

Abstract
Carbonyl reductase-like 20β-hydroxysteroid dehydrogenase (CR/20β-HSD) is an enzyme that converts 17α-hydroxyprogesterone to 17α,20β-dihydroxy-4-pregnen-3-one (the maturation-inducing hormone of salmonid fish). We have previously isolated two types of CR/20β-HSD cDNAs from ovarian follicle of rainbow trout (Oncorhynchus mykiss). Recombinant proteins produced by expression in Escherichia coli in vitro showed that one (type A) had CR and 20β-HSD activity but that the other (type B) did not. Among the three distinct residues between the protein products encoded by the two cDNAs, two residues (positions 15 and 27) are located in the N-terminal Rossmann fold, the coenzyme binding site. To investigate the structure/function relationships of CR/20β-HSDs, we generated mutants by site-directed mutagenesis at the following positions: MutA/I15T, MutB/T15I, and MutB/Q27K. Enzyme activity of wild-type A was abolished by substitution of Ile-15 by Thr (MutA/I15T). Conversely, enzyme activity was acquired by the replacement of Thr-15 with Ile in type B (MutB/T15I). MutB/T15I mutant showed properties similar to the wild-type A in every aspect tested. Mutation MutB/Q27K had only partial enzyme activity, indicating that Ile-15 plays an important role in enzyme binding of cofactor NADPH.
Rainbow trout ovarian follicle carbonyl reductase-like 20β-hydroxysteroid dehydrogenase (CR/20β-HSD) is an enzyme with the potential physiological role of converting 17α-hydroxyprogesterone (17α-HP) into 17α,20β-dihydroxy-4-pregnen-3-one (17α,20β-DP), the endogenous oocyte maturation-inducing hormone of salmonid fishes (1, 2). cDNA cloning studies showed that the protein products of the two rainbow trout CR/20β-HSD cDNAs obtained share high homology of 61% with mammalian CRs, enzymes that can metabolize steroids, prostaglandins, and a variety of carbonyl-containing compounds. CRs belong to the short-chain dehydrogenase/reductases (SDR) superfamily (3), which contains a large number of prokaryotic and eukaryotic enzymes with different specificities for coenzymes and substrates. Although only 15–30% amino acid residue identities are shared by members of the SDR family in general, two domains, the TyrXXXLys catalytic site and a Rossmann fold coenzyme binding domain (4), are strictly conserved. In many CRs, the Rossmann fold coenzyme binding consensus is identified as GlyXXXGlyXGly near the N terminus. Although CRs purified from several mammalian tissues are monomeric and depend on NADPH, enzymes from the lungs of guinea pig, mouse, and pig are in tetrameric form (5–7), with dual coenzyme specificity for NADPH (strongly preferred) and NADH, even though the motif harboring βA and αB has the sequence GlyXXXGlyXGly, whereas GlyXGlyXXGly is usually the sequence for NADH-preferring enzymes. Many studies have shown that Gly in the GlyXGlyXXGly fingerprint is the most important residue for NADH-binding. Recombinant proteins with the Gly pattern removed have no enzyme activity (8). Restoration of the complete Gly pattern led to an increase of activity (9). Little is known about the critical residues involved and the role of the GlyXXXGlyXGly fingerprint in determining NADPH-preference. Thus far, the three-dimensional structure of NADPH-preferring SDRs has been resolved only for mouse lung CR (MLCR) by Tanaka et al. (10). In MLCR, the determinant for NADP(H) preference is the positively charged environment made by a pair of basic residues (Lys-17 and Arg-39), whereas an Asp at the second β-strand of the βαβ fold is responsible for NAD(H)-preference (11, 12).
We have reported two closely related CR/20β-HSD cDNAs obtained from rainbow trout ovarian follicles, which encode two proteins with only three amino acid differences (13). Analysis of activity of recombinant proteins resulting from expression in Escherichia coli showed that one (CR/20β-HSD type A) had both 20β-HSD and CR enzyme activities, but the other (CR/20β-HSD type B) did not possess either CR or 20β-HSD activity. Among their three distinct amino acids, two residues (Ile-15 and Lys-27 in type A) are located in the GlyXXXGlyXGly domain or nearby. They are substituted by Thr and Gln in CR/20β-HSD type B, respectively. Ile-15 in trout CR/20β-HSD type A is well conserved among many CRs, suggesting that substitution at this site could affect the formation of enzyme–coenzyme complex. To test whether these mutations are responsible for abolishing enzyme activities by disturbing the stability of the enzyme–coenzyme complex, a series of studies employing site-directed mutagenesis were conducted and confirmed the role of Ile-15 in coenzyme (NADPH) binding of trout CR/20β-HSD.
Materials and Methods
Site-Directed Mutagenesis.
Point mutations were introduced into the ORF of CR/20β-HSD type A and type B cDNAs according to the oligonucleotide-directed dual amber method by using a Mutant-Express Km kit (Takara Shuzo, Kyoto). The mutagenic oligonucleotides were designed to introduce a NruI or a SacI site without changing the deduced amino acid residues and to change ATA for Ile to ACA for Thr in CR/20β-HSD type A (termed MutA/I15T), ACA for Thr to ATA for Ile in CR/20β-HSD type B (termed MutB/T15I), and CAA for Gln to AAA for Lys in type B (termed MutB/Q27K). The sequences of the mutagenic oligonucleotide are summarized in Table 1. Mutants were identified by the additional NruI or SacI site after restriction digestion. Each ORF fragment of wild types and mutants was amplified to contain a NdeI site at the 5′ end and a BamHI site at the 3′ end by using the following primers: forward primer (5′-GGGCATATGTCAAAAAAAGTGGCAG-3′ and reverse primer (5′-GGGGATCCTACCATTCCTGAACGG-3′).
Table 1.
Primers for the mutagenic reactions
| Mutation | Mutagenic oligonucleotide | Mutated codon | Restriction site |
|---|---|---|---|
| MutA/I15T | 5′-TCGCGAGTCCTGTGCCTTTATTG-3′ | Ile-15 → Thr | NruI |
| MutB/T15I | 5′-TCGCGAGTCCTATGCCTTTATTG-3′ | Thr-15 → Ile | NruI |
| MutB/Q27K | 5′-GTAAATTTTGCCTTACAGAGCTC-3′ | Gln-27 → Lys | SacI |
The amplified products were subcloned into pET21b+ expression vector by using NdeI and BamHI sites. Dideoxynucleotide sequencing was used to ensure the fidelity of the mutant constructs and to prevent any unexpected mutations.
Expression, Purification, and Characterization of Protein Products.
The mutant expression vectors were expressed in E. coli (BL21), and overexpressed proteins were purified as described for wild-type recombinant CR/20β-HSD (13). Because mutation of Ile-15 decreased the solubility of the recombinant protein generated by E. coli, cultures were carried out in a final concentration of 0.1 mM isopropyl β-d-thiogalactoside (IPTG; 1/10 of that used for wild-type A) at 15°C overnight to improve the recovery efficiency. The supernatants from 12,000 × g centrifugation of the homogenates of the cells were purified with FPLC. The molecular mass of recombinant products from wild types and mutants and protein purity were analyzed by SDS/12.5% PAGE. Protein concentrations were determined by the method of Bradford (14) (Bio-Rad protein assay) by using BSA as the standard.
Enzyme Kinetic Analysis of Wild Types and Mutants.
[3H]17α-HP (1.48–2.22 TBq/mmol; NEN) was used as substrate to measure 20β-HSD activity. The reaction products were separated by using thin-layer chromatography and further confirmed by the recrystallization method as described (13). For CR activity, enzyme assay was performed with 4-nitrobenzaldehyde as substrate, and enzyme activity was measured on a Beckman Du7400 spectrophotometer by observing the rate of change in absorbance of NADPH (0.08 mM) at 340 nm in a 1-ml volume at 25°C. Purified recombinant protein (20 μg) was added to 1 ml of assay mixture containing 20 mM Tris⋅HCl/1 mM EDTA buffer (pH 8.0) with 4-nitrobenzaldehyde (0.5 mM). Reactions were initiated by the addition of NADPH, and the absorbance was measured against a blank containing all components except coenzyme. Absorbance of reactions was followed from 1 min to 5 min after addition of NADPH. Velocities were calculated from the slopes of the zero order portion of the kinetics obtained and were corrected for the control absorbance, and 1 unit of enzyme activity was defined as that amount catalyzing oxidation of 1 μmol of NADPH/min at 25°C.
Determination of Binding Constants for NADPH by Fluorescence Titration.
Measurement of the binding constants for NADPH to wild-type CR/20β-HSD type A, type B, MutA/I15T, MutB/T15I, and MutB/Q27K was accomplished by titration of the intrinsic protein fluorescence (excitation at 290 nm) on a fluorometer (Shimadzu, FDU-3) after the incremental addition of NADPH (0–2.4 μM) in 20 mM Tris⋅HCl/1 mM EDTA buffer (pH 8.0) by using described methods (15, 16). Untransformed fluorescence data were plotted as percentage of change in fluorescence (%ΔF) at emission λmax (336 nm) vs. NADPH concentration ([NADPH]). These data were fitted to a saturation absorption isotherm, which provided an estimate of the binding constants (Kd) and the associated standard error. These data were transformed with the Lineweaver–Burk equation to generate a linear plot of 1/%ΔF vs. 1/[NADPH].
Results
Construction, Site-Directed Mutagenesis, and Expression of Wild-Type and Mutant CR/20β-HSDs.
The N-terminal secondary structures of trout CR/20β-HSD type A and type B proteins predicted with the dnasis program are depicted in Fig. 1. Compared with CR/20β-HSD type A and other CRs, the α-helix of type B is predicted to start from Leu-17 rather than from Ile-15, resulting in the lack of involvement of the second and third Gly of GlyXXXGlyXGly in the α-helix structure. Because Gly residues in GlyXXXGlyXGly harboring the turn of the β-sheet and α-helix are known to play an important role in the formation of enzyme–cofactor complex (17), it is likely that a less favorable configuration for NADPH binding would be formed in CR/20β-HSD type B with the substitution of Ile and Lys to Thr and Gln, respectively.
Figure 1.

Sequence alignment of mammalian CRs and trout CR/20β-HSDs at the N terminus. Three Gly residues of the Rossmann fold (GXXXGXG) are boxed. The predicted α-helix and β-sheet are displayed by α and β, respectively. The α-sheet on the bottom line corresponds to the position in trout CR/20β-HSD type B. ratiCR, rat inducible carbonyl reductase; ratnCR, rat noninducible carbonyl reductase.
To test this hypothesis, we performed site-directed mutagenesis to create several mutants as shown in Table 1. An NruI site or a SacI site with no change in the deduced codons was cointroduced with a mutated codon. Mutant clones were isolated by identifying the additional NruI site or SacI site, and the authenticity of nucleotide sequences was confirmed. Mutants and wild-type ORFs were subcloned into pET21b+ expression vector to yield pET21b+rcA, pET21b+rcB, pET21b+rcMutA/I15T, pET21b+rcMutB/T15I, and pET21b+rcMutB/Q27K. The wild-type and mutant recombinants were overexpressed in BL21 and purified by FPLC to yield 50–500 μg of each expressed protein. SDS/PAGE analysis confirmed the purity of each enzyme. Coomassie brilliant blue staining indicated that the correct protein had been purified, even when the mutant enzyme had no detectable activity in a spectrophotometric assay (Fig. 2).
Figure 2.

Gel electrophoretic patterns of purified recombinants of CR/20β-HSDs and mutants. SDS/PAGE electrophoresis was performed in 12.5% gels. Proteins were stained with Coomassie brilliant blue. The molecular mass (in kDa) of marker proteins is shown at the left side.
Enzyme Activity of Mutants for Oxidoreduction of Steroid and Other Carbonyl Compounds.
To assess the effect of each mutation on steroid reduction, 3H-labeled 17α-HP was used as a substrate to detect the production of 17α,20β-DP by using thin layer chromatography. Wild-type A converted 17α-HP into 17α,20β-DP, whereas the MutA/I15T, MutB/Q27K, as well as wild-type B failed to produce 17α,20β-DP, as shown in Fig. 3. However, enzyme activity was recovered in the MutB/T15I in which Thr-15 was replaced by Ile. This observation is consistent with the results from spectrophotometric assay with 4-nitrobenzaldehyde as a substrate, which is a typical substrate for CR (Table 2). Enzyme specific activity of MutB/T15I was similar to that of wild-type A (1.57 ± 0.08 units/mg and 1.64 ± 0.11 units/mg, respectively). However, catalytic activity of MutA/I15T and wild-type B were not detectable. MutB/Q27K showed weak activity compared with type A (12% of type A).
Figure 3.

Results of thin layer chromatography after incubation of wild-type and mutant proteins with 3H-labeled 17α-HP. Arrows indicate the products from incubation with wild-type A or MutB/T15I recombinant proteins that comigrated with standard 17α,20β-DP.
Table 2.
Kinetic parameters of recombinants of wild type and mutants of CR/20β-HSD
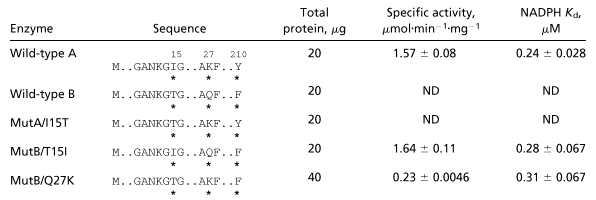 |
Asterisks below the sequences indicate different amino acid residues; numbers above the sequences indicate relative position. ND, not detectable.
The Binding of NADPH and Formation of Enzyme–Cofactor–Substrate Complex.
It was unclear whether the abolition of enzyme activity by substitution of Ile-15 to Thr was caused by the inhibition of binding of cofactor or of substrate. To address this question, the binding constants of wild types and mutants for NADPH were measured. The intrinsic fluorescence at 336 nm (excitation at 290 nm) of the enzyme can be quenched by the addition of NADPH if the enzyme binds with NADPH (14). The fluorescence of recombinant protein products from wild-type A was quenched by the addition of NADPH in a dose-related fashion. In contrast to wild-type A, the protein fluorescence of wild-type B did not change after addition of various concentrations of NADPH (0–2.4 μM). The fluorescence emission spectrum of mutant A/I15T was similar to that of wild-type B, indicating that the mutant A/I15T could not bind NADPH. However, the fluorescence emissions from MutB/T15I and MutB/Q27K were gradually quenched by titration with NADPH, suggesting that the binding of NADPH occurred. The percentage of change in fluorescence at λ336 was plotted against NADPH concentration as shown in Fig. 4. The wild-type enzyme CR/20β-HSD type A and the MutB/T15I had similar binding constants (Kd) of 0.24 ± 0.028 μM and 0.289 ± 0.067 μM, respectively (Table 2). The Kd of MutB/Q27K was 0.31 ± 0.067 μM.
Figure 4.

Measurement of binding constants of NADPH to wild-type CR/20β-HSDs and mutants. The binding of NADPH to enzymes was determined by measuring the decrease in fluorescence emission at 336 nm (excitation wavelength of 290 nm) in 20 mM Tris⋅HCl/1 mM EDTA buffer (pH 8.0). Wild-type and mutant proteins (20 μg) were used, and the data are plotted as percentage of change of fluorescence against NADPH concentration, with maximum change in fluorescence of wild-type A defined as 100%.
Discussion
To investigate the role of amino acid residues critical to the protein conformation and enzymatic activity of trout CR/20β-HSDs, we created three mutants by site-directed mutagenesis. Our results show that a point mutation of Ile at position 15 inactivated enzyme activity (by the assay employed), whereas the replacement of Ile at 15 of type B restored enzyme activity to the level of wild-type A. In the SDR family, the tight turn of βαβ at the N terminus, which is the coenzyme binding domain, is strictly conserved and starts the succeeding α-helix from the second glycine. However, based on analysis of the structure of trout CR/20β-HSD type B with the dnasis program, the substitution of Ile-15 to Thr causes the succeeding α-helix to start from the residue (Lys-17) behind the third glycine (Fig. 1), resulting in changes in spatial conformation of the tight βαβ turn. We hypothesized that this conformational change could cause the geometrical rearrangement of the GlyXXXGlyXGly to a less favorable configuration for NADPH binding. The point mutation of Ile-15 to Thr (MutA/I15T) rendered the enzyme inactive in our experiment, supporting the idea that substitution of Ile to Thr in trout CR/20β-HSD type B prevents the enzyme from binding NADPH.
Many CRs have been purified, and many cDNAs encoding proteins have been cloned, allowing determination of primary structures of mammalian CRs; however, the crystal structure of MLCR determined by x-ray crystallography (10) is the only three-dimensional structure of an NADPH-preferring CR described. In MLCR, eight amino acid residues are involved in enzyme interaction with cofactor through hydrogen bonds forming between NADPH and the enzyme or interaction through four water molecules. Lys-17 and Arg-39 are two basic residues critical for making electrostatic interactions with the 2′-phosphate group of NADPH. The side chain of Ile-19, however, is important for coenzyme binding through its direct interaction with the pyrophosphate moiety of NADPH. Any mutation on Ile-19 would destabilize holoenzyme formation (N. Tanaka, personal communication). Ile-19 in MLCR is strictly conserved among various mammalian CRs (Fig. 1), with rabbit CR being the only exception in which Ile-19 is replaced by Val. Ile-19 in MLCR is equivalent to Ile-15 in trout CR/20β-HSD type A. It is replaced by threonine in trout CR/20β-HSD type B. Because valine and isoleucine belong to the nonpolar, aliphatic R group of amino acids, whereas threonine belongs to the polar, uncharged R group, substitution of isoleucine by valine is considered a milder mutation than substitution by threonine. Indeed, the point mutation of I15T in type A caused the enzyme to lose its binding affinity, indicating that the Ile-15 residue plays an important role in NADPH binding. However, because the cofactor binding site involves contacts with at least eight amino acids in MLCR, it is unlikely that a point mutation of Ile-15 can cause gross structural changes in the three-dimensional structure of the enzyme. It is possible that Ile-15 interacts with other residues to keep the enzyme in a favorable configuration for coenzyme binding. It was reported that Thr-12 of 3β/17β-HSD is crucial for keeping the correct position of strand βD and adjacent residues toward the coenzyme NADH (18). Exchange of Thr-12 to Ala caused loss of 3β-HSD activity. MutB/Q27K showed lower activity and had a lower affinity for NADPH compared with wild-type A, suggesting that position 27 is also involved in keeping the enzyme in a favorable configuration. Structural change of the coenzyme binding region by either changing stereoconformation or weakening of some noncovalent bonds may interrupt the correct binding of NADPH. Further investigation is necessary to analyze the consequences of mutation of Ile and other candidate residues on coenzyme binding.
In the SDR family, enzymes are classified into two groups: one is NADH-preferring and the other is NADPH-preferring. NAD(H) is a ubiquitous coenzyme involved in biological oxidative degradations that yield ATP, whereas NADP(H) is confined, with few exceptions, to reactions of reductive biosynthesis. The only distinction between NAD(H) and NADP(H) is the possession of the extra phosphate group by NADP+ that permits biosynthetic enzymes to recognize it. Monomeric CRs purified and cloned from many mammalian tissues are NADPH-dependent SDRs with a conserved fingerprint of GlyXXXGlyXGly for cofactor binding. However, some tetrameric CRs show dual cofactor preference. Lys-17 and Arg-39 in these fingerprints are crucial for NADPH preference. Substitution of Thr-38 to Asp switched MLCR preference to NADH (12, 19, 20). Lys-17, Arg-39, and Thr-38 in MLCR are equivalent to Lys-13, Arg-35, and Ala-34, respectively, in trout CR/20β-HSDs. Lys and Arg residues are strictly conserved in trout CR/20β-HSD type A and type B, whereas Thr is replaced by Ala in trout CR/20β-HSDs. The kinetic profile from catalytic analysis indicated that trout CR/20β-HSD type A recombinant protein could catalyze its substrate only in the presence of NADPH. There was no enzyme activity in the reaction with NADH (data not shown), supporting the idea that trout CR/20β-HSD preferentially uses NADPH as cofactor.
This article reports the importance of Ile-15 in cofactor binding of CRs. To our knowledge, no reports on the effects of point mutation in the GlyXXXGlyXGly fingerprint on enzyme activity exist, with the exception of a report on Drosophila alcohol dehydrogenase, a mutant generated by ethyl methanesulfonate treatment that contains an aspartic acid substitution at position 14 instead of glycine (21). This mutant shows no alcohol dehydrogenase activity and has a low affinity for NADH, indicating that Gly-14 is important for alcohol dehydrogenase activity.
In conclusion, this study showed that the lack of activity of trout CR/20β-HSD type B is due to the amino acid substitution of Ile-15 by Thr. Enzyme activity could be restored by point mutation of T15I in type B. It was further shown that Ile-15 plays an important role in permitting the enzyme to bind with its coenzyme, NADPH. The biological significance of this point mutation will remain unclear until the biological functions and differential expression of trout CR/20β-HSDs are fully elucidated.
Acknowledgments
This work was supported in part by Grant-in-Aid JSPS-RFTF 96L100401 for research for the future from the Japan Society for the Promotion of Science; by Grants-in-Aid 07283104, 08454266, and 10440247 for scientific research from the Ministry of Education, Science, Culture, and Sports (Japan); and by funds from the Bio Design Program from the Ministry of Agriculture, Forestry, and Fisheries (Japan). G.Y. is grateful for partial support from the Royal Society of New Zealand Marsden Fund (contract U00815).
Abbreviations
- CR
carbonyl reductase
- 20β-HSD
20β-hydroxysteroid dehydrogenase
- 17α-HP
17α-hydroxyprogesterone
- 17α,20β-DP
17α,20β-dihydroxy-4-pregnen-3-one
- SDR
short-chain dehydrogenase/reductase
- MLCR
mouse lung CR
- IPTG
isopropyl β-d-thiogalactoside
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.040548697.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.040548697
References
- 1.Nagahama Y, Adachi S. Dev Biol. 1985;109:425–435. doi: 10.1016/0012-1606(85)90469-5. [DOI] [PubMed] [Google Scholar]
- 2.Nagahama Y. In: Hormones and Reproduction in Fishes, Amphibians, and Reptiles. Norris D O, Jones R E, editors. New York: Plenum; 1987. pp. 171–202. [Google Scholar]
- 3.Jornvall H, Persson B, Krook M, Atrian S, Gonzalez-Duarte R, Jeffery J, Ghosh D. Biochemistry. 1995;34:6003–6013. doi: 10.1021/bi00018a001. [DOI] [PubMed] [Google Scholar]
- 4.Rossmann M G, Liljas A, Branden C I, Banaszak L J. In: The Enzymes. Boyer P D, editor. New York: Academic; 1975. pp. 62–102. [Google Scholar]
- 5.Nakayama T, Hara A, Sawada H. Arch Biochem Biophys. 1982;217:564–573. doi: 10.1016/0003-9861(82)90538-0. [DOI] [PubMed] [Google Scholar]
- 6.Nakayama T, Sawada H. Biochem Biophys Acta. 1986;882:220–227. doi: 10.1016/0304-4165(86)90158-3. [DOI] [PubMed] [Google Scholar]
- 7.Oritani H, Deyashiki Y, Nakayama T, Hara A, Sawada H, Matsuura K, Bunai Y, Ohya I. Arch Biochem Biophys. 1992;292:539–547. doi: 10.1016/0003-9861(92)90028-u. [DOI] [PubMed] [Google Scholar]
- 8.Chen Z, Lu L, Shirley M, Lee W R, Chang S H. Biochemistry. 1990;29:1112–1118. doi: 10.1021/bi00457a003. [DOI] [PubMed] [Google Scholar]
- 9.Albalat R, Atrican S, Gonzalez-Duarte R. FEBS Lett. 1994;341:171–176. doi: 10.1016/0014-5793(94)80451-6. [DOI] [PubMed] [Google Scholar]
- 10.Tanaka N, Nonaka T, Nakanishi M, Deyashiki Y, Hara A, Mitsui Y. Structure. 1996;4:33–45. doi: 10.1016/s0969-2126(96)00007-x. [DOI] [PubMed] [Google Scholar]
- 11.Scrutton N S, Berry A, Perham R N. Nature (London) 1990;343:38–43. doi: 10.1038/343038a0. [DOI] [PubMed] [Google Scholar]
- 12.Nakanishi M, Kaibe H, Matsuura K, Kakumoto M, Tanaka N, Nonaka T, Mitsui Y, Hara A. Enzymol Mol Biol Carbonyl Metab. 1996;6:555–561. doi: 10.1007/978-1-4615-5871-2_63. [DOI] [PubMed] [Google Scholar]
- 13.Guan G J, Tanaka M, Todo T, Young G, Yoshikuni M, Nagahama Y. Biochem Biophy Res Commun. 1999;255:123–128. doi: 10.1006/bbrc.1998.0127. [DOI] [PubMed] [Google Scholar]
- 14.Bradford M. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 15.Bohren K M, Von Wartburg J P, Wermuth B. Biochem J. 1987;244:165–171. doi: 10.1042/bj2440165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jez J, Schlegel B, Penning T. J Biol Chem. 1996;271:30190–30198. doi: 10.1074/jbc.271.47.30190. [DOI] [PubMed] [Google Scholar]
- 17.Tanaka N, Nonaka T, Nakanishi M, Deyashiki Y, Hara A, Mitsui Y. J Biochem. 1995;118:871–873. doi: 10.1093/jb/118.5.871. [DOI] [PubMed] [Google Scholar]
- 18.Oppermann U, Filling C, Berndt K, Persson B, Benach J, Ladenstein R, Jornvall H. Biochemistry. 1997;36:34–40. doi: 10.1021/bi961803v. [DOI] [PubMed] [Google Scholar]
- 19.Nakanishi M, Matsuura K, Kaibe H, Tanaka N. J Biol Chem. 1997;202:2218–2222. doi: 10.1074/jbc.272.4.2218. [DOI] [PubMed] [Google Scholar]
- 20.Chen Z, Lee W R, Chang S H. Eur J Biochem. 1991;202:263–267. doi: 10.1111/j.1432-1033.1991.tb16371.x. [DOI] [PubMed] [Google Scholar]
- 21.Thatcher D R. Biochem J. 1980;187:875–883. doi: 10.1042/bj1870875. [DOI] [PMC free article] [PubMed] [Google Scholar]