diacetone galactose 84 under standard reaction conditions in BTF to give pure fluorous disaccharide 85 after triphasic extraction. Fluorous compound 85 was debenzylated by catalytic hydrogenation with H2 and Pd(OH)2 in FC-72. After another triphasic extraction, product 86 in the organic phase was acylated to give disaccharide 82.
2.5.6. Fluorous Bfp
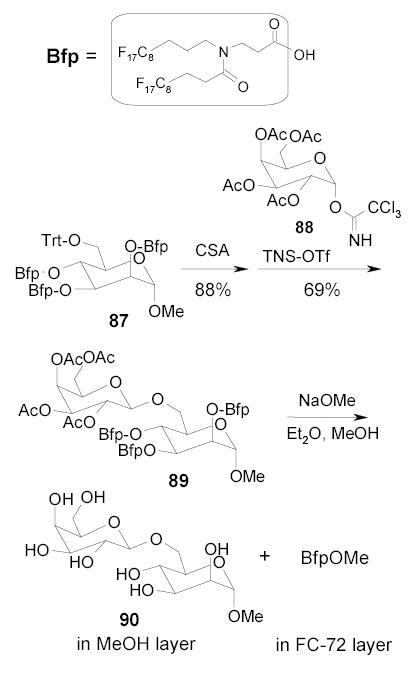
The Inazu group employed a fluorous propanoyl (Bfp) containing two C8F17 chains to protect three hydroxyl groups of a mannose derivative (Scheme 40).99–101 The triphenylmethyl (Trt) group of 87 was selectively removed by treatment with 10-camphorsulfonic acid (CSA). The deprotected hydroxyl group was coupled with galactose derivative 88 to give fluorous disaccharide 89. Deprotection of both the acetyl and Bfp groups followed by FC-72/MeOH extraction gave disaccharide 90 in MeOH layer in 93%. The protection group was recovered from the FC-72 layer as a methyl ester in 92%. A tetrasaccharide was also prepared by a similar approach.
2.6. Fluorous Multicomponent Reactions
Multicomponent reactions have high efficiency in the construction of core structures with variable side chains. Since not all the components are used in equal amounts and since their reactivities may be different, excess or unreacted components in the reaction mixture may complicate the product purification. Fluorous multicomponent reactions can simplify the separation process.
2.6.1. Biginelli Reactions
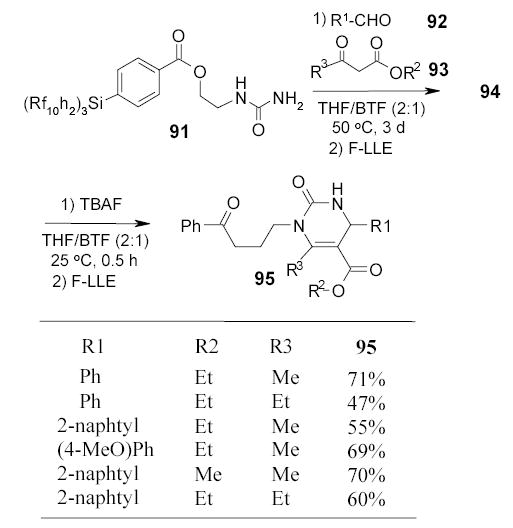
In a collaboration work by the Wipf and Curran groups, a Biginelli reaction was carried out by using fluorous urea 91 as the limiting agent, whereas β-ketone ester 92 and aldehyde 93 were each used in 10-fold excess (Scheme 41).102 The condensed fluorous dihydropyrimidines 94 were easily obtained by FC-84 extraction. The desilylated products 95 with TBAF were isolated in high purity by a second FC-84 extraction.
2.6.2. 1,3-Dipolar Cycloadditions
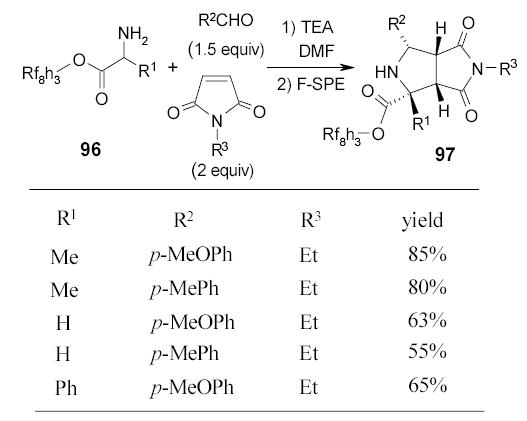
Fluorous amino esters 96 have been used in the synthesis of proline analogs by three component reactions (Scheme 42).103 The 1,3-dipolar cycloaddition products 97 were isolated by F-SPE as single diastereomers. Adducts 97 have been used as key scaffolds in the construction of several highly functionalized tricyclic heterocycles.
2.6.3. Ugi Reactions
See Section 2.7.2.
2.7. Microwave-Assisted Fluorous Reactions
The combination of microwave reaction and fluorous separation can speed up both the reaction and separation process.6 Since fluorous-tags are thermally stable and the tagged molecules have solution-phase character, fluorous synthesis is believed to be superior to solid-phase synthesis under microwave heating.
2.7.1. Stille Couplings
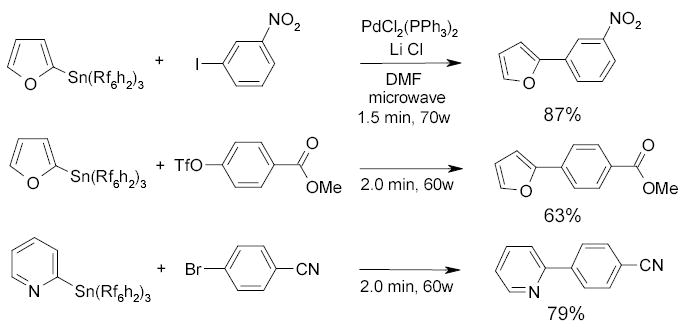
The Curran and Hallberg groups employed a single-mode microwave reactor to promote fluorous reactions. A similar fluorous tin hydride-based reaction described in Scheme 7 was finished within 6 min under microwave. Scheme 43 shows that fluorous Stille couplings can be done in less than 2 min.104,105
2.7.2. Ugi Reactions
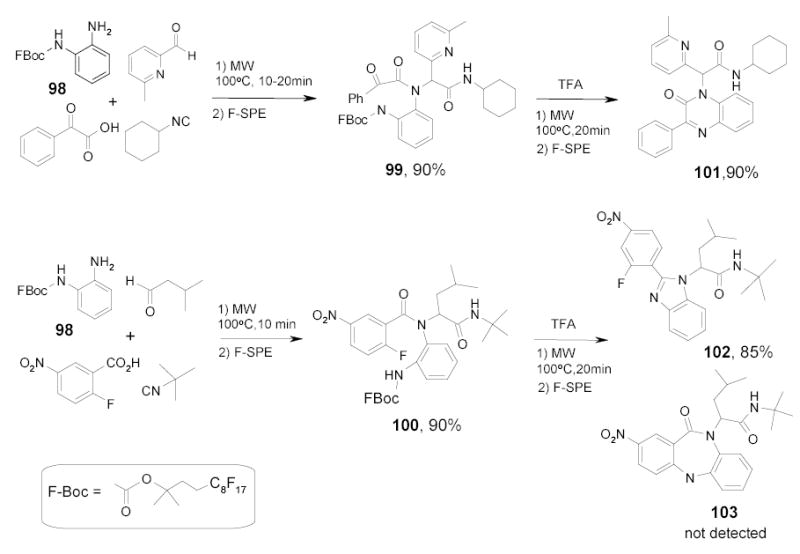
Hulme and coworkers reported a nice Ugi/de-Boc/cyclization sequence in the synthesis of different heterocyclic cores including quinoxalinone, benzazepine, and benzimidazole.106–109 The reactions gave excellent yields, but the Ugi reactions were slow (36–48h) and the condensation products were purified by double scavenging with immobilized tosylhydrazide and diisopropylethylamine to remove excess aldehydes and unreacted acids. The deprotection of the F-Boc group with TFA required 4–24 h. These reactions were recently modified by using fluorous Boc protected aniline 98. The reaction times for both steps were reduced to less than 20 min under microwave conditions (Scheme 44).110 The Ugi condensation products 99 and 100 were purified by F-SPE without scavenging. After the deprotection of 100, benzimidazole 102 was isolated as a single product in good yield, whereas in the originally reported thermo Boc deprotection, benzazepine 103 was also detected.
2.7.3. Perfluorosulfonate-Based Cross Couplings
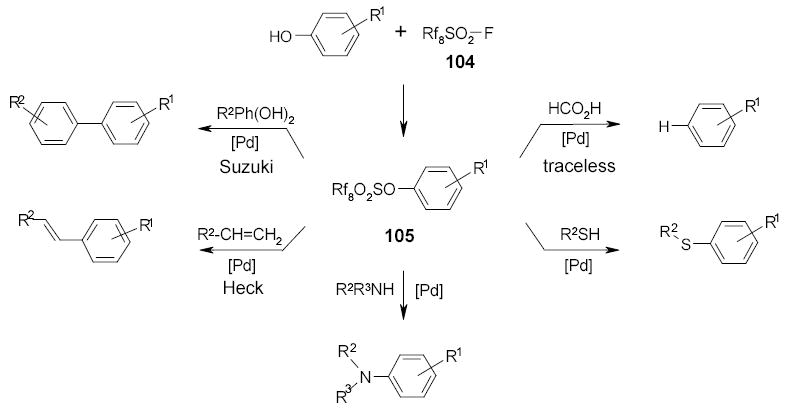
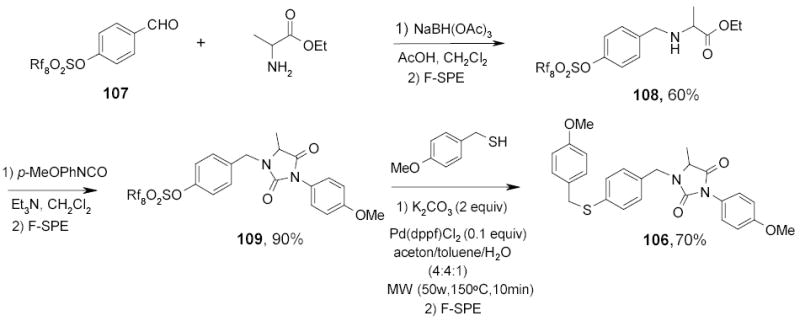
A series of fluorous sulfonate-based Pd-catalyzed reaction under microwave conditions has been explored (Scheme 45).111 The fluorous sulfonate tag 104 was also used in the multi-step synthesis of heterocycles. An example of synthesis of substituted hydantoin 106 is outlined in Scheme 45. Intermediate 108 was prepared by reductive amination of 107. This compound was then reacted with an isocyanate to form substituted hydantoin 109. A standard palladium-catalyzed cross coupling was carried out under microwave irradiation to convert F-sulfonates 107 to the sulfide 106.112
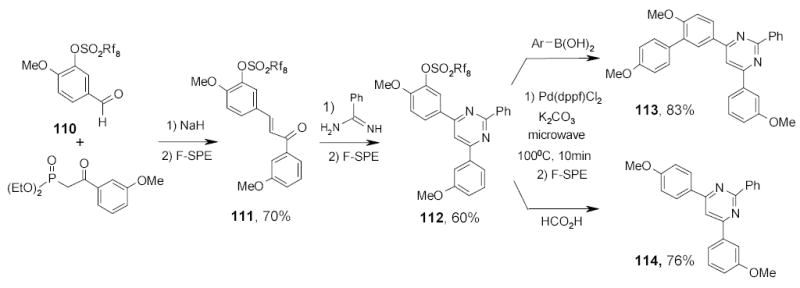
In another fluorous sulfonate-based multistep synthesis, tagged substrate 110 was taken through two transformations before the microwave reactions.111 Fluorous sulfonate 112 was reacted with boronic acids to generate the C-C bond of biaryl compounds 113 or reacted with HCO2H to give traceless detag product 114 (Scheme 47).
2.8. Fluorous Solid-Phase Synthesis (F-SPS)
2.8.1. Oligomer Synthesis
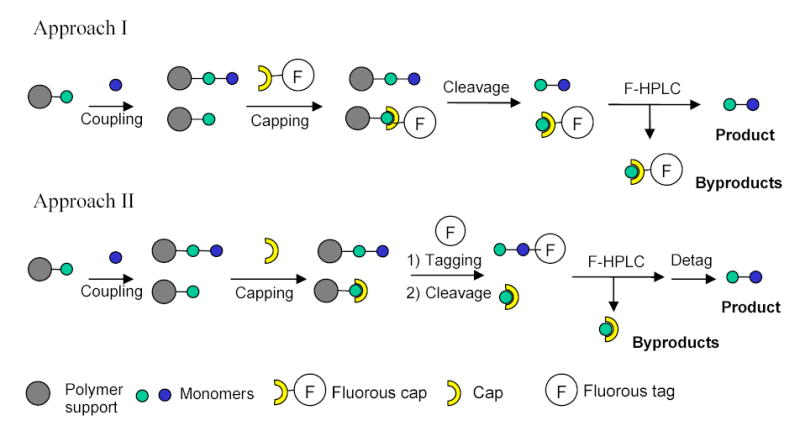
The fluorous tagging strategy has been used in the solid-phase synthesis of oligosaccharides and peptides.113–115 Two general fluorous approaches in oligomer synthesis are shown in Scheme 48. The first approach employs fluorous material to cap the deletion sequences after each coupling reactions. At the end of the synthesis, all sequences are cleaved from the resin. Since only desired product is nonfluorous, it can be separated from the fluorous byproduct by F-HPLC. In the second approach, organics are used to cap the deletion sequences, while the desired sequence is captured by a fluorous tag after the last coupling. After F-HPLC purification and detagging, the target molecule is obtained. Inazu employed this method in peptide synthesis.115
2.8.2. Small Molecule Synthesis
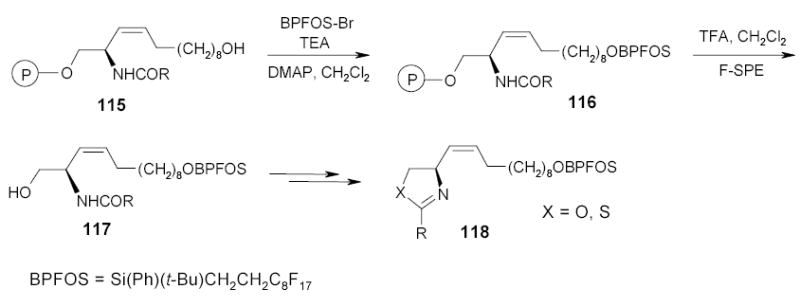
Wipf and Rover introduced fluorous tagging strategy into the solid-phase synthesis of small molecules (Scheme 49).116 Intermediates 115 prepared on the resin over several steps were attached to a fluorous silyl group (BPFOS). The fluorous molecule 116 was then cleaved from the resin together with nonfluorous byproducts resulting from previous solid-phase reaction steps. The fluorous tagged product 116 was isolated by F-SPE and subjected to additional transformations to afford desired oxazoles and thioazoles 118 as curacin analogs.
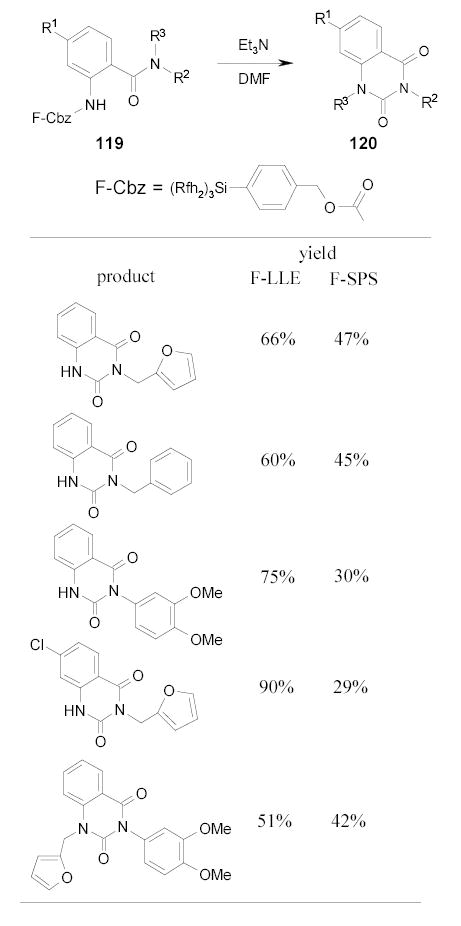
Bannwarth and coworkers recently modified the F-SPS of quinazoline-2,4-diones described in Section 2.5.3.117 The new method eliminated F-LLE and hence the fluorous solvent. The heavy fluorous Cbz-tagged intermediates 119 were mixed with fluorous silica gel in the organic solvent. The fluorous molecules were believed to be absorbed by the silica gel through strong fluorine-fluorine interactions. After the cyclization reaction, products 120 were released, whereas the cleaved tags still adsorbed onto the silica gel. The products were isolated by simple filtration. The comparison results using F-LLE and F-SPS are listed in Scheme 50.
2.9. Triphasic Reactions
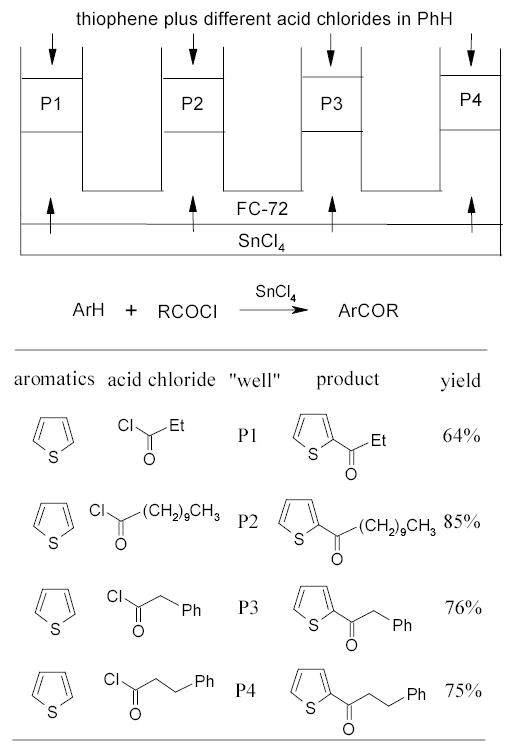
The Curran and Ryu groups developed the highly innovative triphasic reaction systems. The mechanism of triphasic systems for detagging and phase-vanishing reactions has been described in Section 1.4.3. Ryu and coworkers recently employed the phase-vanishing method in Friedel-Crafts acylation of thiophene with SnCl4 as Lewis acid.118 Reactions were carried out in parallel U-tubes charged with three layers of liquids with different density (Scheme 51). The heavier SnCl4 was at the bottom, FC-72 in the middle, and a benzene solution of thiophene and four different acid chlorides floating on the top of each U-tube (P1 to P4). The SnCl4 layer was gently stirred without mixing of three layers until this layer was disappeared in about 30 min. At the same time, the benzene layer was gradually turned to dark purple indicating the transfer in of SnCl4. After an additional 2.5 h, the reaction was over and products were harvested from the benzene layers at the top of each “well” of the U-tube. No cross contamination was detected. This experiment demonstrates that the traditional organic synthesis using a dropping funnel can be accomplished in a triphasic system with chemical control of addition rates.
2.10. Fluorous Mixture Synthesis (FMS)
FMS has been developed based on predictable and reliable F-HPLC for intermediate analysis and product demixing.8,33,119 This is the first highly efficient solution–phase mixture synthesis technique to make individual pure compound libraries. A schematic overview using three components FMS as an example is show in Figure 11. Three analogous starting materials are paired with three different fluorous tags. The tagged substrates are mixed together and taken through a multi-step synthesis to incorporate new diversities. After the synthetic sequence is over, each mixture is demixed by F-HPLC followed by detagging to give the individually pure final products.
Figure 11.
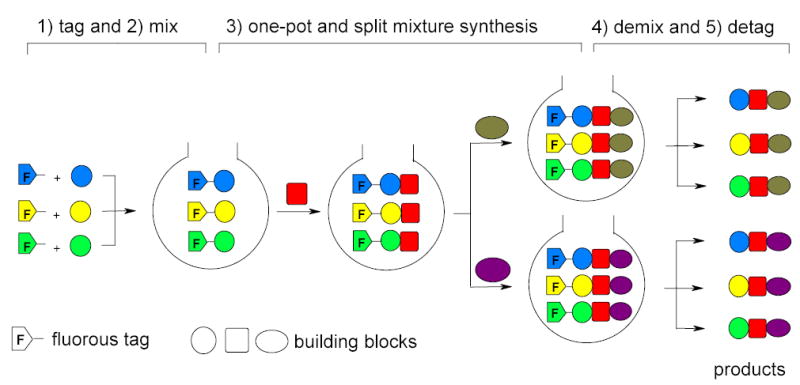
Schematic diagram of the concept of FMS
2.10.1. Library Synthesis
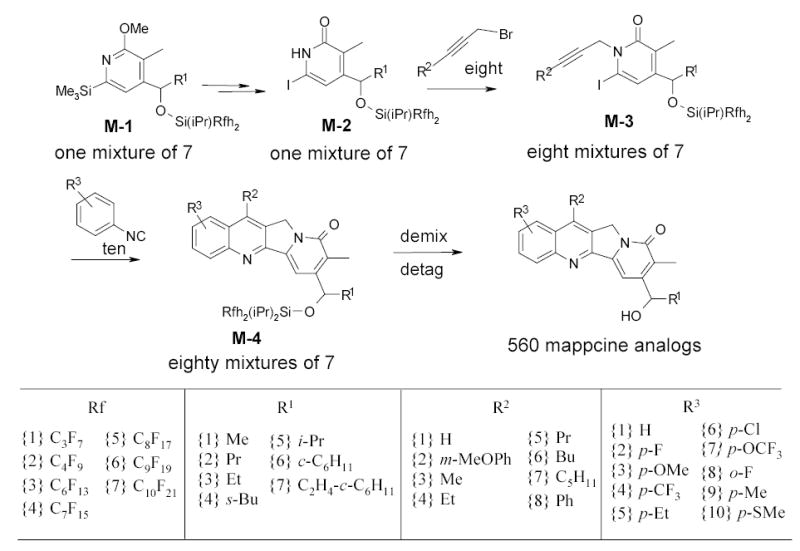
The power of FMS has been demonstrated by preparation of a 560-memberd mappicine library (Scheme 52).8,33 A mixture of seven pyridines M-1 (7 different R1 groups) was carried through a 4-step reaction sequence including two one-pot reactions and two split-parallel reactions. The first split of M-2 to 8 portions for N-propargylation (8 different R2 groups) was followed by second split to 10 portions for the radical tandem annulation with isonitriles (10 different R3 groups). The FMS ended up with eighty mixtures M-4 which were demixed by F-HPLC followed by detagging with HF-pyridine to give a 560-member mappicine library (Figure 3).
The quality control on reaction intermediates is a unique feature of FMS. The reaction mixtures can be analyzed by F-HPLC and purified by normal flash column chromatography to remove impurity in a mixed mode. Figure 12 demonstrates the intermediate purification at the alkylation step. The propaglation of M-2 resulted in two sets of mixtures each having 7-components, one set from N-alkylation and another set from O-alkylation. Seven desired N-alkylation products were separated from seven O-alkylation byproducts M-5 by normal flash column chromatography based on the different polarities of the O- and N-alkylated compounds. The synthesis of this 560-membered library required only 90 reactions (not including the detagging step) and 90 chromatography steps (including F-HPLC demixing).
Figure 12.
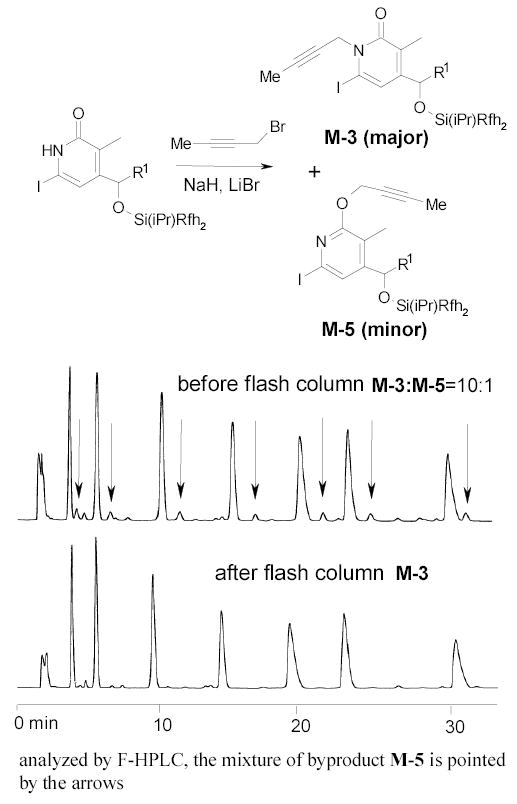
Analysis of a mixture of propargylation products before and after standard flash column chromatography purification
2.10.2. Enantiomer Synthesis
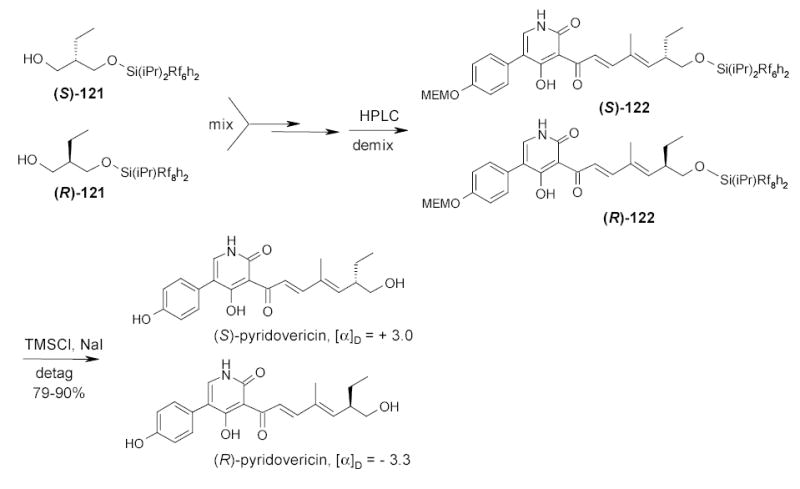
Parallel synthesis of both enantiomerically pure products is a common strategy in the determination of absolute configuration of a chiral natural product. Two enantiomerically pure products now can be made by one-pot quasiracemic FMS. In the synthesis of enantiomers of pyridovericin,120 Curran and coworkers used two different fluorous silanes (Rf = C6F13 and C8F17) to tag (S)- and (R)-enantiomeric pure starting materials 121. The combined quasienantiomeric mixture was then taken through a multi-step synthesis followed by F-HPLC demixing and detagging to afford two enantimerically pure pyridovericins (Scheme 53). Quasiracemic synthesis is the simplest version of FMS which has only two mixture components and without splitting involved in the synthesis.
In another application of quasienantiomeric FMS conducted by the Curran group and FTI, the (d)- and (l)-phenylalanines were tagged with newly developed fluorous Cbz-OSu 113 with different length of Rf groups (C6F13 and C8F17), respectively.121 The mixture of these two quasienantiomers 114a and 114b was then coupled with tetrahydroisoquinoline under standard conditions (Scheme 54). The crude product was both purified and resolved into its quasienantiomeric components 115a and 115b by fluorous HPLC.
2.10.3. Diastereomer Synthesis
Curran and coworkers also employed FMS technique to synthesize (+)-murisoline and its diastereomers.122 The murisoline family of acetogenins has six diastereocenters and this research focused on the rapid synthesis of sixteen stereoisomers of the dihydoxy tetrahydofuran fragment (shown in the box) with the 4(R) and 34(S) centers fixed.
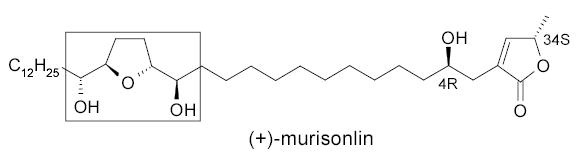
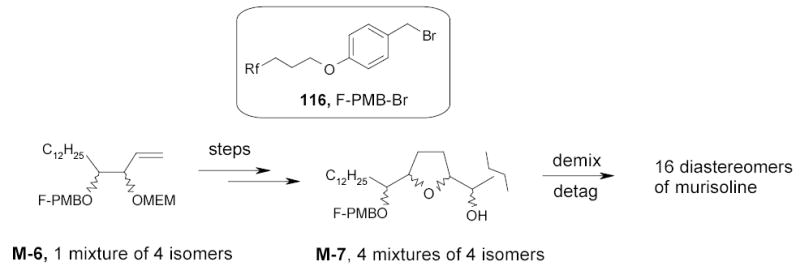
The FMS approach started with M-6, a mixture of four enantiomerically pure compounds, each tagged with a PMB group of differing fluorine content (Rf) (Scheme 55). This mixture was then taken through multiple synthetic steps, including two split and parallel syntheses to provide M-7, which contains four mixtures of four tagged products (16 products in total). Fluorous HPLC demixing of the four mixtures based on tag fluorine content followed by detagging provided all sixteen of the desired diastereomers of murisoline. Since this fluorous mixture synthesis has a total of 39 steps, compared to 156 steps that would be required to accomplish the same transformations using tradition, non-mixture techniques, the efficiency advantage is obvious.
3. Conclusions
Fluorous technologies have become a new synthetic method to fill the gap between traditional solution-phase and solid-phase syntheses. Since the birth in the early 1990’s, fluorous technologies have been growing into their “adolescence” and will assume a more important role in heterocyclic synthesis and other areas of organic and medicinal chemistries.
Scheme 1.
Scheme 2.
Scheme 3.
Scheme 4.
Scheme 5.
Scheme 6.
Scheme 7.
Scheme 8.
Scheme 9.
Scheme 10.
Scheme 11.
Scheme 12.
Scheme 13.
Scheme 14.
Scheme 15.
Scheme 16.
Scheme 17.
Scheme 18.
Scheme 19.
Scheme 20.
Scheme 21.
Scheme 22.
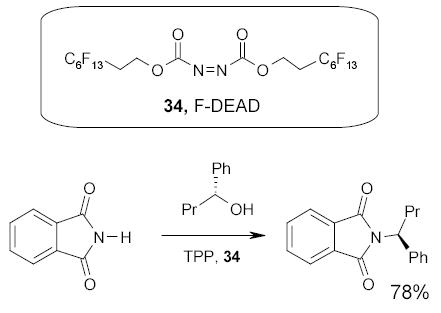
Scheme 23.
Scheme 24.
Scheme 25.
Scheme 26.
Scheme 27.
Scheme 28.
Scheme 29.
Scheme 30.
Scheme 31.
Scheme 32.
Scheme 33.
Scheme 34.
Scheme 35.
Scheme 36.
Scheme 37.
Scheme 38.
Scheme 39.
Scheme 40.
Scheme 41.
Scheme 42.
Scheme 43.
Scheme 44.
Scheme 45.
Scheme 46.
Scheme 47.
Scheme 48.
Scheme 49.
Scheme 50.
Scheme 51.
Scheme 52.
Scheme 53.
Scheme 54.
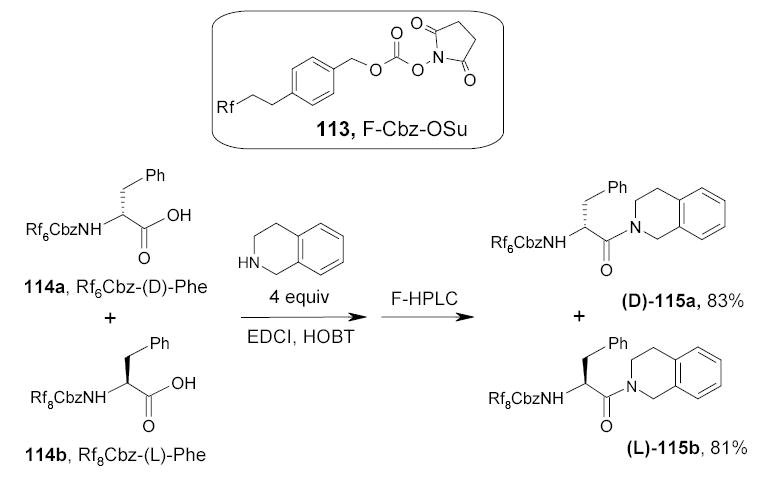
Scheme 55.
Acknowledgments
I thank Prof. Dennis Curran for introducing me to the area of fluorous chemistry and my coworkers at FTI for their research contributions. I appreciate the National Institutes of General Medical Sciences for generous SBIR and STTR funding (1R43GM062717-01, 2R44GM062717-02, 1R43GM066415-01, 1R43GM067326-01, and 1R41AI54198-01).
References
- 1.Studer A, Hadida S, Ferritto SY, Kim PY, Jeger P, Wipf P, Curran DP. Science. 1997;275:823. doi: 10.1126/science.275.5301.823. [DOI] [PubMed] [Google Scholar]
- 2.Curran DP. Angew Chem Int Ed Eng. 1998;37:1175. [Google Scholar]
- 3.Vogt, M. Ph.D. Thesis, Technische Hochschule Aachen, 1991
- 4.Zhu DW. Synthesis. 1993:953. [Google Scholar]
- 5.Horvath IT, Rabai T. Science. 1994;266:72. doi: 10.1126/science.266.5182.72. [DOI] [PubMed] [Google Scholar]
- 6.Zhang W. Tetrahedron. 2003;59:4475. [Google Scholar]
- 7.Curran DP, Hadida S, He M. J Org Chem. 1997;62:6714. [Google Scholar]
- 8.Luo Z, Zhang Q, Oderaotoshi Y, Curran DP. Science. 2001;291:1766. doi: 10.1126/science.1057567. [DOI] [PubMed] [Google Scholar]
- 9.Nakamura H, Linclau B, Curran DP. J Am Chem Soc. 2001;123:10119. doi: 10.1021/ja011716c. [DOI] [PubMed] [Google Scholar]
- 10.Wende M, Gladysz JA. J Am Chem Soc. 2003;125:5861. doi: 10.1021/ja029241s. [DOI] [PubMed] [Google Scholar]
- 11.Curran DP. Chemtracts-Org Chem. 1996;9:75. [Google Scholar]
- 12.Horvath IT. Acc Chem Res. 1998;31:641. [Google Scholar]
- 13.de Wolf E, van Koten G, Deelman BJ. Chem Soc Rev. 1999;28:37. [Google Scholar]
- 14.Cavazzinni M, Montannari F, Pozzi G, Quici S. J Fluorine Chem. 1999;94:183. [Google Scholar]
- 15.BarthelRosa L, Gladysz JA. Coord Chem Rev. 1999;192:587. [Google Scholar]
- 16.Curran DP. Med Res Rev. 1999;19:432. doi: 10.1002/(sici)1098-1128(199909)19:5<432::aid-med8>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 17.Curran DP. Pure Appl Chem. 2000;72:1649. [Google Scholar]
- 18.Curran, D. P. In Stimulating Concepts in Chemistry; Stoddard, F.; Reinhoud, D.; Shibasaki, M. Eds.; Wiley-VCH: New York, 2000; p25.
- 19.Gladysz JA. Actualite Chimique. 2000;9:47. [Google Scholar]
- 20.Curran, D. P.; Hadida, S.; Studer, A.; He, M.; Kim, S.-Y.; Luo, Z.; Larhed, M.; Hallberg, M.; Linclau, B. In Combinatorial Chemistry: A Practical Approach; H. Fenniri, Ed.; Oxford University Press: Oxford, 2001; Vol. 2; p 327.
- 21.Curran DP, Lu Z. Green Chem. 2001;3:G3. [Google Scholar]
- 22.Curran DP. Synlett. 2001:1488. [Google Scholar]
- 23.For a special issue on fluorous chemistry seeGladysz JA, Curran DP.Tetrahedron 200253823
- 24.Tzschucke CC, Markert C, Bannwarth W, Roller S, Hebel A, Haag R. Angew Chem Int Ed. 2002;41:3964. doi: 10.1002/1521-3773(20021104)41:21<3964::AID-ANIE3964>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 25.Dobbs AP, Kimberley MR. J Fluorine Chem. 2002;118:3. [Google Scholar]
- 26.Yoshida J-I, Itami K. Chem Rev. 2002;102:3693. doi: 10.1021/cr0103524. [DOI] [PubMed] [Google Scholar]
- 27.Pozzi G, Shepperson I. Coord Chem Rev. 2003;24:115. [Google Scholar]
- 28.The Handbook on Fluorous Chemistry, Gladysz, J. A.; Hovath, I.; Curran. D. Eds; Wiley-VCH: in press.
- 29.Web address. www.fluorous.com
- 30.For more information on F-SPE log on to http://fluorous.com/download/an_spe.pdf
- 31.Curran DP, Luo Z. J Am Chem Soc. 1999;121:9069. [Google Scholar]
- 32.Curran DP, Oderaotoshi Y. Tetrahedron. 2001;57:5243. [Google Scholar]
- 33.Zhang W, Luo Z, Chen CHT, Curran DP. J Am Chem Soc. 2002;124:10443. doi: 10.1021/ja026947d. [DOI] [PubMed] [Google Scholar]
- 34.http//www.keystonescientific.com
- 35.Gerendsen GE, de Galan L. J Liquid Chromatogr. 1978;1:403. [Google Scholar]
- 36.Gerendsen GE, Pikaart KA, de Galan L, Olieman C. Anal Chem. 1980;52:1990. [Google Scholar]
- 37.Billiet HAH, Schoenmakers PJ, de Galan L. J Chromatogr. 1981;218:443. [Google Scholar]
- 38.Sadek PC, Carr PW. J Chromatogr. 1984;288:25. [Google Scholar]
- 39.Nakamura Y, Takeuchi S, Okumura K, Ohgo Y, Matsuzawa H, Mikami K. Tetrahedron Lett. 2003;44:6221. [Google Scholar]
- 40.Matsuzawa H, Mikami K. Tetrahedron Lett. 2003;44:6227. [Google Scholar]
- 41.Matsuzawa H, Mikami K. Synlett. 2003:1607. [Google Scholar]
- 42.Fluorous silica gel cartridges for flash chromatography are available from Biotage and Isco.
- 43.Luo Z, Swaleh SM, Theil F, Curran DP. Org Lett. 2002;4:2585. doi: 10.1021/ol026232f. [DOI] [PubMed] [Google Scholar]
- 44.Theil F, Sonnenschein H. Pharma Chem. 2003;2(5):32. [Google Scholar]
- 45.Ryu I, Matsubara H, Yasuda S, Nakamura H, Curran DP. J Am Chem Soc. 2003;124:12945. doi: 10.1021/ja027965y. [DOI] [PubMed] [Google Scholar]
- 46.Arnold LD, Assil HI, Vederas J. J Am Chem Soc. 1989;111:3973–3976. [Google Scholar]
- 47.Tunoori AR, Dutta D, Georg GI. Tetrahedron Lett. 1998;39:8751–8754. [Google Scholar]
- 48.Starkey GW, Parlow JJ, Flynn DL. Bioorg Med Chem Lett. 1998;8:2384. doi: 10.1016/s0960-894x(98)00431-4. [DOI] [PubMed] [Google Scholar]
- 49.Kiankarimi M, Lowe R, McCarthy JR, Whitten JP. Tetrahedron Lett. 1999;40:4497. [Google Scholar]
- 50.Pelletier JC, Kincaid S. Tetrahedron Lett. 2000;41:787. [Google Scholar]
- 51.Barrett AGM, Roberts RS, Schroder J. Org Lett. 2000;2:2999. doi: 10.1021/ol006313g. [DOI] [PubMed] [Google Scholar]
- 52.Dandapani S, Curran DP. Tetrahedron. 2002;58:3855. [Google Scholar]
- 53.The use of fluorous DEAD combined with normal phosphine, seeDobbs AP, McGregor-Johnson C.Tetrahedron Lett 2002432807 [Google Scholar]
- 54.Zhang W, Curran DP, Chen CHT. Tetrahedron. 2002;58:3871. [Google Scholar]
- 55.Zhang W, Chen CHT, Nagashima T. Tetrahedron Lett. 2003;44:2065. [Google Scholar]
- 56.Lindsley CW, Zhao Z, Leister WH. Tetrahedron Lett. 2002;43:4225. [Google Scholar]
- 57.Lindsley CW, Zhao Z, Leister WH, Strauss KA. Tetrahedron Lett. 2002;43:6319. [Google Scholar]
- 58.Werner S, Curran DP. Org Lett. 2003:3293. doi: 10.1021/ol035214a. [DOI] [PubMed] [Google Scholar]
- 59.Zhang, W.; Chen, C. H.-T. unpublished results.
- 60.Lindsley CW, Zhao Z, Newton RC, Leister W, Strauss KA. Tetrahedron Lett. 2002;43:4467. [Google Scholar]
- 61.Grigg R, York M. Tetrahedron Lett. 2000;41:7255. [Google Scholar]
- 62.Schneider S, Bannwarth W. Angew Chem Int Ed. 2000;39:4142. doi: 10.1002/1521-3773(20001117)39:22<4142::aid-anie4142>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 63.Osswald T, Schneider S, Wang S, Bannwarth W. Tetrahedron Lett. 2001;42:2965. [Google Scholar]
- 64.Schneider S, Bannwarth W. Helv Chim Acta. 2001;84:735. [Google Scholar]
- 65.De Campo F, Lastercoueres D, Vincent JM, Verlhac JB. J Org Chem. 1999;64:4969. doi: 10.1021/jo990134z. [DOI] [PubMed] [Google Scholar]
- 66.Yao, Q.; Zhang, Y. J. Am. Chem. Soc.2003, ASAP article.
- 67.Hadida S, Super MS, Beckman EJ, Curran DP. J Am Chem Soc. 1997;119:7406. [Google Scholar]
- 68.Curran DP, Hadida S, Kim SY, Luo Z. J Am Chem Soc. 1999;121:6607. [Google Scholar]
- 69.Ryu I, Niguma T, Minakata S, Komatsu M, Hadida S, Curran DP. Tetrahedron Lett. 1997;38:7883. [Google Scholar]
- 70.Mulholland, K. 16th Process Development Symposium, SCI, December 3, 1998, London, UK.
- 71.Curran DP, Hoshino M. J Org Chem. 1996;61:6480. doi: 10.1021/jo961309x. [DOI] [PubMed] [Google Scholar]
- 72.Hoshino M, Degenkolb P, Curran DP. J Org Chem. 1997;62:8341. doi: 10.1021/jo9709413. [DOI] [PubMed] [Google Scholar]
- 73.Curran DP, Hadida S, Kim SY. Tetrahedron. 1999;55:8997. [Google Scholar]
- 74.Hao X, Yamazaki O, Yoshida A, Nishikido J. Green Chem. 2003;5:534. [Google Scholar]
- 75.Yoshida A, Hao X, Yamazaki O, Nishikido J. Green Chem. 2003;5:554. [Google Scholar]
- 76.Yamazaki O, Hao X, Yoshida A, Nishikido J. Tetrahedron Lett. 2003;44:8791. [Google Scholar]
- 77.Crich D, Hao X, Lucas MA. Org Lett. 1999;1:269. [Google Scholar]
- 78.Crich D, Hao X, Lucas Tetrahedron. 1999;55:14261. [Google Scholar]
- 79.Crich D, Barba GR. Org Lett. 2000;2:989. doi: 10.1021/ol005669p. [DOI] [PubMed] [Google Scholar]
- 80.Crich D, Neelamkavil S, Sartillo-Piscil F. Org Lett. 2000;2:4029. doi: 10.1021/ol0066532. [DOI] [PubMed] [Google Scholar]
- 81.Brink GJ, Vis JM, Arends IWCE, Sheldon RA. Tetrahedron. 2002;58:3977. [Google Scholar]
- 82.Barthelemy S, Schneider S, Wang S, Bannwarth W. Tetrahedron Lett. 2002;43:807. [Google Scholar]
- 83.Lindsley, C. W.; Zhou, Z. in The Handbook on Fluorous Chemistry, Gladysz, J.; Hovath, I.; Curran. D. Eds; Wiley-VCH: in press.
- 84.Wipf P, Reeves JT, Balachandran R, Giuliano KA, Hamel E, Day BW. J Am Chem Soc. 2000;122:9391. [Google Scholar]
- 85.Wipf P, Methot JL. Org Lett. 1999;1:1253. [Google Scholar]
- 86.Zhang W, Lu Y. Org Lett. 2003;5:2555. doi: 10.1021/ol034854a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lu, Y.; Zhang, W. unpublished results.
- 88.Zhang W. Org Lett. 2003;5:1011. doi: 10.1021/ol027469e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chen CHT, Zhang W. Org Lett. 2003;5:1015. doi: 10.1021/ol0274864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cioffi, C. L.; Berlin, M. L.; Herr, R. J. (Albany Molecular Research, Inc), personal communication.
- 91.Wolfe JP, Åhman J, Sadighi JP, Singer RA, Buchwald SL. Tetrahedron Lett. 1997;38:6367. [Google Scholar]
- 92.Studer A, Curran DP. Tetrahedron. 1997;53:6681. [Google Scholar]
- 93.Mazoni L. Chem Commun. 2003:2930. doi: 10.1039/b311448a. [DOI] [PubMed] [Google Scholar]
- 94.Luo Z, Williams J, Read RW, Curran DP. J Org Chem. 2001;66:4261. doi: 10.1021/jo010111w. [DOI] [PubMed] [Google Scholar]
- 95.Schwinn D, Bannwarth W. Helv Chim Acta. 2002;85:255. [Google Scholar]
- 96.Read R, Zhang C. Tetrahedron Lett. 2003;44:7045. [Google Scholar]
- 97.Unpublished results of Read, R. and Zhang, C., University of New South Wales.
- 98.Curran DP, Ferritto R, Hua Y. Tetrahedron Lett. 1998;39:4937. [Google Scholar]
- 99.Miura T, Hirose Y, Ohmae M, Inazu T. Org Lett. 2001;3:3947. doi: 10.1021/ol016838o. [DOI] [PubMed] [Google Scholar]
- 100.Miura T, Inazu T. Tetrahedron Lett. 2003;44:1819. [Google Scholar]
- 101.Miura T, Goto K, Hosaka D, Inazu T. Angew Chem Int Ed. 2003;42:2047. doi: 10.1002/anie.200250531. [DOI] [PubMed] [Google Scholar]
- 102.Studer A, Jeger P, Wipf P, Curran DP. J Org Chem. 1997;62:2917. doi: 10.1021/jo970095w. [DOI] [PubMed] [Google Scholar]
- 103.Unpublished results of Zhang, W.; Lu, Y. and Chen; C. H.-T.
- 104.Larhed M, Hoshino M, Hadida S, Curran DP, Hallberg A. J Org Chem. 1997;62:5583. [Google Scholar]
- 105.Olofeeson K, Kim SY, Larhed M, Curran DP, Hallberg A. J Org Chem. 1999;64:4539. [Google Scholar]
- 106.Hulme C, Gore C. Curr Med Chem. 2003;10:51. doi: 10.2174/0929867033368600. [DOI] [PubMed] [Google Scholar]
- 107.Tempest P, Ma V, Kelly MG, Jones W, Hulme C. Tetrahedron Lett. 2001;42:4963. [Google Scholar]
- 108.Nixey T, Tempest P, Hulme C. Tetrahedron Lett. 2002;43:1637. [Google Scholar]
- 109.Tempest P, Pettus L, Gore V, Hulme C. Tetrahedron Lett. 2003;44:1947. [Google Scholar]
- 110.Unpublished results of Zhang, W. and Tempest, P. (Amgem).
- 111.Unpublished results of Zhang, W.; Nagashima, T.; Chen, C. H.-T. and Lu, Y.
- 112.Zhang W, Lu Y, Chen CHT. Mol Diversity. 2003;7:199. doi: 10.1023/b:modi.0000006825.12186.5f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Filippov DV, van Zoelen DJ, Oldfield SP, van der Marel GA, Overkleeft HS, Drijfhout JW, van Boom JH. Tetrahedron Lett. 2002;43:7809. [Google Scholar]
- 114.Palmacci ER, Hewitt MC, Seeberger PH. Angew Chem Int Ed. 2001;40:4433. doi: 10.1002/1521-3773(20011203)40:23<4433::aid-anie4433>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 115.Miura T, Hirose Y, Ohmae M, Inazu T. Org Lett. 2001;3:3947. doi: 10.1021/ol016838o. [DOI] [PubMed] [Google Scholar]
- 116.Wipf, P.; Jon, R.; Rover, S. US Patent 06673539.
- 117.Schwinn D, Glatz H, Bannwarth W. Helv Chim Acta. 2003;86:188. [Google Scholar]
- 118.Matsubara H, Yasuda S, Ryu I. Synlett. 2003:247. [Google Scholar]
- 119.Zhang W. Chim Oggi. 2003;21(3–4):22. [Google Scholar]
- 120.Zhang Q, Rivkin A, Curran DP. J Am Chem Soc. 2002;124:5774. doi: 10.1021/ja025606x. [DOI] [PubMed] [Google Scholar]
- 121.Curran DP, Amatore M, Campbell M, Go E, Guthrie D, Luo Z. J Org Chem. 2003;68:4643. doi: 10.1021/jo0344283. [DOI] [PubMed] [Google Scholar]
- 122.Zhang Q, Lu H, Richard C, Curran DP. J Am Chem Soc. 2004;126:36. doi: 10.1021/ja038542e. [DOI] [PubMed] [Google Scholar]