

Abstract
Rapid amplification of cDNA ends (RACE) is widely utilized to determine the 5′- and 3′-terminal nucleotide sequences of genes. Many different RACE methods have been developed to meet various requirements, but none address the difficult problems that arise when trying to isolate the ends of extremely GC-rich genes. In this study, we found that we were unable to isolate the correct 5′ or 3′ ends of an insect gene, which appeared to include extremely GC-rich sequences, using current RACE methods. Thus, we developed a new RACE method that can be used for this purpose. This new method entails first strand cDNA synthesis at 70°C with Thermo-X™ reverse transcriptase in the presence of homoectoine, followed by a polymerase chain reaction with 98°C denaturation steps and Phusion™ DNA polymerase in the presence of 1M betaine and 5% DMSO. The use of these conditions yielded 5′- and 3′-RACE products that were about 80% GC over 213 and 162 bp, respectively, and included shorter, internal regions of 82–89% GC.
Introduction
Rapid amplification of cDNA ends (RACE) is an important method that can be used to isolate cDNA fragments derived from the 5′- and 3′-ends of genes for subsequent determination of their nucleotide sequences [1]. Previous reports have described a wide array of different RACE methods [2–4]. Generally, these methods involve the addition of a defined sequence, or “anchor” to the 5′- or 3′- end of first-strand cDNAs, which sets the stage for subsequent polymerase chain reactions (PCRs) using a primer complementary to the anchor sequence together with one complementary to a known, internal, gene-specific sequence. However, a problem with these basic 5′-RACE methods was that they could not distinguish between cDNA products derived from full-length or degraded mRNA templates. Thus, other RACE methods were developed to address this problem and to exclusively amplify the 5′ ends of full-length cDNAs [5–13].. While these relatively newer 5′-RACE methods are now routinely used to amplify the 5′ ends of full-length mRNAs, new RACE methods have not been designed to address the additional problems encountered when working with extremely GC-rich genes. We recently encountered these problems in our efforts to use current RACE methods to determine the correct 5′- and 3′-terminal sequences of an extremely GC-rich insect gene. Thus, we developed a new RACE method, which ultimately allowed us to successfully isolate the correct ends of this gene for subsequent nucleotide sequence analysis.
Materials and methods
mRNA isolation
mRNA was isolated either from second instar Trichoplusia ni larvae (Chesapeake-PERL, Savage, MD) or from the Trichoplusia ni cell line, BTI-Tn5B1-4 (High Five®; [14]). Briefly, total RNA was isolated from one million BTI-Tn5B1-4 cells or from one second instar larva using 1 mL of Tri-Reagent® (MRC, Cincinnati, OH) according to the manufacturer’s instructions. Subsequently, total RNA was subjected to oligo(dT)-cellulose fractionation using the Poly(A) Purist™ mRNA purification kit (Ambion, Austin, TX) according to the manufacturer’s instructions. The eluted mRNA was spectrophotometrically quantified, aliquoted, and stored at −85°C.
The new 5′-RACE method (Fig. 1A)
Figure 1.
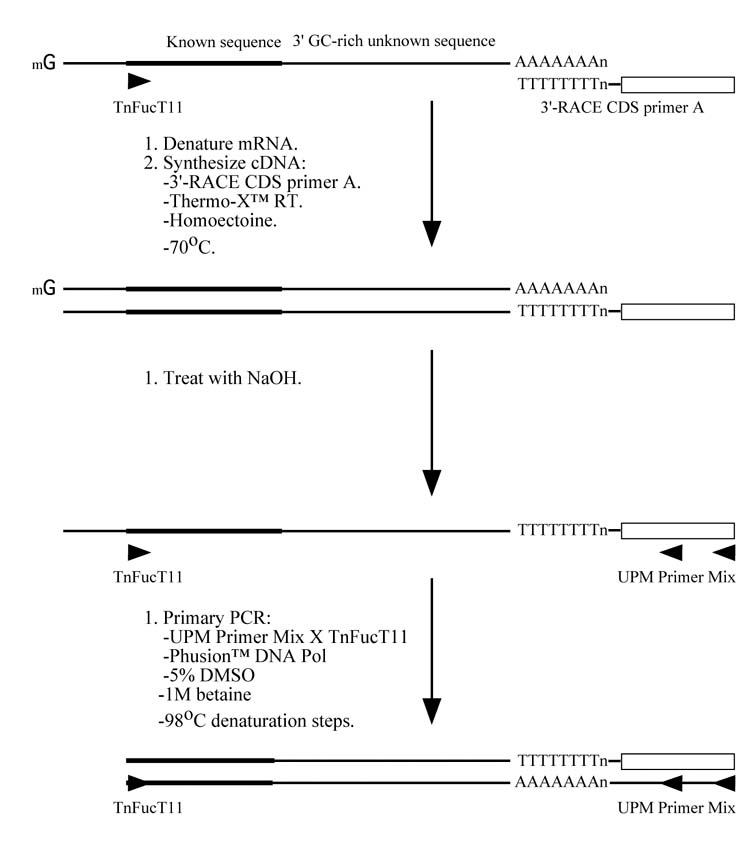
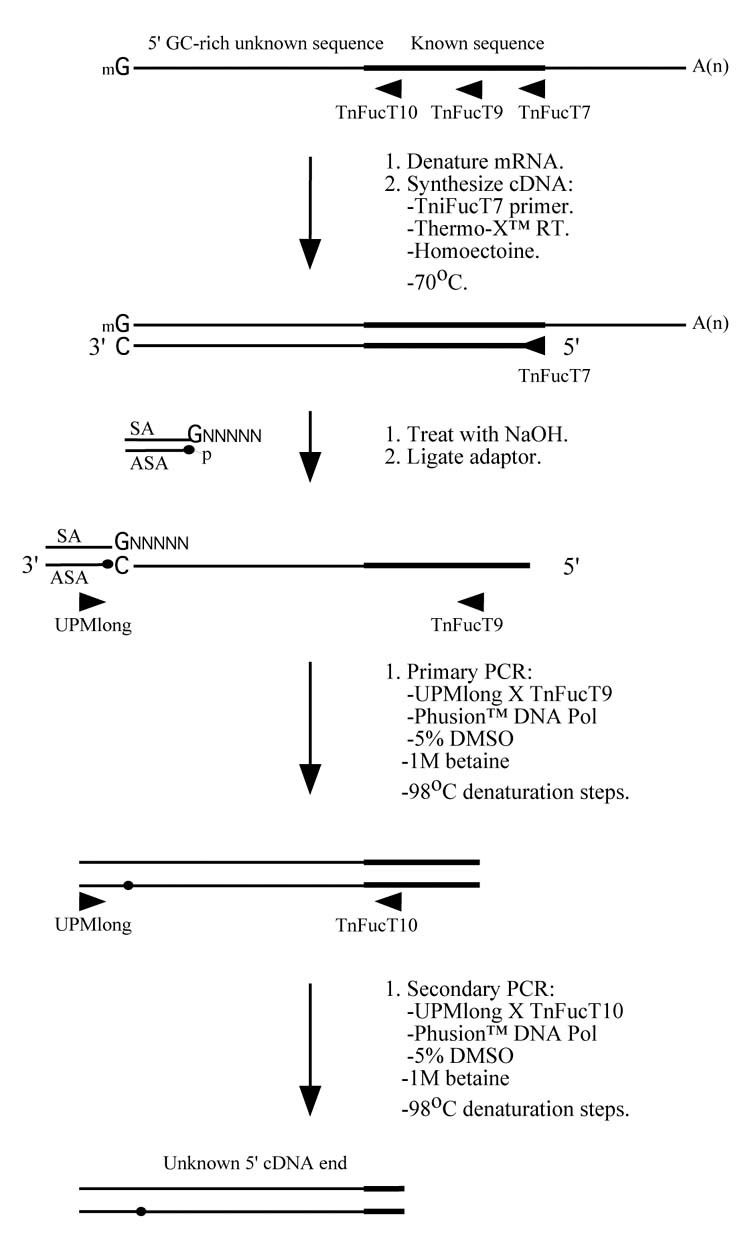
A new RACE method for extremely GC-rich genes. (A) For 5′-RACE, the first strand cDNA is synthesized at 70°C using Thermo-X reverse transcriptase in the presence of 0.4875 M homoectoine and a gene specific primer (in this case, FucT7). The mRNA is then hydrolyzed and the cDNA is denatured in preparation for the addition of an adaptor, which provides an anchor sequence that can be used for subsequent semi-nested PCRs. The primary and secondary PCRs are performed using nested internal gene specific primers (in this case, TniFucT9 and TniFucT10), a primer complementary to the antisense adaptor sequence, ASA (in this case, a commercial primer called UPMlong), Phusion™ DNA polymerase, and a denaturation temperature of 98°C. (B) For 3′-RACE, the first strand cDNA is synthesized at 70°C using Thermo-X reverse transcriptase in the presence of 0.4875 M homoectoine and a primer complementary to the poly-A tail of the mRNA (in this case, a commercial primer called 3′-RACE CDS primer A). The mRNA is then hydrolyzed and a PCR is performed using a gene specific primer (in this case, TnFucT11) and a primer complementary to the specific portion of the oligonucleotide used to prime the reverse transcription reaction (in this case, a commercial primer called UPM primer mix). The other conditions used for the PCR are identical to those used for 5′-RACE and nested PCR may be used, if required.
cDNA synthesis
One microgram of Trichoplusia ni mRNA was denatured at 70°C for 2 min in the presence of 0.35 μM of a gene-specific primer (TniFucT7; Table 1), 0.70 mM of each dNTP, and 0.67 M homoectoine (courtesy of Dr. Erwin A. Galinski, Institute of Microbiology & Biotechnology, University of Bonn) in a total reaction volume of 14.5 μL. The mixture was immediately quenched on ice for 2 min, and then 4 μL of 5X Thermo-X™ reaction buffer and 1.5 μL of Thermo-X™ reverse transcriptase (Invitrogen, Carlsbad, CA) were added. After a very brief microcentrifugation, the mixture was incubated for 5 min at 55°C, for 45 min at 70°C, and then for 15 min at 75°C in a thermal cycler (Eppendorf MasterCycler epgradient S; Brinkmann Instruments, Inc., New York, NY). The reaction was subsequently treated with an equal volume of 0.6 N NaOH for 20 min at 65°C to hydrolyze the mRNA and denature the first strand cDNA, and then the cDNA was precipitated by adjusting the solution to 0.5 M NaCl and adding 2.5 times the total volume of 100% ethanol. After a 2 h incubation at −80°C, the cDNA was pelleted by microcentifugation for 30 min at 13,000 × g, washed once with 70% ethanol, and dissolved in 10 μL of 10 μM of a commercial “ligation adaptor”, which was prepared as described below.
Table 1.
Oligonucleotides used in this study
| Name | Tm | Sequence (5′ to 3′) |
|---|---|---|
| UPMlong | 77°C | CTAATACGACTCACTATAGGGCAAGCAGTGGTATCAACGCAGAGT |
| UPMshort | 53°C | CTAATACGACTCACTATAGGGC |
| Sense Adaptor (SA) | 71°C | AAGCAGTGGTATCAACGCAGAGTGNNNNN |
| Antisense Adaptor (ASA) | 62°C | p-ACTCTGCGTTGATACCACTGCTT (5′-phosphorylated) |
| 3′-RACE CDS Primer A | 62°C | AAGCAGTGGTATCAACGCAGAGTAC(T)30VN |
| TniFucT7 | 70°C | GGATGTGCCTGCTGAGCTGCC |
| TniFucT9 | 74°C | GCGCCAAAACCGTTCGCAAGGAG |
| TniFucT10 | 73°C | CCTGGTGCGCCTTCAGGGAACAG |
| TniFucT11 | 61°C | GATGCTCGACAGGGACTACAAG |
Adaptor ligation
The “ligation adaptor” was prepared by mixing two commercial oligonucleotides, termed “sense adaptor” and “antisense adaptor” (Table 1; Invitrogen), together at final concentrations of 10 μM each in an annealing buffer consisting of 10 mM Tris-HCl (pH 8.0) and 50 mM NaCl. The mixture was placed in a boiling water bath, which was then allowed to slowly cool to room temperature, and the resulting adaptor was added to the first strand cDNA, as described above. Subsequently, a ligation reaction was performed using 3 μL of the cDNA-adaptor mixture, 1.0 μL of 10X T4 DNA ligase buffer, and 1.0 μL of T4 DNA ligase (New England Biolabs, Beverly, MA) in a total volume of 10 μL. This mixture was incubated overnight at room temperature and then the adaptor-ligated cDNA was diluted 5-fold with ddH2O prior to being used as the template for subsequent PCRs.
PCR amplification
All PCRs were performed in an Eppendorf MasterCycler epgradient S cycler (Brinkmann) equipped with a silver block. Primary PCR mixtures included 0.5 μL of Phusion™ DNA polymerase (New England Biolabs), the commercial Phusion™ GC Buffer, all four dNTPs at final concentrations of 0.2 mM each, 1.0 μL of the diluted adaptor-ligated first strand cDNA described above, a gene-specific primer (TniFucT9; Table 1) at a final concentration of 0.2 μM, a primer complementary to the 3′ end of the adaptor sequence (UPMlong, BD Biosciences; Table 1) at a final concentration of 0.2 μM, 5% (v/v) DMSO (Sigma-Aldrich, St. Louis, MO), and 1 M betaine (Sigma-Aldrich) in a total volume of 50 μL. Touchdown cycling conditions [15] were used for the primary amplification, with an initial denaturation step for 3 min at 98°C, 5 PCR cycles each at three different annealing temperatures (70°C, 63°C, and 56°C), and then 30 PCR cycles with an annealing temperature of 50°C. Each PCR cycle included a 30 second denaturation step at 98°C, a 15 second annealing step at the temperatures indicated above, and a complex extension step involving the use of increasing times and temperatures (15 seconds at 72°C, followed by 20 seconds at 75°C, followed by 25 seconds at 78°C), which was designed to progressively melt any secondary structures that might have formed during the preceding annealing step.
Semi-nested secondary PCRs were performed using 1 μL of the spent primary PCR together with a nested gene-specific primer (TnFucT10; Table 1) and the commercial UPMlong primer used for the primary reaction. Other than the template and the nested gene-specific primer, the secondary PCR components were identical to the primary PCRs. The secondary PCR cycling conditions included an initial 3 min denaturation step at 98°C, followed by 30 cycles of (i) 30 sec denaturation at 98°C, (ii) 15 sec annealing at 64°C, and (iii) increasing extension times and temperatures, as described above.
Other 5′-RACE methods
Additional 5′-RACE methods, which were used in our initial failed attempts to isolate the 5′ end of the insect gene of interest in this project, were performed using SMART-RACE™ (BD Biosciences, Palo Alto, CA), RLM-RACE™ (Ambion), and Gene-Racer™ (Invitrogen) kits, with the reverse transcriptases and thermostable DNA polymerases provided in those kits, according to the manufacturer’s instructions.
The new 3′-RACE method (Fig. 1B)
First strand cDNA synthesis was performed with 1 μg of T. ni mRNA at 70°C with Thermo-X reverse transcriptase (Invitrogen) in the presence of homoectoine and a commercial 3′-RACE primer (3′-RACE CDS Primer A, BD Biosciences; Table 1), as described for 5′-RACE, above. The ethanol-precipitated first strand cDNA pellet was dissolved in 250 uL of a commercial Tricine-EDTA buffer (BD Biosciences), and then 2.5 μL was used as the template for a PCR with Phusion™ DNA polymerase (New England Biolabs), a gene specific forward primer (TnFucT11; Table 1), and a commercial reverse primer mix UPM, which is a mixture of UPMlong and UPMshort in a 1:5 ratio (BD Biosciences; Table 1). Except for the primers, the PCR components were the same as those described for 5′-RACE, above. The reaction conditions included an initial denaturation for 3 min at 98°C, followed by 5 cycles of amplification at each of the following annealing temperatures: 72°C, 68°C, and 65°C. In each case, the annealing time was 30 sec, extension was for 1 min at 72°C, and denaturation was for 30 sec at 98°C. These cycles were followed by an additional 30 cycles of (i) denaturation for 30 sec at 98°C, (ii) annealing for 30 sec at 52°C, and (iii) extension for 1 min at 72°C.
Other 3′-RACE method
The following describes the 3′-RACE method used as a representative “standard” method in the final phase of this study. First strand cDNA synthesis was performed with 1 μg of T. ni mRNA at 42°C with PowerScript™ reverse transcriptase (BD Bioscience) and a commercial 3′-RACE primer (3′-RACE CDS Primer A, BD Biosciences; Table 1) under the conditions recommended by the manufacturer. The ethanol-precipitated first strand cDNA pellet was dissolved in 250 uL of a commercial Tricine-EDTA buffer (BD Biosciences), and then 2.5 μL was used as the template for a PCR with PfuUltra™ DNA polymerase (Stratagene, La Jolla,CA), a gene specific forward primer (TnFucT11; Table 1), and a commercial reverse primer mix (UPM, BD Biosciences; Table 1). All other PCR components were those recommended by Stratagene for 3′-RACE with PfuUltra™ DNA polymerase. The reaction conditions included an initial denaturation for 2 min at 94°C, followed by 5 cycles of denaturation for 30 sec at 94°C and annealing/extension for 6 min at 72°C, 5 cycles of denaturation for 30 sec at 94°C and annealing/extension for 6 min at 70°C, and 16 cycles of touchdown PCR that included (i) denaturation for 30 sec at 94°C, (ii) annealing for 30 sec at temperatures ranging from 68°C to 60°C, with a decrease of 0.5°C per cycle, and (iii) extension for 6 min at 72°C. The final touchdown step was followed by an additional 25 cycles under the same conditions as those used for the final touchdown step.
Results and Discussion
As part of a broader research program in insect glycobiology, we undertook efforts to clone a core α1,3 fucosyltransferase gene from the lepidopteran insect, Trichoplusia ni. A previously described degenerate PCR approach [16] was used to successfully isolate an internal fragment of this gene (R.L. Harrison and D.L. Jarvis, unpublished results), which was designated TnFT3. This fragment was cloned and sequenced and the nucleotide sequence data were used to design subsequent 3′RACE experiments, which yielded a putative 3′ nucleotide sequence for the TnFT3 gene (data not shown). In stark contrast, however, multiple attempts to use a variety of previously described 5′-RACE methods failed to amplify the 5′ end of this gene.
Fortunately, some of these latter experiments yielded partial 5′-RACE products, which extended the TnFT3 sequence by a small distance in the 5′ direction. These results revealed that the presumed gene-specific sequence ended abruptly with short, highly GC-rich sequences, followed by sequences that bore no recognizable resemblance to known core α1,3 fucosyltransferase sequences (data not shown). These results suggested that the 5′ end of the TnFT3 gene might have an extremely high GC content, which allowed the mRNA template to form a highly stable hairpin structure(s) that was not accessible to the reverse transcriptase under any conditions utilized to this point in our study. Thus, the reverse transcriptase would copy the TnFT3 mRNA until encountering the hairpin(s), at which point it would jump to template sequences located upstream of the hairpin(s). Presumably, these latter template sequences would be far enough upstream of the coding and 5′-untranslated regions tied up in secondary structure(s) to be unrecognizable as core α1,3 fucosyltransferase sequences in our data analyses.
In theory, 5′-RACE can fail when applied to extremely GC-rich genes due to problems with either the first-strand cDNA synthesis reaction or the subsequent PCR amplification. In practice, first strand cDNA synthesis problems are usually assumed to be the major reason for 5′-RACE failures, and this was the focus of our troubleshooting efforts. Because conventional reverse transcriptases are not thermostable, the reverse transcription reactions are usually performed at a relatively low temperature (42°C), which cannot melt secondary structures that can form in GC-rich mRNAs. We attempted to address this problem by using recently commercialized thermostable reverse transcriptases for first strand cDNA synthesis reactions at 50°C (SuperScriptIII™) or 65°C (Thermo-X™), but with no success (data not shown). Thus, we turned to homoectoine, which has an exceptional ability to decrease the melting temperature of DNA and has been used as a potent PCR enhancer [17]. It seemed likely that homoectoine might be able to decrease the melting temperature of our extremely GC-rich mRNA template and, if it could do so without inactivating the reverse transcriptase, it should facilitate the reverse transcription step in our 5′-RACE method.
We examined this possibility by performing a first strand cDNA synthesis reaction using Thermo-X™ reverse transcriptase at 70°C in the presence of 0.4875 M homoectoine, as described in Materials and Methods and illustrated in Fig. 1A. To anchor the 3′ end of the first strand cDNA for subsequent PCR amplification steps, we used an adaptor ligation approach similar to a previously described single-strand linker ligation method (Fig. 1), which had been originally developed for full-length cDNA library construction [18]. The sense strand of the adaptor (SA) included a 3′ overhang sequence (GNNNNN) designed to anneal to the 3′ end of the first strand cDNA and the antisense strand of the adaptor (ASA) included a free 5′ phosphate for subsequent ligation of the adaptor molecule to the first strand cDNA. It should be noted that this method was not designed to selectively amplify only full-length templates, as the adaptor could theoretically be linked to any degraded first strand cDNA molecule with a 3′-terminal deoxycytidine residue. We minimized this possibility by using great care to prevent degradation during the isolation of mRNA. Expecting an extremely high GC content in the 3′ end of the first strand cDNA product, we also incorporated special denaturation measures into our method prior to the adaptor addition. Specifically, the first strand cDNA was heated for 20 min at 65°C in a solution of 0.3 N NaOH. The denatured cDNA was then ethanol precipitated, directly re-dissolved in the adaptor solution, and the DNAs were ligated, as described in Materials and Methods and illustrated in Fig. 1A.
We were concerned that secondary structure also might adversely impact the PCR steps in our 5′-RACE procedure. Previous reports have shown that complete denaturation of DNA at 98°C is essential for successful amplification of highly GC-rich sequences [19, 20]. Other previous reports have shown that 5% DMSO and 1 M betaine can be used together to reduce stable secondary structures in DNA templates [21]. Thus, we used a denaturation temperature of 98°C and included both 5% DMSO and 1 M betaine in our PCRs to eliminate putative secondary structures and facilitate 5′-RACE of the adaptor-ligated cDNA preparation (Fig. 1A). Under these conditions, we found that neither Taq nor Pfu DNA polymerases produced specific amplification products (data not shown). Conversely, Phusion™ DNA polymerase was able to produce specific amplification products under these conditions. Finally, the PCR conditions developed as part of our new 5′-RACE method included unusual extension steps with progressively increasing times and temperatures, as described in Materials and Methods. This was designed to progressively melt any secondary structures that might have formed during the preceding annealing step.
A stepwise description of our new 5′-RACE method for extremely GC-rich genes is given in Materials and Methods and illustrated in Fig. 1. The results obtained when we used this new 5′-RACE method to try to isolate the 5′ end of the TnFT3 gene are shown in Fig. 2. Whereas all other conditions utilized in this study failed to produce a full-length 5′-RACE product, the new method described herein produced four major DNA fragments ranging from about 450–800 bp in length, each of which was large enough to accommodate the expected size of the open reading frames of the known core α1,3 fucosyltransferases. Direct sequencing of these 5′-RACE products, followed by BLAST-P [22] and CLUSTAL-W [23] analyses of the theoretical translation products revealed that each was specific, as both translation products were highly similar to known core α1,3 fucosyltransferases (data not shown). The 800 bp fragment extended the putative TnFT3 sequence from the internal primer through a putative translational initiation codon located at about the expected distance upstream of that primer, and well into the 5′ untranslated region. The 450 bp fragment included an identical 3′ sequence, but had a shorter 5′ untranslated region. The two fragments of intermediate size appeared to be specific, as they did not appear in the single primer control lanes, but were not sequenced because they probably arose from mRNA species of intermediate size with 5′-terminal deoxycytidine residues.
Figure 2.
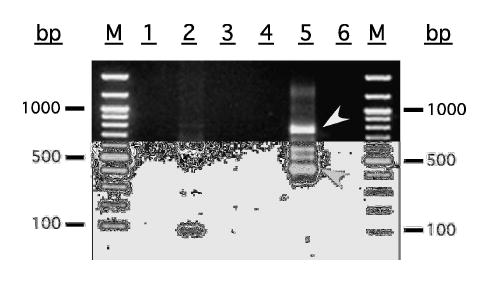
Products obtained using new method to isolate 5′ end of an extremely GC-rich insect gene. Primary PCR products are shown in lanes 1 (UPMlong single primer control), 2 (UPMlong+TniFucT9), and 3 (TniFucT9 single primer control). Secondary PCR products are shown in lanes 4 (UMPlong single primer control), 5 (UPMlong+TniFucT10), and 6 (TniFucT10 single primer control). The two specific TnFT3 5′-RACE products are marked by the arrowheads. The lane marked M contains a 100 bp DNA ladder (New England Biolabs).
A detailed description of the putative TnFT3 gene, together with a functional analysis of the gene product is in progress and will be described elsewhere. However, in the context of this methodological report, it is important to discuss its GC content (data not shown). The overall GC content of the full-length TnFT3 cDNA turned out to be 48% over 3263 bp, which is not significantly biased. On the other hand, the 5′ end of the open reading frame included extremely GC-rich sequences, as predicted. In fact, the TnFT3 sequence begins with a 104 bp sequence in the 5′ untranslated region with a GC content of 68%. This is followed by a 213 bp sequence with a GC content of 78%, which includes the putative initiation codon. Within this latter 213 bp sequence, there are two smaller nucleotide sequences with even higher GC levels. The first is a 63 bp sequence with a GC content of 89% and the second is a 61 bp sequence with a GC content of 87%. Thus, it is likely that the extremely high GC content at the 5′ end of the TnFT3 mRNA and first strand cDNA resulted in the formation of extensive secondary structures that interfered with our ability to isolate the 5′ end for sequence analysis by conventional 5′-RACE methods.
At this point in our study, we believed we had determined the correct 5′ and 3′ ends of the putative TnFT3 gene. Our next goal was to characterize the biochemical properties of the gene product. Thus, we designed PCRs to isolate the sequences encoding the full-length enzyme and its computer-predicted soluble domain for enzyme expression, purification, and activity assays. To avoid potential problems associated with the presence of extremely GC-rich sequences in the TnFT3 gene, we produced the first strand cDNAs and performed the PCRs using the new conditions we had developed for 5′-RACE, as detailed in Fig. 1, except we used gene specific primers and performed only a single round of PCR. This approach yielded amplification products of the expected size (data not shown), which were cloned and sequenced. To our surprise, however, the sequencing results revealed that the 3′ end of the TnFT3 open reading frame included an additional 73 nucleotides, relative to the sequence that we had originally determined using a conventional 3′-RACE method (data not shown). This new sequence had a GC content of 82% and was embedded within a larger sequence of 162 nucleotides with a GC content of 80% (data not shown). Thus, it appeared that we had isolated the incorrect 3′ end of the TnFT3 gene in our original 3′-RACE experiments due, once again, to the presence of an extremely GC-rich sequence. This led us to perform an experiment in which we compared the results obtained using a “standard” 3′-RACE method or a new method using the conditions we had developed for 5′-RACE (Fig. 1B), as described in Materials and methods.
Analysis of the products of these 3′-RACE reactions on an agarose gel with ethidium bromide staining showed that the established 3′-RACE method yielded no detectable product, whereas the new conditions outlined in Fig. 1B yielded a 3′-RACE product of about the expected size (1.9 kb; Fig. 3). Direct sequence analysis of this product confirmed that it was TnFT3-specific and that it contained the 73 bp GC-rich sequence that had apparently been deleted in our original 3′-RACE experiment (data not shown). It is noteworthy that the standard 3′-RACE conditions used in this experiment failed to yield a product, which was inconsistent with the fact that we had obtained a TnFT3-specific product, albeit with a 73 bp deletion, in our initial “standard” 3′-RACE experiments. This apparent discrepancy is easily explained by the fact that different amplification conditions were used for our original experiment and the experiment shown in Fig. 3. Specifically, the original 3′-RACE method involved secondary PCR with nested primers, whereas both of the 3′-RACE methods included in the controlled experiment shown in Fig. 3 involved only primary PCRs. Secondary PCRs were not done in the latter experiment because this was not required to produce the correct 3′ end of the TnFT3 gene using the new 3′-RACE method.
Figure 3.
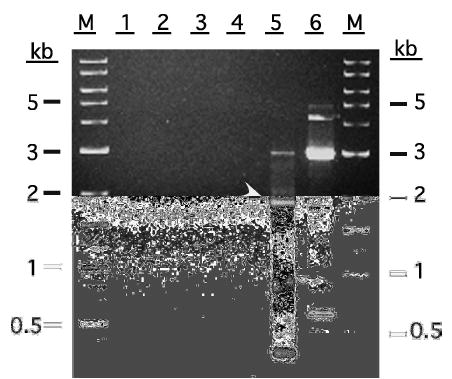
Products obtained using current and improved 3′-RACE methods with an extremely GC-rich gene. The results obtained with a representative “standard” 3′-RACE method described in Materials and methods are shown in lanes 1 (UPM primer control), 2 (UPM+TniFucT11), and 3 (TniFucT11 single primer control). The results obtained using the improved 3′-RACE method described in Materials and methods are shown in lanes 4 (UPM primer control), 5 (UPM+TniFucT11), and 6 (TniFucT11 single primer control). The specific TnFT3 3′-RACE product is marked by the arrowhead. The lane marked M contains a 1 kb and 100 bp DNA ladders (New England Biolabs).
Finally, we note that in addition to using the new RACE method described in this paper to successfully isolate the extremely GC-rich ends of the TnFT3 gene, we also have used the conditions used for the cDNA synthesis portion of our new method to successfully amplify another problematic insect gene. This latter gene included a 125 bp sequence that is 84% GC, which was consistently deleted when the full length coding sequence was amplified using conventional reverse transcription and PCR methods (data not shown). Thus, we anticipate that the new RACE method described in this report, as well as the conditions used for the PCR steps, will be widely applicable for the isolation of genes with extremely high GC content.
Acknowledgments
This study was supported by the National Institute of Standards and Technology Advanced Technology Program (Award #70NANB3H3042) and by the National Institutes of Health (Award GM49734). We thank Dr. Erwin A. Galinski of the Institute of Microbiology & Biotechnology, University of Bonn, for generously providing homoectoine. We also thank Dr. Robert L. Harrison of the USDA-ARS (Beltsville, MD) for his contributions to the early stages of this project.
References
- 1.Schaefer BC. Revolutions in rapid amplification of cDNA ends: new strategies for polymerase chain reaction cloning of full-length cDNA ends. Analyt Biochem. 1995;227:255–273. doi: 10.1006/abio.1995.1279. [DOI] [PubMed] [Google Scholar]
- 2.Frohman MA, Dush MK, Martin GR. Rapid production of full-length cDNAs from rare transcripts: amplification using a single gene-specific oligonucleotide primer. Proc Natl Acad Sci USA. 1988;85:8998–9002. doi: 10.1073/pnas.85.23.8998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Troutt AB, McHeyzer-Williams MG, Pulendran B, Nossal GJ. Ligation-anchored PCR: a simple amplification technique with single-sided specificity. Proc Natl Acad Sci USA. 1992;89:9823–9825. doi: 10.1073/pnas.89.20.9823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schmidt WM, Mueller MW. Controlled ribonucleotide tailing of cDNA ends (CRTC) by terminal deoxynucleotidyl transferase: a new approach in PCR-mediated analysis of mRNA sequences. Nuc Acids Res. 1996;24:1789–1791. doi: 10.1093/nar/24.9.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schmidt WM, Mueller MW. CapSelect: a highly sensitive method for 5′ CAP-dependent enrichment of full-length cDNA in PCR-mediated analysis of mRNAs. Nuc Acids Res. 1999;27:e31. doi: 10.1093/nar/27.21.e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shi X, Karkut T, Chahmanhkah M, Alting-Mees M, Hemmingsen SM, Hegedus D. 5′-RACEing across a bridging oligonucleotide. Biotechniques. 2002;32:480–482. doi: 10.2144/02323bm01. [DOI] [PubMed] [Google Scholar]
- 7.Matz M, Shagin D, Bogdanova E, Britanova O, Lukyanov S, Diatchenko L, Chenchik A. Amplification of cDNA ends based on template-switching effect and step-out PCR. Nuc Acids Res. 1999;27:1558–1560. doi: 10.1093/nar/27.6.1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maruyama K, Sugano S. Oligo-capping: a simple method to replace the cap structure of eukaryotic mRNAs with oligoribonucleotides. Gene. 1994;138:171–174. doi: 10.1016/0378-1119(94)90802-8. [DOI] [PubMed] [Google Scholar]
- 9.Volloch V, Schweitzer B, Rits S. Ligation-mediated amplification of RNA from murine erythroid cells reveals a novel class of beta globin mRNA with an extended 5′-untranslated region. Nuc Acids Res. 1994;22:2507–2511. doi: 10.1093/nar/22.13.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mules EH, Uzun O, Gabriel A. In vivo Ty1 reverse transcription can generate replication intermediates with untidy ends. J Virol. 1998;72:6490–6503. doi: 10.1128/jvi.72.8.6490-6503.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Volloch VZ, Schweitzer B, Rits S. Transcription of the 5′-terminal cap nucleotide by RNA-dependent DNA polymerase: possible involvement in retroviral reverse transcription. DNA Cell Biol. 1995;14:991–996. doi: 10.1089/dna.1995.14.991. [DOI] [PubMed] [Google Scholar]
- 12.Chen D, Patton JT. Reverse transcriptase adds nontemplated nucleotides to cDNAs during 5′-RACE and primer extension. Biotechniques. 2001;30:574–580. doi: 10.2144/01303rr02. 582. [DOI] [PubMed] [Google Scholar]
- 13.Martinez MA, Vartanian JP, Wain-Hobson S. Hypermutagenesis of RNA using human immunodeficiency virus type 1 reverse transcriptase and biased dNTP concentrations. Proc Natl Acad Sci USA. 1994;91:11787–11791. doi: 10.1073/pnas.91.25.11787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wickham TJ, Davis T, Granados RR, Shuler ML, Wood HA. Screening of insect cell lines for the production of recombinant proteins and infectious virus in the baculovirus expression system. Biotechnol Progr. 1992;8:391–396. doi: 10.1021/bp00017a003. [DOI] [PubMed] [Google Scholar]
- 15.Hecker KH, Roux KH. High and low annealing temperatures increase both specificity and yield in touchdown and stepdown PCR. Biotechniques. 1996;20:478–485. doi: 10.2144/19962003478. [DOI] [PubMed] [Google Scholar]
- 16.Girgis SI, Alevizaki M, Denny P, Ferrier GJ, Legon S. Generation of DNA probes for peptides with highly degenerate codons using mixed primer PCR. Nuc Acids Res. 1988;16:10371. doi: 10.1093/nar/16.21.10371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schnoor M, Voss P, Cullen P, Boking T, Galla HJ, Galinski EA, Lorkowski S. Characterization of the synthetic compatible solute homoectoine as a potent PCR enhancer. Biochem Biophys Res Comm. 2004;322:867–872. doi: 10.1016/j.bbrc.2004.07.200. [DOI] [PubMed] [Google Scholar]
- 18.Shibata Y, Carninci P, Watahiki A, Shiraki T, Konno H, Muramatsu M, Hayashizaki Y. Cloning full-length, cap-trapper-selected cDNAs by using the single-strand linker ligation method. Biotechniques. 2001;30:1250–1254. doi: 10.2144/01306st01. [DOI] [PubMed] [Google Scholar]
- 19.Dutton CM, Paynton C, Sommer SS. General method for amplifying regions of very high G+C content. Nuc Acids Res. 1993;21:2953–2954. doi: 10.1093/nar/21.12.2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raychaudhuri A, Tipton PA. Protocol for amplification of GC-rich sequences from Pseudomonas aeruginosa. Biotechniques. 2004;37:752. doi: 10.2144/04375BM08. 754, 756. [DOI] [PubMed] [Google Scholar]
- 21.Kang J, Lee MS, Gorenstein DG. The enhancement of PCR amplification of a random sequence DNA library by DMSO and betaine: application to in vitro combinatorial selection of aptamers. J Biochem Biophys Meth. 2005;64:147–151. doi: 10.1016/j.jbbm.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 22.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 23.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nuc Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]