

Abstract
Retinitis pigmentosa (RP) is a heterogeneous group of hereditary disorders of the retina caused by mutation in genes of the photoreceptor proteins with an autosomal dominant (adRP), autosomal recessive (arRP), or X-linked pattern of inheritance. Although there are over 100 identified mutations in the opsin gene associated with RP, only a few of them are inherited with the arRP pattern. E150K is the first reported missense mutation associated with arRP. This opsin mutation is located in the second cytoplasmic loop of this G protein-coupled receptor. E150K opsin expressed in HEK293 cells and reconstituted with 11-cis-retinal displayed an absorption spectrum similar to the wild type (WT) counterpart and activated G protein transducin slightly faster than WT receptor. However, the majority of E150K opsin showed a higher apparent molecular mass in SDS-PAGE and was resistant to endoglycosidase H deglycosidase. Instead of being transported to the plasma membrane, E150K opsin is partially colocalized with the cis/medial Golgi compartment markers such as GM130 and Vti1b but not with the trans-Golgi network. In contrast to the endoplasmic reticulum-retained adRP mutant, P23H opsin, Golgi-retained E150K opsin did not influence the proper transport of the WT opsin when coexpressed in HEK293 cells. This result is consistent with the recessive pattern of inheritance of this mutation. Thus, our study reveals a novel molecular mechanism for retinal degeneration that results from deficient export of opsin from the Golgi apparatus.
Retinitis pigmentosa (RP)2 refers to a group of inherited degenerative retinal diseases that display heterogeneous genetic backgrounds and clinical phenotypes (1). Many genetic loci have been reported to cause this retinopathy, including mutations in the opsin gene (RetNet, www.sph.uth.tmc.edu/RetNet/). Rod visual pigment rhodopsin (Rho) consists of the apoprotein, opsin, and the covalently bound chromophore, 11-cis-retinal (2, 3). Although more than 100 opsin mutations are associated with RP (the Human Gene Mutation Data base, archive.uwcm.ac.uk/uwcm/mg/ns/1/120347.html), only a few have been reported to be associated with autosomal recessive retinitis pigmentosa (arRP).
The first reported case of arRP associated with a mutation in the opsin gene is a nonsense mutation at codon 249 (4) that eliminates helices VI and VII (containing the retinal binding site Lys296), cytoplasmic helix 8, and the C terminus of opsin (Fig. 1A). Even heterozygous carriers of this mutation had affected electroretinograms, indicating an abnormality in rod photoreceptor function (4). This mutant was partially characterized (5).
FIGURE 1. E150K and other RP mutations in opsin.
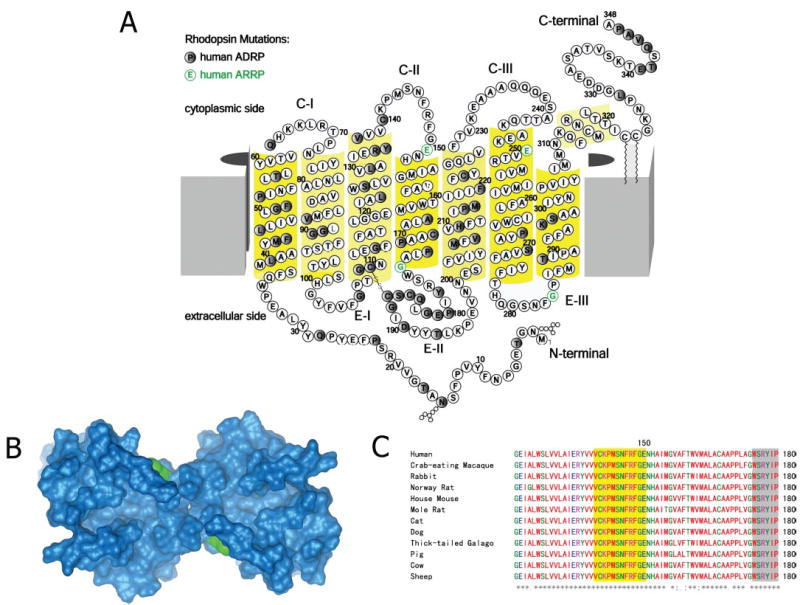
A, the two-dimensional model of opsin with indicated nonsense/missense mutations related to arRP (in green) and adRP (in black). B, three-dimensional model of E150K Rho with the cytoplasmic side facing outside and the mutation in green. C, the amino acid sequence alignment of 12 mammalian opsins around Glu150 (residues 121–180). The yellow background indicates the cytoplasmic loop, the white background indicates the transmembrane helices, and the gray background indicates the intradiscal loop.
E150K is missense opsin mutation associated with arRP (6); however, prior to this work the effects of the mutation had not been biochemically characterized. Glu150 is located in the proximity of the C-terminal region of the second cytoplasmic loop, embedded at the edge of the phospholipid bilayer (Fig. 1, A and B). Based on the crystal structure of Rho (7), this residue may have electrostatic interaction with Arg69 on C-I. In general, residues on the cytoplasmic side, especially in C-II, C-III, and helix 8, play important roles in the activation of a cognate G protein, transducin (Gt) (8–12, 24, 25). Another arRP mutation, G284S, is listed in www.sph.uth.tmc.edu/RetNet/ without any further characterization.
We focused our research on the E150K mutant of opsin to understand the biochemical mechanisms underlying arRP and further characterize opsin biosynthesis by studying this recombinant opsin mutant in vitro. We explored whether the mutation interferes with phototransduction by affecting the binding and activation of Gt as suggested previously (6) or causes disruption of the cellular trafficking.
EXPERIMENTAL PROCEDURES
Materials
11-cis-Retinal was a gift from Dr. R. Crouch (University of South Carolina) through a contract with the National Institutes of Health. Human eyes were obtained from Lions Eye Bank (Seattle, WA and Portland, OR). The mouse anti-Rho (C-terminal) monoclonal antibody 1D4 was purchased from the University of British Columbia (Dr. R. Molday). The mouse anti-Rho (N-terminal) monoclonal antibody B6–30N was a generous gift from Dr. P. Hargrave (University of Florida). The rabbit anti-Rho IgG was a generous gift from Dr. E. L. Kean (Case Western Reserve University). The anti-Gtα subunit (Gtα) monoclonal antibody was a generous gift from Dr. H. E. Hamm (Vanderbilt University Medical Center). The anti-Gtβ subunit (Gtβ) polyclonal antibody was a generous gift from Dr. O. G. Kisselev (St. Louis University School of Medicine). Rabbit anti-calreticulin IgG, Cy3-conjugated anti-FLAG, and fluorescein isothiocyanate-conjugated anti-c-Myc mouse monoclonal antibodies were purchased from Sigma-Al-drich. Mouse monoclonal anti-GM130, anti-P230, and anti-Vti1b monoclonal antibodies were purchased from BD Biosciences (Franklin Lakes, NJ). Cy3-conjugated goat anti-mouse IgG or goat anti-rabbit IgG were purchased from Jackson ImmunoResearch Laboratories, Inc (West Grove, PA). Hoechst33342 dye and Alexa 488-conjugated goat anti-rabbit or mouse IgG were purchased from Invitrogen. N-Dodecyl-β-D-maltoside (DM) was purchased from Anatrace Inc. (Maumee, OH). GTPγS was purchased from Sigma-Aldrich. Endoglycosidase H (Endo H) and peptide: N-glycosidase F (PNGase F) were purchased from New England Biolabs. Dithiobis(succinimidyl propionate) (DSP) was purchased from Pierce. Brefeldin A was obtained from EMD Biosciences (San Diego, CA). The synthetic 1D4 peptide was ordered from United Biochemical Research, Inc. (Seattle, WA). Other resins and columns for chromatography were purchased from Amersham Biosciences.
Cell Lines and Growth Conditions
Tetracycline-inducible HEK293 cells stably transfected with constructs encoding human WT and E150K opsins were generated according to the manufacturer’s procedure (T-REx-293TM; Invitrogen), grown in the Dulbecco’s modified Eagle’s medium containing high glucose (Invitrogen) at 37 °C in the presence of 5% CO2. Unless otherwise mentioned, the cells were harvested after 48 h of tetracycline induction (1 μg/ml). Brefeldin A was added to the stable cell lines 1 h after tetracycline induction, at a final concentration of 2.5 μg/ml, and the cells were fixed 1 h after the brefeldin A treatment or left to recover for another 10 h before fixation. To add FLAG or c-Myc to the C terminus of the protein, WT or E150K opsin constructs were cloned in frame into the pCMV-Tag4 or pCMV-Tag5 vectors, respectively (Stratagene). After selection of the cloned constructs and confirmation of the sequence, HEK293 cells were cotransfected with 50% of each vector using Lipofectamine (Invitrogen). Two days post-transfection (48 h), the cells were then fixed with 2% of paraformaldehyde for 5 min.
Electrophoresis and Immunoblotting
All of the protein separations were performed on 12% SDS-PAGE gels. Silver staining and immunoblotting (Immobilon-P polyvinylidene difluoride; Millipore) were carried out according to standard protocols. Sample buffer without dithiothreitol (DTT) was used with DSP cross-linked samples for the nonreducing condition. Anti-Rho 1D4 and anti-Rho B6–30N antibodies were used to detect the corresponding C-terminal and N-terminal epitopes. Alkaline phosphatase-conjugated goat anti-mouse IgG or goat anti-rabbit IgG (Promega) were used as secondary antibodies. Protein bands were visualized with the nitro blue tetrazolium chloride (NBT)/5-bromo-4-chloro-3′-indolyl-phosphate p-toluidine salt (BCIP) color development substrate (Promega).
Pigment Reconstitution
Harvested cells expressing opsins were homogenized with a glass-glass homogenizer on ice and then centrifuged at 2,000 × g in a bench top centrifuge for 2 min to remove nuclei and cellular debris. The cell membranes were then pelleted by centrifugation at 14,000 × g for 5 min. All of the procedures employing reconstituted pigment or retinoids were performed under dim red light unless mentioned otherwise. The membranes were washed three times with 37 mM NaCl, 5.4 mM Na2HPO4, 2.7 mM KCl, and 1.8 mM KH2PO4, pH 7.4, and incubated with a final concentration of 50 μM 11-cis-retinal in the presence of protease inhibitors (protease inhibitor mixture; Sigma-Aldrich) at room temperature overnight to generate Rho.
Purification of Opsin
Opsin or Rho was purified using anti-Rho C-terminal 1D4 antibody (13) immobilized on CNBr-activated SepharoseTM 4B (14, 15). Briefly, a 4.6 × 12-mm column was packed with 300 μl of 2 mg 1D4/ml Sepharose beads. The incubated cell membranes were pelleted and homogenized with PBS, using a glass-to-glass homogenizer. Soluble proteins in the supernatant were removed by centrifugation at 14,000 × g for 5 min, and the pellet was then solubilized in buffer A (20 mM DM, 10 mM 1,3-bis(tris(hydroxymethyl)methylamino)propane (BTP), pH 7.5, containing 500 mM NaCl). The supernatant was loaded onto the 1D4 immunoaffinity column after a 20-min centrifugation at 125,000 μg at 4 °C, followed by a thorough washing with buffer B (2 mM DM, 10 mM BTP, pH 7.5, and containing 500 mM NaCl) at a flow rate of 0.5 ml/min. The purified protein was eluted with buffer C (500 μM TETSQVAPA in buffer B, pH 6.0) at room temperature. The protein concentration was determined from absorption at 280 and 500 nm using a Hewlett-Packard 8452A UV-visible spectrophotometer.
Chemical Cross-linking with DSP
The cross-linking experiments were performed as described previously (16, 17). Briefly, the cell membranes were suspended in 100 mM sodium phosphate buffer, pH 8.0. DSP in Me2SO was added to reach a final concentration of 50 μM, and the reaction was allowed to proceed on ice for 30 min. The concentration of Me2SO did not exceed 2% of the reaction volume. To quench the reaction, 1 M Tris-HCl, pH 7.5, was added to a final concentration of 100 mM. The cell membranes were then washed with phosphate buffer and pelleted for opsin purification.
Deglycosylation of Opsin
Approximately 3 μg of immunoaffinity-purified opsin was incubated with 100 units of Endo H or PNGase F at room temperature for 1 h (deglycosylation of opsin purified from cells) or at 4 °C for 24 h (for purified DSP cross-linked opsin), and the products were analyzed by SDS-PAGE.
Photosensitivity of the Visual Pigment
UV-visible absorption spectra were measured with freshly purified Rho at 20 °C (15). In brief, photoactivation spectra were taken at intervals of 5, 10, 20, 30, and 180 s of exposure to light (applied with a long pass wavelength filter, > 490 nm, at a distance of 20 cm). To trap the protonated retinylidene Lys296, concentrated H2SO4 was added to adjust the final pH to 1.9–2.0 after bleaching the sample for 3 min. The rate of photoactivation was determined using a plot of A496 nm against bleaching time.
Rates of Meta II Decay
Rho (10 nM) in 2 mM DM, 10 mM BTP, pH 6.0, containing 100 mM NaCl was used to measure Meta II decay. The increase in intrinsic Trp fluorescence results from hydrolysis of the protonated Schiff base and release of all-trans-retinal from Rho (18). Immunoaffinity-purified Rho was bleached by a Fiber-Lite illuminator (> 490-nm filter applied) for 30 s from a distance of 15 cm, immediately followed by fluorescence measurements. The fluorometer slit was set to 2.5 μm at 295 nm for excitation and 8 μm at 330 nm for emission on a PerkinElmer Life Sciences LS 50B luminescence spectrophotometer. A thermostat was applied to stabilize the temperature of the cuvette at 20 °C during the measurement. Both fitting curves had R2 values of over 0.99.
Gt Binding and Activation Assay
Gt was purified as described previously (16). In the binding assay, affinity-purified Rho (WT or E150K) was mixed with a molar equivalent amount of Gt in 20 mM BTP, pH 7.5, containing 120 mM NaCl, 1 mM MgCl2, 1 mM DTT, and 2 mM DM. After bleaching Rho, the mixture was incubated for 15 min on ice to allow the complex to form. Gt dissociation was induced by the addition of 100 μM GTPγS (Sigma-Aldrich) on ice for 30 min before loading onto a size exclusion column (15), using 20 mM BTP, pH 7.5, containing 120 mM NaCl, 1 mM MgCl2, and 1 mM DTT.
In the fluorescence assay, the ratio of Gt to Rho was adjusted to 10:1 with Rho at ~2.5 nM. The bleached sample was incubated for 10 min at 20 °C with continuous low speed stirring. The fluorescence was measured as described by Farrens et al. and others (19, 20), employing excitation and emission wavelengths at 300 and 345 nm, respectively.
Immunocytochemistry
HEK293 cells were cultured on 35-mm glass-bottomed dishes (MatTek Corporation), and opsin expression was induced by 1 μg/ml tetracycline for 48 h after overnight attachment. The cells were then fixed with 2% paraformaldehyde in PBS (11.4 mM sodium phosphate, pH 7.4, containing 136 mM NaCl) for 5 min and washed with PBS. To block nonspecific labeling, the cells were incubated with 1.5% normal goat serum in PBST (PBS with 0.1% Triton X-100) for 15 min at room temperature. The cells were then incubated overnight at 4 °C with purified mouse anti-Rho 1D4 or rabbit anti-Rho antibody with PBST, along with one of the rabbit anti-calreticulin IgG, mouse anti-GM130, anti-Golgin84, anti-P230, or anti-Vti1b monoclonal antibodies for double labeling. The cells were then rinsed in PBST and incubated with Cy3-conjugated goat anti-mouse IgG or goat anti-rabbit IgG for detection of opsin immunofluorescence and with Hoechst 33342 dye for DNA staining. For detection of other antigens, the cells were also incubated with Alexa 488-conjugated goat anti-rabbit or mouse IgG. For transient transfected cells, FLAG-tagged WT opsin was labeled with Cy3-conjugated anti-FLAG antibody, whereas E150K opsin was labeled with fluorescein isothiocyanate-conjugated anti-c-Myc antibody along with the DNA stain. After labeling with secondary antibodies, the cells were rinsed in PBST and mounted with 50 μl 2% 1,4-diazabicyclo (2,2,2)-octane in 90% glycerol to retard bleaching. For confocal imaging, the cells were analyzed on a Zeiss LSM510 laser scanning microscope (Carl Zeiss, Inc.). Image contrast and brightness were adjusted by Adobe Photoshop CS.
Modeling
A phospholipid enhanced molecular model of Rho in the oligomeric state was constructed using the Rho network structure 1N3M from the Protein Data Bank. A three-component lipid bilayer mimicking the rod disc membranes was composed of phospholipids with phosphatidylcholine head groups on the intradiscal side and phosphatidylethanolamine together with phosphatidylserine head groups on the cytoplasmic side. All three types of phospholipids contained the saturated stearoyl chain (18:0) in the sn1 position and the polyun-saturated docosahexaenoyl chain (22:6) in the sn2 position. The detailed procedure of the membrane embedding, solvation, and equilibration of the protein-membrane-water system was described previously (21). The equilibration phase was performed at a constant temperature of 300 K and a constant pressure of 1013 hPa for 1 ns. Opsin mutants were subjected to additional molecular dynamics simulation for a subsequent 500 ps.
RESULTS
E150K Opsin Exhibits a Different Glycosylation Pattern than WT Opsin
The arRP-related E150K Rho and WT human and bovine Rho as controls were immunoaffinity-purified from HEK293 cells and analyzed by UV-visible spectroscopy, SDS-PAGE, and immunoblotting. After normalization for the number of cells, the expression level of the mutant was ⅛th that of WT Rho as determined by immunoblotting (supplemental Fig. S1) and based on the amount of isolated mutant and WT Rho. The spectra of the human mutant and WT Rho were indistinguishable from each other (λmax = 496 nm) but shifted from bovine Rho (λmax = 498 nm) (Fig. 2A). Both proteins were purified to similar levels as judged by A280 nm/A500 nm ratio, which was 1.6–1.8 in most preparations (Fig. 2A, inset). In SDS-PAGE analysis, the monomeric form of WT Rho migrated as a broad band (~40–55 kDa), because of heterogenous N-linked glycosylation in HEK293 cells. However, the majority of monomeric E150K opsin migrated with an apparent molecular mass of ~50–58 kDa (Fig. 2B), which is slightly higher than that of WT opsin. These differences in the apparent molecular mass could result from altered post-translational glycosylation. Hence, Endo H and PNGase F were used to assess the status of glycosylation of the mutant and WT opsins. Endo H cleaves only the ER products containing high mannose structures and some hybrid oligosaccharides at the GlcNAc-GlcNAc bond of its chitobiose core from N-linked glycoproteins (22). Mature proteins are resistant to Endo H once they exit the ER and enter the Golgi. The amidase PNGase, cleaves the innermost GlcNAc-Asn bond from N-linked glycoproteins (22). Both silver-stained gels (Fig. 2C) and immunoblots (Fig. 2D) showed that E150K and WT opsins were mostly resistant to deglycosylation by Endo H. These data suggest that the majority of E150K opsin passed the ER processing and underwent early trimming such as the cleavage of the high mannose form of the N-linked glycans in the Golgi apparatus. These properties distinguish E150K mutants from the most extensively studied adRP opsin mutant, P23H, which is sensitive to Endo H cleavage (23). The fact that after PNGase F treatment the two E150K opsin bands merged into one with the same apparent molecular mass as deglycosylated monomeric WT opsin (~35 kDa) (Fig. 2, C and D) excluded the possibility that the lower molecular mass resulted from partial degradation. E150K opsin formed more SDS-resistant oligomers than WT opsin, suggesting a higher propensity of the mutant to aggregate (Fig. 2B). The significance of this observation is unclear. The difference in apparent molecular masses between WT and E150K opsins was also observable for the SDS-induced dimer forms.
FIGURE 2. Spectra, SDS-PAGE migration, and deglycosylation of E150K Rho.
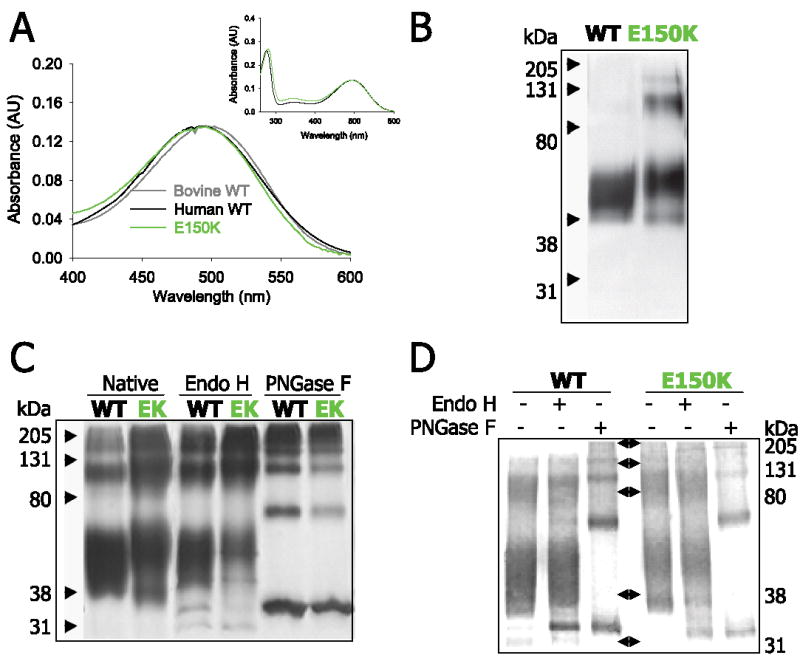
A, the visible absorbance spectra of immunoaffinity-purified bovine (gray, 498 nm), human (black, 496 nm) WT, and human E150K mutant (green, 496 nm) Rho. Inset, an extended UV-visible range of the spectrum. B, silver staining of the SDS-PAGE gel of immunoaffinity-purified WT opsin on the left and E150K opsin on the right. The molecular mass is shown by the arrows on the left side (kDa). The results of deglycosylation are shown in C and D. WT and E150K opsins were deglycosylated by either Endo H or PNGase F as indicated. C shows silver staining of the native or deglycosylated WT/E150K opsin as marked. D shows the immunoblot of deglycosylated WT/E150K opsin probed with anti-Rho 1D4 antibody. Molecular mass markers (in kDa) are 205, 131, 80, 38, and 31.
E150K Mutant Rho and Gt Activation
The ability of the mutant to couple to Gt was investigated by size exclusion chromatography and fluorescence assays (Fig. 3, A and B). Upon light activation, Rho* bound Gtα βγ, and the Rho*·Gtα βγcomplex was detected by gel filtration, where Rho and Gt were eluted in the same fractions (Fig. 3A, left panels). Upon the addition of nonhydrolyzable GTP analog, GTPγS, Rho, and Gt were eluted in different fractions (Fig. 3A, right panels) indicating that Gtα ·GTP (Gtα ·GTPγS) had dissociated to free Gtβ γand Rho*. In the direct intrinsic fluorescence assay (18), E150K Rho* activated Gtα with an initial activation rate of k0 ~0.112 min−1, a rate that was faster than the activation by human WT Rho, k0 ~0.071 min−1, but similar to activation by bovine WT Rho k0 ~0.114 min−1. No changes in fluorescence were observed in the absence of Gt, and no constitutive Gt activation was detected with purified E150K opsin in the absence of light or chromophore (Fig. 3C) in the presence or absence of phospholipids to stabilize opsins (26). No Gt activity was observed using the whole membrane preparations (data not shown). These results suggest that mutated E150K did not affect G protein binding and activation. The increase in the Gt activation for the E150K mutant could be a result of modified photoactivation properties of the mutant.
FIGURE 3. Gt binding, activation, and Meta II decay.
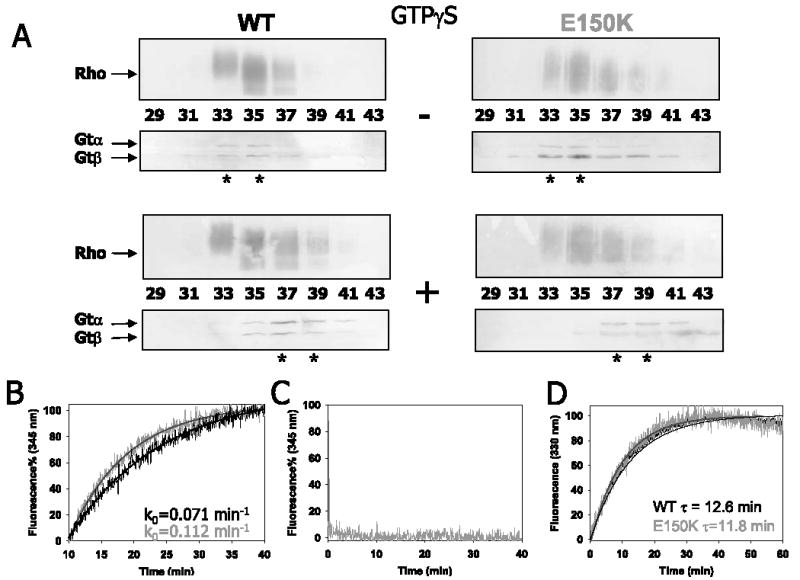
A, Gt binding assay of WT/E150K Rho by size exclusion chromatography. Immunoblots are shown using anti-Rho (top panels), and the anti-Gtα or anti-Gtβ (bottom panels), with or without the addition of GTPγS. The number of fractions is listed. B, Gt activation by WT or E150K Rho monitored by the increase in fluorescence at 345 nm. The initial rates calculated as a pseudo first order reaction for WT and E150K are 0.071 and 0.112 min−1, respectively. C, E150K opsin showed no constitutive Gt activation. D, Meta II decay of WT/E150K measured by fluorescence emission at 330 nm. The absolute fluorescence intensity changes were comparable (in arbitrary units) 82– 86 for WT Rho and 85– 89 for the E150K mutant. The relaxation time (τ) for WT and E150K Rho is 13.6 and 11.8 min, respectively.
The active form of Rho, Meta II (or Rho*), had a faster decay rate (τE150K = 11.8 min) than that of WT Rho (τWT = 13.6 min), indicating that the release rate of all-trans-retinal from the binding pocket was faster (Fig. 3D). Both Rho proteins were photoactivated in DM, but again the decrease of A498 nm was faster for the mutant (supplemental Fig. S2), consistent with the rate of decay of Meta II. Additionally, DM-solubilized E150K opsin regenerated with 11-cis-retinal ~50% more slowly than its WT counterpart. This observation could be a result of lower stability of the mutant in the detergent. Collectively, these data suggest that E150K Rho is correctly folded at the protein core around the binding pocket and undergoes similar, but not identical, photoactivation processes.
Chemical Cross-linking of Opsin with DSP
WT opsin forms covalently linked dimers when expressed in HEK293 cells and treated with DSP (16, 17). Based on the crystal structure of bovine Rho (7, 27), there are several Lys residues from each Rho molecule of the dimer that lie within the accessible range of the DSP spacer (1.2 nm). When the membranes from cells expressing E150K opsin or WT opsin were treated with DSP, the formation of dimers was observed (Fig. 4). We used DTT in control experiments to reduce the disulfide bond within the cross-linker to exclude the possibility of nonspecific oligomerization. Similar results were obtained from both WT and E150K opsins, suggesting that the mutation did not induce dramatic changes in either the global folding of individual mutant opsin or the intermolecular interactions between opsins within dimers.
FIGURE 4. Chemical cross-linking of E150K opsin.
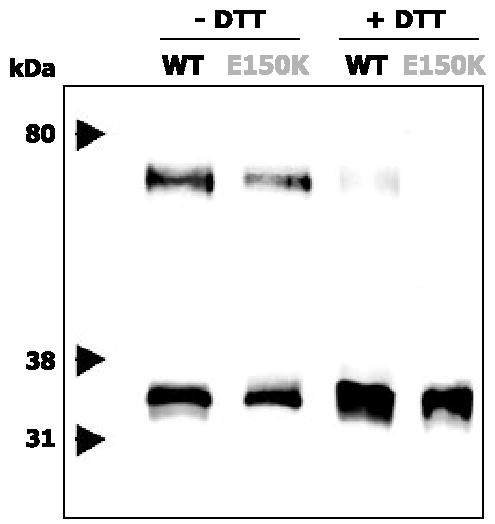
Immunoblots of purified cross-linked WT or E150K opsins deglycosylated with PNGase F. WT or E150K opsins were cross-linked in the membranes before immunoaffinity purification. The corresponding samples in lanes 1 and 2 are reduced in lanes 3 and 4 by 100 mM DTT for 5 min before SDS-PAGE.
E150K Opsin Partially Colocalizes with cis/Medial Golgi Marker
Confocal laser scanning microscopy was used to study the localization of WT and mutant opsins in HEK293 cells. WT opsin was properly folded and transported to the plasma membrane as previously reported (Fig. 5; see also supplemental Fig. S3) (14). The adRP-related opsin mutation, P23H, is known to be ubiquitinylated and targeted for degradation by the ubiquitin-proteasome system while expressed in HEK293 or COS-7 cells (23, 28, 29). These studies are consistent with our observations of the colocalization of P23H opsin with the ER marker calreticulin (30). P23H opsin was retained in the ER when the expression level was low (less than 24 h after induction), whereas ubiquitinated aggresomes started to appear with accumulation of the mutant opsin (> 24 h) (data not shown). E150K opsin, when expressed in HEK293 cells, was not efficiently transported to the plasma membrane. The expression level of the mutant was about 8-fold lower than that of WT Rho (supplemental Fig. S1). The mutant opsin was also not retained in the ER, like P23H opsin, as shown by the lack of colocalization with calreticulin (Fig. 5). Only when the image shown in Fig. 5 was captured with longer integration time (pixel dwell time), a low level of this mutant in the calreticulin positive compartment was observed (data not shown). Instead, the majority of E150K opsin localized together with the cis-Golgi compartment marker GM130 and the cis/medial Golgi compartment marker Vti1b (Fig. 6). E150K opsin did not colocalize with the trans-Golgi network-specific protein, P230 (31) (Fig. 6). This observation is consistent with the results of deglycosy-lation of E150K opsin. Thus, E150K opsin has matured beyond the ER and obtained the Endo H-resistant oligosaccharide modification in the Golgi apparatus (32). When cells stably expressing E150K opsin were treated with brefeldin A (33), which induces disassembly of Golgi stacks and redistribution of Golgi proteins to the ER, the aggregation morphology of the mutant was no longer evident (data not shown). This observation strengthens the immunocytochemical results on the Golgi localization of E150K opsin. Together, these results indicate that the mutant opsin passes the quality control system in the ER and is defective in its transport from cis-Golgi compartment or medial Golgi compartment to trans-Golgi network.
FIGURE 5. Intracellular colocalization of opsin with calreticulin, an ER marker.
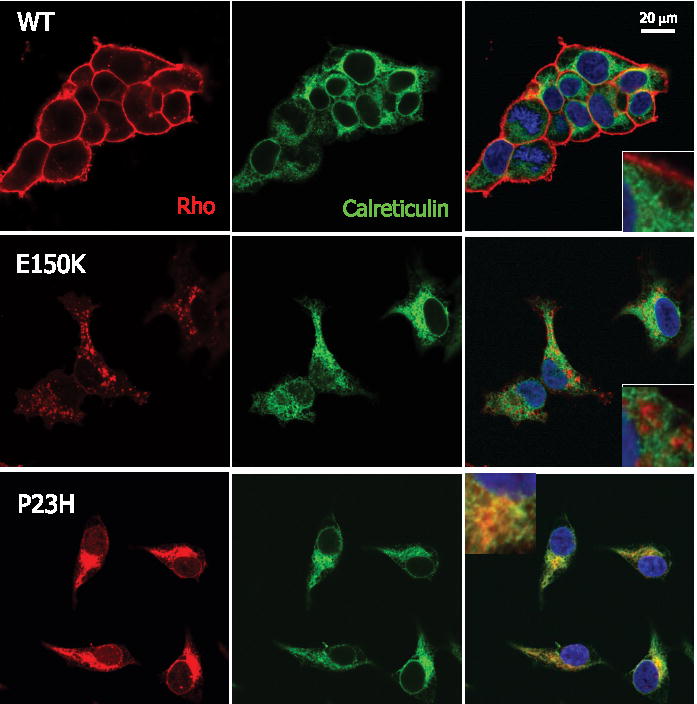
HEK293 cells expressing WT (top row), E150K (middle row), or P23H (bottom row) opsin are stained with both anti-Rho 1D4 (red) and anti-calreticulin (green). The nuclei are stained by Hoechst 33342 (blue). The right column shows the merged images. The insets in the right column are high magnification images of the merged images. The scale bar represents 20 μ m.
FIGURE 6. Intracellular colocalization of E150K opsin with Golgi markers.
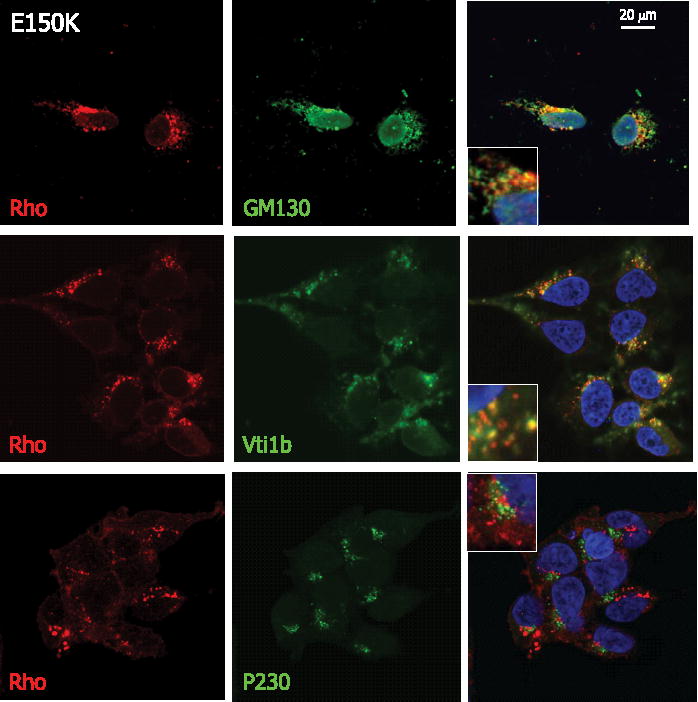
From top to bottom, anti-Rho 1D4 is used together with anti-GM130 (cis), anti-Vti1b (cis/medial), or anti-P230 (trans) to examine the intracellular colocalization of E150K opsin (red) and the Golgi apparatus (green).
Subcellular Localization of WT Opsin Is Not Influenced by Coex-pression of E150K Opsin
When WT and E150K opsins were cotransfected into HEK293 cells, regardless of the expression levels, the majority of WT opsin was properly transported to the plasma membrane. Interestingly, different subcellular localizations of E150K opsin were observed (Fig. 7). When the ratio of WT opsin construct transfected was much higher than that of the mutant (WT/E150K = 10/3), E150K opsin was rescued by the coexpression of WT opsin, and the majority of E150K opsin also reached the plasma membrane. However, when the ratio of WT opsin to E150K opsin was 3/10, the localization of E150K opsin was not influenced by WT opsin expression. This difference observed in HEK293 cells is most likely due to the different ratio of the total amount of WT versus E150K opsin transiently expressed in the individual cells. The fluorescent intensity correlated well with the intended expression level based on the amount of DNA. When more WT opsin was coexpressed, hetero-oligomerization of WT opsin with E150K opsin allowed cotrans-port to the plasma membranes. Thus, proper transport of the mutant was achieved in the presence of WT opsin.
FIGURE 7. Cotransfection of WT and E150K opsins.
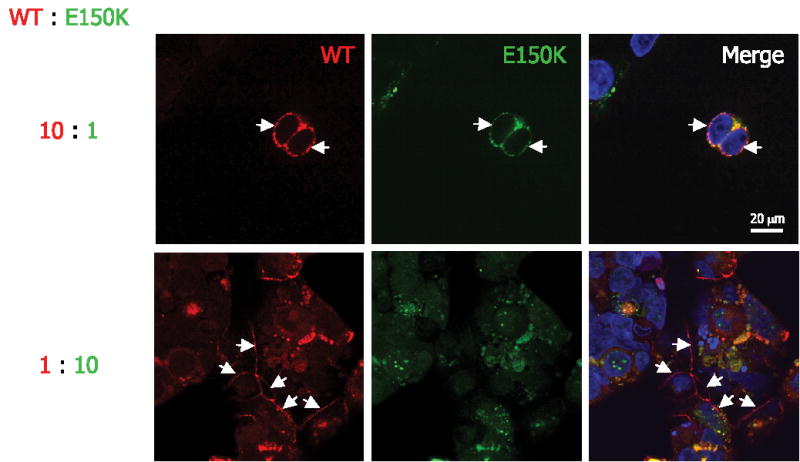
Different ratios of WT and E150K construct, 10/1 and 1/10, respectively, were used to cotransfect HEK293 cells. WT opsin was labeled with Cy3-conjugated anti-FLAG (red) antibody, and E150K opsin was labeled with fluorescein isothiocyanate-conjugated anti-c-Myc antibody (green). The nuclei were stained by Hoechst 33342 dye (blue). The bar represents 20 μm. The arrows indicate the plasma membrane expression of the opsins.
DISCUSSION
The Glu150 residue is located in the second cytoplasmic loop of opsin (Fig. 1). This loop is important role in the proper folding of opsin in addition to the binding and activation of Gt. Although a different deletion mutant lacking residues 143–150 and a hybrid opsin containing 140–152 residues replaced by unrelated sequences are able to form a visual pigment with normal spectral properties, both mutants behaved aberrantly in their interactions with Gt (10, 34, 35). These mutations can cause large changes in the conformation of cytoplasmic loop II. Ridge et al. (36) replaced residues 136–150 individually, and the Cys substitution mutants formed the characteristic visual pigments when regenerated with 11-cis-retinal (λmax ~500 nm), displayed normal photoactivation characteristics, and activated Gt in a light-dependent manner. In addition, chicken E150A mutant (37) showed properties similar to bovine E150C opsin. In our study, the replacement of Glu150 with an oppositely charged, larger amino acid, Lys residue did not induce dramatic biochemical changes of mutant opsin as compared with WT opsin. However, it should be noted that increased activation of Gt and faster decay of Meta II was observed for the E150K mutant. These differences likely resulted from modified photo-activation property of the mutant. Similarly, another loop II V137M mutant of Rho associated with autosomal dominant RP displayed enhanced activation of Gt (38). Faster activation of Gt was proposed to be the molecular mechanism that should theoretically lead to adRP. However, because E150K mutant is associated with autosomal recessive RP, we sought alternative mechanisms underlying the pathogenesis of arRP caused by this mutation.
Intracellular Transport of Opsin
Several key factors have been found to be important in the intracellular transport of opsin-like GPCRs: 1) chaperone proteins are involved in the proper folding including calnexin (39), BiP (40), and GRP94 (41); 2) the well conserved motifs proximal to the C terminus including the DXE motif and the di-Leu motifs (LXnL, n = 0, 2, or 3 depending on specific receptors) were shown to influence ER export (reviewed in Ref. 41); and 3) the C-terminal sequence plays an indispensable role in Rho vectorial transport in photo-receptor cells, as exemplified by the mislocalization of the truncated Gln344ter Rho (42) or Ser334ter Rho (where ter is termination) (43).
In the ER GPCRs acquire the first sugar chain (44). Ras-like Rab GTPases and glycosylation play a key role in the exporting of GPCRs to the cell surface by regulating protein transport from the ER to the Golgi (41) and from the Golgi to the plasma membrane (45). The E150K opsin mutant has passed the quality control system of the ER and reached the Golgi apparatus, suggesting that there are no major changes in the overall structure of the protein. The Golgi apparatus is a complex structure and plays a key role in the intracellular sorting, processing, and secretion of glycolipids and glycoproteins, including the GPCRs (46). Enzymatic modification of oligosaccharide chains on glycoproteins in the Golgi stack is necessary for increasing the protein stability as well as, in some cases, for their proper function (46). The E150K mutation of opsin leads to impaired traffic, and as a consequence, the mutant does not acquire mature glycans like WT opsin. This mutant is the first example among GPCR mutants, to our knowledge, that is aberrantly retained in the Golgi rather than the more typically observed retention in the ER (35, 47, 48).
This retention of the mutant Rho within the Golgi apparatus observation is unexpected because the mutation is in a portion of Rho exposed to the cytoplasm, whereas the glycosylation occurs in the extracellular region of Rho. Thus, the changes imposed by the mutation must be transmitted across the entire transmembrane segment of the receptor. Interestingly, slight alterations in plasma membrane protein structures have been shown to lead to retention or retarded traffic in the Golgi (49). Changes on the cytoplasmic side of the mutant opsin may affect the interaction with proteins involved in vesicular transport.
Based on our results, it is possible that in the photoreceptor cells heterozygous for the mutant, E150K opsin is either chaperoned to the rod outer segment or efficiently degraded in the inner segment. WT Rho will reach the rod outer segment and maintain the normal rod cell functions similar to heterozygote opsin knock-out mice (21). In the homozygous state, insufficient amounts of E150K opsin would reach the rod outer segment, resembling the phenotype of opsin knock-out mice (50, 51). Additionally, the accumulation of the mutant opsin within the Golgi apparatus would increase cellular stress, leading to eventual apoptosis and secondary cone degeneration (52). This pathology is consistent with the faster disease progression in the patients carrying the recessive E150K mutation compared with those with the P23H mutation (6).
Dimerization of Opsin and E150K Opsin
Protein oligomerization may play an important role in Golgi retention, and the cytoplasmic or luminal sequences of GPCRs also play a role in this process. Rho undergoes dimerization as shown by atomic force microscopy and transmission electron microscopy (21, 53, 54), biochemical assays (16, 55, 56), fluorescent techniques with reconstituted phospholipids (57), in chemical cross-linking studies (16, 17), and Cys cross-linking (16, 58). The Class II opsin mutants that are deficient in proper folding, including P23H opsin, were shown to have a negative effect on the coexpressed WT opsin, namely enhancing the degradation of WT opsin by the ubiquitin proteasome system (59). This effect can occur via direct interaction between these two proteins, which takes place during or shortly after their biosynthesis in the ER (41, 59, 60). This mechanism was not observed when WT and E150K opsins were coexpressed in HEK293 cells. This observation agrees with the autosomal recessive inheritance pattern of the E150K mutation. The location of the E150K mutation at the interface of Rho molecules can affect the stability of oligomerization because of weakened electrostatic or hydrogen bond interactions between Rho molecules in the oligomeric structure (Fig. 8). Cross-linking experiments indicate that the formation of dimers seems to be unaffected, which explains the lack of influence of the mutation on the activation of Gt. This rearrangement can promote nonspecific oligomerization of the mutant protein and aberrant glycosylation. The possibility of Rho/opsin being transported in the form of dimers or oligomers from the ER to the cell membrane is also in agreement with the results of the coexpression experiments. When the amount of WT opsin expressed was increased, more of the E150K opsin mutant was observed to be transported to the plasma membrane along with WT opsin (Fig. 7). The increased formation of WT/E150K hetero-oligomers rather than the E150K/E150K homo-oligomers is a reasonable explanation. This hypothesis, although requiring in vivo verification and biophysical confirmation, appears to be a logical interpretation of the results described in the study.
FIGURE 8. Molecular modeling of Rho tetramer in the membrane.
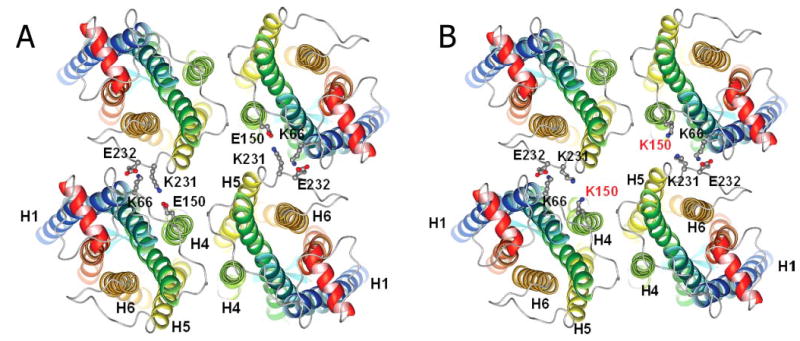
View from cytoplasmic side. Rho dimers are located horizontally, so mutation does not prevent formation of dimers (interface H4-H5) but rather longer oligomers. A, molecular model of WT Rho tetramer. B, molecular model of E150K Rho tetramer.
In summary, we have characterized a missense arRP mutant of opsin that has abberant traffic properties through the Golgi apparatus. The biochemical results in HEK293 cells suggest that the mutant is not properly glycosylated and is retained in the Golgi apparatus. Future in vivo experiments are needed for conformation of the observation made in HEK293 cells and will help to develop a possible intervention to rescue vision in RP patients carrying the E150K mutation in the opsin gene.
Acknowledgments
We thank Drs. R Stenkamp, D. Lodowski, and A. Moise and Philip Kiser for comments on the manuscript.
Footnotes
This work was supported in part by United States Public Health Service Grants EY08061 and P30 EY11373 from the NEI, National Institutes of Health.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3.
The abbreviations used are: RP, retinitis pigmentosa; arRP, autosomal recessive RP; adRP, autosomal dominant RP; BTP, 1,3-bis(tris(hydroxymethyl)-methylamino)propane; DM, n-dodecyl-β-D-maltoside; DSP, dithiobis(succinimidyl propionate); Endo H, endoglycosidase H; Gt, transducin, rod photoreceptor G protein; GPCR, G protein-coupled receptor; Meta II (or Rho*), photoactivated Rho; PNGase F, peptide:N-glycosidase F; Rho, rhodopsin; WT, wild type; ER, endoplasmic reticulum; GTPγS, guanosine 5′-O-(thio)triphosphate; DTT, dithiothreitol; PBS, phosphate-buffered saline.
References
- 1.Dryja TP, Li T. Hum Mol Genet. 1995;4:1739–1743. doi: 10.1093/hmg/4.suppl_1.1739. [DOI] [PubMed] [Google Scholar]
- 2.Filipek S, Stenkamp RE, Teller DC, Palczewski K. Annu Rev Physiol. 2003;65:851–879. doi: 10.1146/annurev.physiol.65.092101.142611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Palczewski K. Annu Rev Biochem. 2005;75:743–767. doi: 10.1146/annurev.biochem.75.103004.142743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rosenfeld PJ, Cowley GS, McGee TL, Sandberg MA, Berson EL, Dryja TP. Nat Genet. 1992;1:209–213. doi: 10.1038/ng0692-209. [DOI] [PubMed] [Google Scholar]
- 5.Ridge KD, Lee SS, Abdulaev NG. J Biol Chem. 1996;271:7860–7867. doi: 10.1074/jbc.271.13.7860. [DOI] [PubMed] [Google Scholar]
- 6.Kumaramanickavel G, Maw M, Denton MJ, John S, Srikumari CR, Orth U, Oehlmann R, Gal A. Nat Genet. 1994;8:10–11. doi: 10.1038/ng0994-10. [DOI] [PubMed] [Google Scholar]
- 7.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 8.Hamm HE. Proc Natl Acad Sci U S A. 2001;98:4819–4821. doi: 10.1073/pnas.011099798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Filipek S, Krzysko KA, Fotiadis D, Liang Y, Saperstein DA, Engel A, Palczewski K. Photochem Photobiol Sci. 2004;3:628–638. doi: 10.1039/b315661c. [DOI] [PubMed] [Google Scholar]
- 10.Franke RR, Konig B, Sakmar TP, Khorana HG, Hofmann KP. Science. 1990;250:123–125. doi: 10.1126/science.2218504. [DOI] [PubMed] [Google Scholar]
- 11.Natochin M, Gasimov KG, Moussaif M, Artemyev NO. J Biol Chem. 2003;278:37574–37581. doi: 10.1074/jbc.M305136200. [DOI] [PubMed] [Google Scholar]
- 12.Abdulaev NG, Ridge KD. Proc Natl Acad Sci U S A. 1998;95:12854–12859. doi: 10.1073/pnas.95.22.12854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Molday RS, MacKenzie D. Biochemistry. 1983;22:653–660. doi: 10.1021/bi00272a020. [DOI] [PubMed] [Google Scholar]
- 14.Oprian DD, Molday RS, Kaufman RJ, Khorana HG. Proc Natl Acad Sci U S A. 1987;84:8874–8878. doi: 10.1073/pnas.84.24.8874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu L, Jang GF, Jastrzebska B, Filipek S, Pearce-Kelling SE, Aguirre GD, Stenkamp RE, Acland GM, Palczewski K. J Biol Chem. 2004;279:53828 –53839. doi: 10.1074/jbc.M408472200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jastrzebska B, Maeda T, Zhu L, Fotiadis D, Filipek S, Engel A, Stenkamp RE, Palczewski K. J Biol Chem. 2004;279:54663–54675. doi: 10.1074/jbc.M408691200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suda K, Filipek S, Palczewski K, Engel A, Fotiadis D. Mol Membr Biol. 2004;21:435–446. doi: 10.1080/09687860400020291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Farrens DL, Khorana HG. J Biol Chem. 1995;270:5073–5076. doi: 10.1074/jbc.270.10.5073. [DOI] [PubMed] [Google Scholar]
- 19.Farrens DL, Altenbach C, Yang K, Hubbell WL, Khorana HG. Science. 1996;274:768–770. doi: 10.1126/science.274.5288.768. [DOI] [PubMed] [Google Scholar]
- 20.Heck M, Schadel SA, Maretzki D, Bartl FJ, Ritter E, Palczewski K, Hofmann KP. J Biol Chem. 2003;278:3162–3169. doi: 10.1074/jbc.M209675200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liang Y, Fotiadis D, Maeda T, Maeda A, Modzelewska A, Filipek S, Saperstein DA, Engel A, Palczewski K. J Biol Chem. 2004;279:48189–48196. doi: 10.1074/jbc.M408362200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maley F, Trimble RB, Tarentino AL, Plummer TH., Jr Anal Biochem. 1989;180:195–204. doi: 10.1016/0003-2697(89)90115-2. [DOI] [PubMed] [Google Scholar]
- 23.Illing ME, Rajan RS, Bence NF, Kopito RR. J Biol Chem. 2002;277:34150–34160. doi: 10.1074/jbc.M204955200. [DOI] [PubMed] [Google Scholar]
- 24.Ernst OP, Hofmann KP, Sakmar TP. J Biol Chem. 1995;270:10580–10586. doi: 10.1074/jbc.270.18.10580. [DOI] [PubMed] [Google Scholar]
- 25.Ernst OP, Meyer CK, Marin EP, Henklein P, Fu WY, Sakmar TP, Hofmann KP. J Biol Chem. 2000;275:1937–1943. doi: 10.1074/jbc.275.3.1937. [DOI] [PubMed] [Google Scholar]
- 26.Fasick JI, Lee N, Oprian DD. Biochemistry. 1999;38:11593–11596. doi: 10.1021/bi991600h. [DOI] [PubMed] [Google Scholar]
- 27.Teller DC, Okada T, Behnke CA, Palczewski K, Stenkamp RE. Biochemistry. 2001;40:7761–7772. doi: 10.1021/bi0155091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chapple JP, Grayson C, Hardcastle AJ, Saliba RS, van der Spuy J, Cheetham ME. Trends Mol Med. 2001;7:414–421. doi: 10.1016/s1471-4914(01)02103-7. [DOI] [PubMed] [Google Scholar]
- 29.Saliba RS, Munro PM, Luthert PJ, Cheetham ME. J Cell Sci. 2002;115:2907–2918. doi: 10.1242/jcs.115.14.2907. [DOI] [PubMed] [Google Scholar]
- 30.Krause KH, Michalak M. Cell. 1997;88:439–443. doi: 10.1016/s0092-8674(00)81884-x. [DOI] [PubMed] [Google Scholar]
- 31.Kjer-Nielsen L, Teasdale RD, van Vliet C, Gleeson PA. Curr Biol. 1999;9:385–388. doi: 10.1016/s0960-9822(99)80168-7. [DOI] [PubMed] [Google Scholar]
- 32.Colley KJ. Glycobiology. 1997;7:1–13. doi: 10.1093/glycob/7.1.1-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lippincott-Schwartz J, Yuan LC, Bonifacino JS, Klausner RD. Cell. 1989;56:801–813. doi: 10.1016/0092-8674(89)90685-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Franke RR, Sakmar TP, Graham RM, Khorana HG. J Biol Chem. 1992;267:14767–14774. [PubMed] [Google Scholar]
- 35.Nathans J. Biochemistry. 1992;31:4923–4931. doi: 10.1021/bi00136a001. [DOI] [PubMed] [Google Scholar]
- 36.Ridge KD, Zhang C, Khorana HG. Biochemistry. 1995;34:8804–8811. doi: 10.1021/bi00027a032. [DOI] [PubMed] [Google Scholar]
- 37.Imai H, Kojima D, Oura T, Tachibanaki S, Terakita A, Shichida Y. Proc Natl Acad Sci U S A. 1997;94:2322–2326. doi: 10.1073/pnas.94.6.2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Andres A, Garriga P, Manyosa J. Biochem Biophys Res Commun. 2003;303:294–301. doi: 10.1016/s0006-291x(03)00328-0. [DOI] [PubMed] [Google Scholar]
- 39.Siffroi-Fernandez S, Giraud A, Lanet J, Franc JL. Eur J Biochem. 2002;269:4930–4937. doi: 10.1046/j.1432-1033.2002.03192.x. [DOI] [PubMed] [Google Scholar]
- 40.Rozell TG, Davis DP, Chai Y, Segaloff DL. Endocrinology. 1998;139:1588–1593. doi: 10.1210/endo.139.4.5881. [DOI] [PubMed] [Google Scholar]
- 41.Duvernay MT, Filipeanu CM, Wu G. Cell Signal. 2005;17:1457–1465. doi: 10.1016/j.cellsig.2005.05.020. [DOI] [PubMed] [Google Scholar]
- 42.Sung CH, Makino C, Baylor D, Nathans J. J Neurosci. 1994;14:5818–5833. doi: 10.1523/JNEUROSCI.14-10-05818.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Green ES, Menz MD, LaVail MM, Flannery JG. Investig Ophthalmol Vis Sci. 2000;41:1546–1553. [PubMed] [Google Scholar]
- 44.Dempski RE, Jr, Imperiali B. Curr Opin Chem Biol. 2002;6:844–850. doi: 10.1016/s1367-5931(02)00390-3. [DOI] [PubMed] [Google Scholar]
- 45.Deretic D, Williams AH, Ransom N, Morel V, Hargrave PA, Arendt A. Proc Natl Acad Sci U S A. 2005;102:3301–3306. doi: 10.1073/pnas.0500095102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ungar D, Oka T, Krieger M, Hughson FM. Trends Cell Biol. 2006;16:113–120. doi: 10.1016/j.tcb.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 47.Kaushal S, Khorana HG. Biochemistry. 1994;33:6121–6128. doi: 10.1021/bi00186a011. [DOI] [PubMed] [Google Scholar]
- 48.Sung CH, Davenport CM, Nathans J. J Biol Chem. 1993;268:26645–26649. [PubMed] [Google Scholar]
- 49.Low SH, Tang BL, Wong SH, Hong W. J Biol Chem. 1994;269:1985–1994. [PubMed] [Google Scholar]
- 50.Humphries MM, Rancourt D, Farrar GJ, Kenna P, Hazel M, Bush RA, Sieving PA, Sheils DM, McNally N, Creighton P, Erven A, Boros A, Gulya K, Capecchi MR, Humphries P. Nat Genet. 1997;15:216–219. doi: 10.1038/ng0297-216. [DOI] [PubMed] [Google Scholar]
- 51.Lem J, Krasnoperova NV, Calvert PD, Kosaras B, Cameron DA, Nicolo M, Makino CL, Sidman RL. Proc Natl Acad Sci U S A. 1999;96:736–741. doi: 10.1073/pnas.96.2.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mohand-Said S, Hicks D, Leveillard T, Picaud S, Porto F, Sahel JA. Prog Retin Eye Res. 2001;20:451–467. doi: 10.1016/s1350-9462(01)00006-4. [DOI] [PubMed] [Google Scholar]
- 53.Fotiadis D, Liang Y, Filipek S, Saperstein DA, Engel A, Palczewski K. FEBS Lett. 2004;564:281–288. doi: 10.1016/S0014-5793(04)00194-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fotiadis D, Liang Y, Filipek S, Saperstein DA, Engel A, Palczewski K. Nature. 2003;421:127–128. doi: 10.1038/421127a. [DOI] [PubMed] [Google Scholar]
- 55.Medina R, Perdomo D, Bubis J. J Biol Chem. 2004;279:39565–39573. doi: 10.1074/jbc.M402446200. [DOI] [PubMed] [Google Scholar]
- 56.Jastrzebska B, Fotiadis D, Jang GF, Stenkamp RE, Engel A, Palczewski K. J Biol Chem. 2006;281:11917–11922. doi: 10.1074/jbc.M600422200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mansoor SE, Palczewski K, Farrens DL. Proc Natl Acad Sci U S A. 2006;103:3060–3065. doi: 10.1073/pnas.0511010103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kota P, Reeves PJ, Rajbhandary UL, Khorana HG. Proc Natl Acad Sci U S A. 2006;103:3054–3059. doi: 10.1073/pnas.0510982103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rajan RS, Kopito RR. J Biol Chem. 2005;280:1284–1291. doi: 10.1074/jbc.M406448200. [DOI] [PubMed] [Google Scholar]
- 60.Salahpour A, Angers S, Mercier JF, Lagace M, Marullo S, Bouvier M. J Biol Chem. 2004;279:33390 –33397. doi: 10.1074/jbc.M403363200. [DOI] [PubMed] [Google Scholar]