

Abstract
Although most cellular glutathione (GSH) is in the cytoplasm, a distinctly regulated pool is present in mitochondria. Inasmuch as GSH synthesis is primarily restricted to the cytoplasm, the mitochondrial pool must derive from transport of cytoplasmic GSH across the mitochondrial inner membrane. Early studies in liver mitochondria primarily focused on the relationship between GSH status and membrane permeability and energetics. Because GSH is an anion at physiological pH, this suggested that some of the organic anion carriers present in the inner membrane could function in GSH transport. Indeed, studies by Lash and colleagues in isolated mitochondria from rat kidney showed that most of the transport (>80%) in that tissue could be accounted for by function of the dicarboxylate carrier (DIC, Slc25a10) and the oxoglutarate carrier (OGC, Slc25a11), which mediate electroneutral exchange of dicarboxylates for inorganic phosphate and 2-oxoglutarate for other dicarboxylates, respectively. The identity and function of specific carrier proteins in other tissues is less certain, although the OGC is expressed in heart, liver, and brain and the DIC is expressed in liver and kidney. An additional carrier that transports 2-oxoglutarate, the oxodicarboxylate or oxoadipate carrier (ODC; Slc25a21), has been described in rat and human liver and its expression has a wide tissue distribution, although its potential function in GSH transport has not been investigated. Overexpression of the cDNA for the DIC and OGC in a renal proximal tubule-derived cell line, NRK-52E cells, showed that enhanced carrier expression and activity protects against oxidative stress and chemically induced apoptosis. This has implications for development of novel therapeutic approaches for treatment of human diseases and pathological states. Several conditions, such as alcoholic liver disease, cirrhosis or other chronic biliary obstructive diseases, and diabetic nephropathy, are associated with depletion or oxidation of the mitochondrial GSH pool in liver or kidney.
Keywords: Glutathione, Mitochondria, Transport, Dicarboxylates, Dicarboxylate carrier, Oxoglutarate carrier, Oxidative stress, Diabetic nephropathy, Alcoholic liver disease
1. Introduction
Glutathione (GSH) is the primary low-molecular-weight thiol in all aerobic cells. Its functional importance lies in the presence of the thiol group on the cysteinyl residue (Fig. 1). The thiol enables GSH to function as both a reductant and a nucleophile. Synthesis of GSH occurs by two sequential, ATP-dependent reactions in the cytoplasm and is catalyzed by γ-glutamylcysteine synthetase and GSH synthetase. The available evidence indicates that most if not all of the biosynthetic activity resides in the cytoplasm with little if any activity in other organelles, including the mitochondria [1,2]. Virtually all mammalian cells have some capacity to synthesize GSH and activity is widely distributed. Degradation of GSH is mediated by a separate pathway that, in contrast to the synthetic pathway, exhibits a discrete tissue distribution [3]. Thus, the initial step in the breakdown of GSH, which involves cleavage of the γ-glutamyl-peptide bond by γ-glutamyltransferase (GGT), is localized in luminal membranes of epithelial cells, such the renal proximal tubule and enterocytes, and is largely absent from cells such as hepatocytes or cardiac myocytes.
Fig. 1.
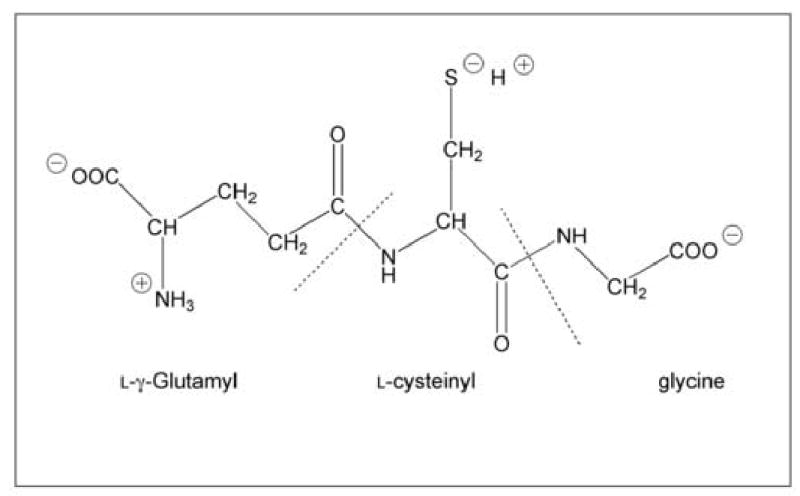
Structure of GSH.
As a reductant, GSH maintains intracellular, sulfhydryl-containing proteins in the reduced and active form by either the reduction of potentially toxic peroxides or by the action of thiol-disulfide exchange reactions. The first process is mediated by the selenium-containing GSH peroxidase and the second process is mediated by thioltransferases. The GSH–glutathione disulfide (GSSG) redox pair has a midpoint electrochemical potential at pH 7.0 (Em,7) of −240 mV, which places it well between the most reduced redox couple, H+/H2 (−420 mV) and the most oxidized redox couple, O2/H2O (+820 mV). Accordingly, the GSH/GSSG redox couple can readily interact with most of the physiologically relevant redox couples, undergoing reversible oxidation or reduction reactions, thereby maintaining the appropriate redox balance in the cell.
As a nucleophile, GSH serves a critical function in the cell by reacting with electrophiles that are generated as a consequence of metabolic processes involving both endogenous compounds and xenobiotics. While GSH can react non-enzymatically with electrophiles, rates of nucleophilic addition reactions are greatly enhanced by the catalytic action of a family of enzymes called the GSH S-transferases (GSTs). Most of the GST isoforms are present in the cytoplasm, although isoforms are also present in the endoplasmic reticulum and mitochondrial matrix.
The mitochondrion is an excellent example of a subcellular organelle whose function is closely linked to maintenance of redox balance. As mitochondria are the primary intracellular sites of oxygen consumption, they may also be primary sites of generation of reactive oxygen species (ROS). Although normal electron transport in mitochondria involves four-electron reduction of molecular oxygen to water, partial reduction reactions occur even under physiological conditions, causing release of superoxide anion and hydrogen peroxide. Toxic or pathological conditions, such as oxidative stress, that lead to an impairment of mitochondrial function, can increase release of ROS. A large number of mitochondrial enzymes, including dehydrogenases and transport ATPases, contain critical sulfhydryl groups that must be maintained in the reduced form for proper function [4]. Furthermore, redox-sensitive components in the electron transport chain, such as iron ions on heme prosthetic groups and iron-sulfur centers, may be oxidized during a redox imbalance, thereby producing mitochondrial dysfunction.
The redox status of GSH and other thiols has long been known to be critical for proper mitochondrial function [5–8]. Alterations in GSH concentration and redox status have been associated with oxidative stress induced by peroxides and other oxidants in mitochondria from kidney, liver, brain, and tumor cells [2,9–15], regulation of mitochondrial Ca2+ ion distribution and pyridine nucleotide oxidation status [16–21], damage to mitochondrial DNA [22,23], and induction of the membrane permeability transition [21,24–29]. More recently, changes in mitochondrial GSH status have been associated with activation of signaling pathways and expression of genes that regulate apoptosis [30–37] and cell growth and differentiation [38,39]. Besides ROS, recent attention has focused on the role of reactive nitrogen species, in particular nitric oxide (NO) and peroxynitrite (ONOO−), in the regulation of mitochondrial and cellular function and in mediating certain forms of chemically induced and pathological injury. Although NO may interact directly with proteins and other cellular macromolecules, effects of NO may also be mediated by formation of S-nitrosoglutathione (GSNO) [40–43]. Thus, GSH may react with NO to release ROS (Fig. 2) or it may serve as an NO donor via formation of GSNO (Fig. 3). The normally high concentration of GSH in the mitochondrial matrix and the presence of a constitutively expressed nitric oxide synthase in the organelle suggest that formation of GSNO plays an important physiological role.
Fig. 2.
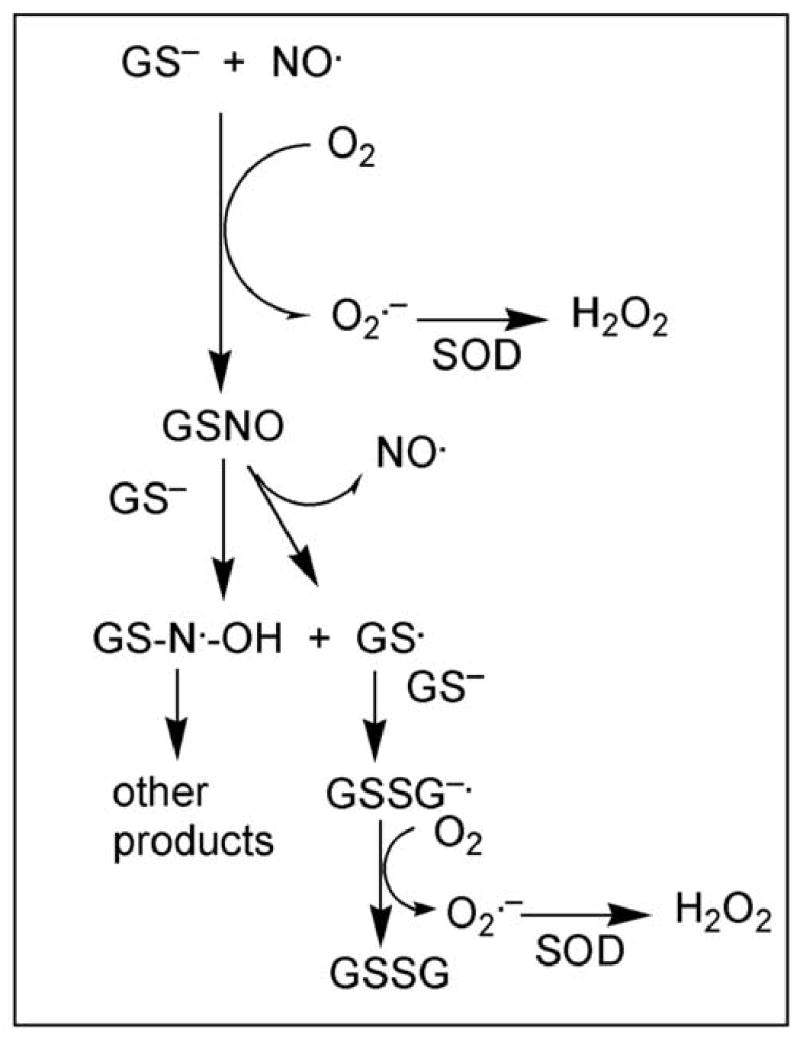
Reactions by which GSH reacts with NO to form GSNO. GSH, as the thiolate, reacts with NO in the presence of O2 and forms GSNO. GSNO can release NO (function of GSNO as an NO donor) or it may react in the presence of the thiolate to form a species that can glutathionylate protein sulfhydryl groups. SOD, superoxide dismutase. Adapted from [43].
Fig. 3.
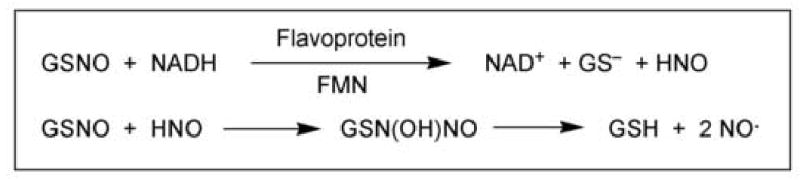
NO formation from GSNO. GSNO acts as an NO donor in two reactions, the first of which is mediated by a flavoprotein containing FMN. The HNO generated from the first reaction can react with another molecule of GSNO to form an intermediate that decomposes to GSH and NO.
It is, therefore, clear that maintenance of adequate concentrations of GSH within the mitochondrial matrix is essential for regulation and proper function of numerous critical processes. This review will focus on the current state of knowledge about how the mitochondrial pool of GSH is determined and regulated and some approaches that the author’s laboratory has taken to modulate this pool to alter susceptibility to chemically induced injury or disease.
2. Early studies on mitochondrial GSH homeostasis and energetics
The earliest studies that focused specifically on the mitochondrial GSH transport process studied the relationships between mitochondrial respiratory state and transport activity in mitochondria from rat liver. In 1990, Kurosawa et al. [44] found that GSH was transported into rat liver mitochondria at highest rates under state 4 conditions and that transport was diminished by a protonophore, by antimycin A, or under state 3 conditions. Fasted rats were used in their studies, indicating that mitochondria were likely in an energy-depleted state. In that same year, Martensson et al. [45] defined two kinetic components for uptake of GSH by isolated mitochondria from rat liver: A high-affinity component (Km = 60 μM, Vmax = 0.5 nmol/min per mg protein) and a low-affinity component (Km = 5.4 mM, Vmax = 5.9 nmol/min per mg protein). Both components were inhibited by a protonophore, glutamate, and by the GSH-analogue ophthalmic acid. Fasted rats were also used in their studies. In 1995, Kaplowitz and colleagues [46] expressed GSH transport activity in mitochondria of Xenopus laevis oocytes that were microinjected with total liver mRNA. The transport activity exhibited similar properties to those observed in mitochondria from rat liver and was distinct from those present in the canalicular or sinusoidal plasma membranes. Fractionation of poly(A)+ RNA identified a single mRNA species of 3 to 3.5 kb. There is some concern, however, about the identity of the activity measured because the oocytes exhibit some endogenous GSH transport activity and no attempt was made to identify the potential function of specific carriers in GSH uptake.
Using rat kidney mitochondria, Schnellmann [47] concluded that GSH was taken up by both a carrier-mediated process and by diffusion. The carrier-mediated process was modestly (30%) inhibited by glycine, serine, and ophthalmic acid, but not by glutamate, cysteine, γ-glutamylglutamate or proline.
Other studies that are described below provided insight into the relationships between mitochondrial GSH status and several pathological or disease states. Because we know that transport of cytoplasmic GSH into the mitochondrial matrix is the primary, if not sole, determinant of GSH status in the organelle, the implication from these studies is that GSH transport is inhibited or somehow defective. Direct evidence for this suggestion, however, has only recently become available. Although these studies and those described above provided mostly indirect information about GSH transport activity under various physiological or pathological conditions, they did not address the crucial questions of what specific carrier protein(s) mediate(s) the transport process and the specific energy sources that are used to maintain the tightly regulated mitochondrial GSH pool. In the section that follows, we present summaries of the studies that identified and confirmed the function of specific carrier proteins in the mitochondrial inner membrane of rat kidney in transport of cytoplasmic GSH into the matrix.
3. Role of anion carriers in GSH uptake
3.1. GSH as an organic anion: Potential carriers in mitochondrial inner membrane
The charged nature of the GSH molecule suggests that it cannot passively diffuse across the mitochondrial inner membrane. Rather, because mitochondria possess a membrane potential with the matrix space negative relative to the cytoplasm and because GSH is a negatively charged molecule at physiological pH, GSH must be transported actively or in exchange for another anion. Possible net charges for GSH are determined by four functional groups (cf. Fig. 1): There is one free amino group (pKa = 8.6; predominantly protonated = +1), two free carboxyl groups (pKa = 3.53 and 2.12; predominantly deprotonated = −2), and the thiol group (pKa = 9.2; predominantly protonated = 0). Whereas the pH of the cytoplasm is typically near 7.0, that of the mitochondrial matrix is slightly alkaline (approximately 7.8). Hence, more of the −SH groups will be deprotonated in the matrix. Additionally, a microenvironment may exist near the active sites of the carrier proteins that effectively lowers the pKa of the thiol group so that a much higher proportion of GSH molecules would be in the thiolate form. Overall then, the GSH pool available for mitochondrial carriers is likely to have a net charge of between −1 and −2.
Considering the anionic nature of GSH, our first approach to determining the function of specific, inner membrane carrier proteins in mitochondrial GSH transport was to examine the potential activity of known carriers [2]. Eight known anion carriers are present in the mitochondrial inner membrane that could conceivably play a role in the uptake of GSH from the cytoplasm (Table 1). These carriers are involved in the translocation of citric acid cycle intermediates, amino acids, and gluconeogenesis precursors across the mitochondrial inner membrane and thus play critical roles in mitochondrial and cellular energetics [48,49]. High activity of these carriers is expected in cells such as those of the renal proximal tubule because of high rates of mitochondrial respiration, active transport, and gluconeogenesis. These carriers are presumably expressed in mitochondria from all tissues, although tissue-specific differences in expression levels and activities also presumably exist (see section 3.2.4 below).
Table 1.
Mitochondrial anion transporters
| Carrier | Function | Charge Transfer | Inhibitors |
|---|---|---|---|
| Adenine nucleotide translocase (AAC1–3; Slc25a4–6) | ADP3− in, ATP4− out | Electrogenic | Atractyloside, Carboxyatractyloside Bongkrekic acid |
| Phosphate (PiC; Slc25a3) | H2PO4− in, OH− out | Electroneutral | SH-reagents |
| Dicarboxylate (DIC; Slc25a10) | Malate2− in, HPO42− out | Electroneutral | Butylmalonate |
| 2-Oxoglutarate (OGC; Slc25a11) | 2-Oxoglutarate2− in, malate2− out | Electroneutral | Phenylsuccinate |
| Glutamate–Aspartate | Glutamate− + H+ in, | Electrogenic | — |
| (AGC1/2; Slc25a12/13) | Aspartate− out | ||
| Glutamate–Hydroxide (GC1/2; Slc25a22/18) | Glutamate− in, OH− out | Electroneutral | — |
| Tricarboxylate / Citrate (CIC; Slc25a1) | Citrate3− + H+ in, malate2− out | Electroneutral | 1,2,3-Benzenetricarboxylate, triethyl citrate |
| Monocarboxylate(MCC) | Pyruvate− in, OH− out | Electroneutral | Cyanohydroxycinnamate |
Based on substrate specificities, potential candidates among these carriers for a role in GSH uptake are the monocarboxylate (MCC), dicarboxylate (DIC; Slc25a10), 2-oxoglutarate (OGC; Slc25a11), tricarboxylate or citrate (CIC; Slc25a1), glutamate–hydroxide (GC1/2;Slc25a22/18), and glutamate-aspartate (AGC1/2;Slc25a12/13) carriers. Because the adenine nucleotide translocase and phosphate–hydroxide carriers have fairly restricted substrate specificities, they are not likely to catalyze GSH transport. Although the MCC, DIC, OGC, and CIC all differ with respect to substrate specificity and inhibitor sensitivity, they share a common ~30 kDa molecular weight subunit and are believed to belong to a carrier “superfamily” [50]. Each of these carriers are electroneutral, meaning that they catalyze exchange of anions or a combination of anions and a proton, so that there is no net transfer of charge across the inner membrane. The glutamate–hydroxide carrier is similarly electroneutral, exchanging glutamate for an hydroxide ion. In contrast, the glutamate–aspartate carrier is electrogenic, catalyzing net transfer of one positive charge into the mitochondrial matrix. Although the two glutamate carriers are not likely candidates to mediate transport of GSH, the presence of a glutamyl residue on the GSH molecule suggests the possibility that GSH may interact with these carriers.
3.2. Identification of the DIC and OGC as major GSH transporters
3.2.1. Keys to accurate measurement of mitochondrial GSH transport
One of the most straightforward and simple approaches to determining the potential function of individual membrane carriers in the transport of a given substrate across the inner membrane is to use the experimental model of suspensions of freshly isolated mitochondria [51]. The mitochondrial suspensions are prepared in a standard buffer, mixed with the appropriate substrate solutions, incubated for various time periods, typically at temperatures no higher than 25°C, and then the intramitochondrial and extramitochondrial compartments are separated by either rapid filtration under vacuum or rapid centrifugation procedures. Although measurement of transport activity in this experimental model is technically simple, the procedures used are subject to a large number of potential artifacts that may compromise the accuracy of measurements [48,52]. Some of these potential artifacts are specific to studies involving GSH, some of specific to studies involving suspensions of isolated mitochondria, and others are relevant to any transport study regardless of the experimental model being used. To convey the complexity of the mitochondrial transport process despite the simplicity of the assay methods, a brief discussion of these artifacts and suggested approaches to minimize or otherwise account for them are presented. We have previously discussed these considerations with regard to accurate measurement of mitochondrial GSH transport [52,53].
Potential artifacts in measurement of mitochondrial GSH transport that are important for any transport study, regardless of substrate, tissue of origin, or specific experimental model being used, include the following: 1) Loss of transported substrate during sample processing; or 2) contamination of compartment of interest with substrate from outside the compartment. The first concern, that of potential loss of transported substrate during sample processing, can be minimized by processing in such a way that the compartments are rapidly separated and metabolic and transport processes inactivated. This is typically accomplished by methods such as filtration under vacuum or rapid centrifugation through a medium that separates compartments and delivers the compartment of interest into a medium that inhibits metabolic and transport processes. Examples of such a stop medium include a buffer that contains a high concentration of an inhibitor of metabolism or transport and/or an acid to effect precipitation of proteins. The second concern, that of contamination of material in the outside of the compartment, can be handled methodologically by the filtration or centrifugation methods just described. In spite of these methods, however, a small fraction of fluid from outside the compartment can still contaminate the sample. This is particularly significant for uptake measurements because of the high concentration of substrate in the medium and the small fraction of total solution volume occupied by the compartment of interest. Such contamination can be monitored by the use of radiolabeled markers of the extra- and intracompartmental space. Typically, an impermeant molecule, such as [14C]-sucrose or [3H]-inulin, is used as a marker for the extracompartmental space and 3H2O is used as a measure of total volume. In this manner, corrections can be made for carry-over of extracompartmental substrate that occurs during sample processing.
Potential artifacts that are unique to studies with isolated mitochondria include: 1) Changes in mitochondrial matrix volume that occur during the transport incubation; and 2) induction of the membrane permeability transition (MPT) during transport incubation. The MPT is defined as a voltage-dependent, cyclosporine A (CsA)-sensitive, high-conductance inner membrane channel. Pore opening is favored by increases in matrix calcium ion concentrations and is strongly promoted by a diverse array of agents, including many that oxidize pyridine nucleotides and thiols. Regarding the first potential artifact, radiolabeled markers of extra- and intramitochondrial space can be used to quantitatively determine matrix volume, similar to the approaches used to account for contamination with extracompartmental medium. Additionally, mitochondrial incubations can be performed in the presence of antimycin A (typically 1–5 μM). Caution must be exercised in the use of antimycin A because it can produce oxidative stress and mitochondrial injury under certain conditions [54,55]. Regarding the second potential artifact, a role for the MPT can be assessed by use of the permeability transition pore inhibitor CsA (e.g., at 0.5 nmol/mg protein); inhibition of apparent uptake by CsA would indicate involvement of the MPT. Similarly, one can distinguish between the mechanism of apparent inhibition of transport by a specific compound as occurring by competitive inhibition or by induction of the MPT by use of CsA. For example, in our study of substrate specificity of GSH uptake in suspensions of mitochondria isolated from rat renal cortex ([53]; see below), we found that the apparent inhibition of GSH uptake by phosphoenolpyruvate, which is a substrate for the CIC, was not due to competitive inhibition but was due to its well known ability to induce the MPT as its effect was eliminated by CsA.
Finally, a potential artifact that is specific to measurement of GSH transport is degradation of GSH during the transport process or during sample processing by contaminating GGT. As explained above, GGT is the sole enzyme that cleaves the γ-glutamyl peptide bond of GSH, thereby initiating its turnover. GGT is present on the luminal membrane, with its active site facing the extracellular space, of many epithelial cells, including the renal proximal tubule, choroid plexus, retinal pigment epithelium, and small-intestinal jejunal epithelium. Although there is a considerable degree of variation in activity levels among species, GGT activity is always highest in the renal proximal tubule [3]. In fact, GGT activity on the luminal or brush-border plasma membrane of renal proximal tubular cells is so high that even a very modest (e.g., < 1%) contamination of an isolated mitochondria preparation with brush-border membranes can result in a significant capacity to degrade GSH that is comparable to rates of transport. Consequently, it is critical to inhibit GGT activity as completely as possible. Although several compounds have been used to inhibit GGT, the most widespread and effective inhibitor is acivicin (l-(αS,5S)-α-amino-3-chloro-4,5-dihydro-5-isoxazoleacetic acid) [56].
3.2.2. Substrate specificity, sensitivity to inhibitors, and energetics of GSH transport
In our initial study of mitochondrial GSH transport [2], we found that uptake of GSH by rat kidney mitochondria was saturable (Km = 1.3 mM, Vmax = 5.59 nmol/min per mg protein), was markedly inhibited by γ-glutamylglutamate, certain S-alkyl derivatives of GSH, glutamate, and by dicarboxylates, but was not inhibited by monocarboxylates. Furthermore, the rate of GSH uptake was not altered by uncouplers or a protonophore. These findings suggested that GSH uptake in rat kidney mitochondria is mediated by an electroneutral exchange with dicarboxylates. More recent studies of ours using isolated mitochondria from rat renal cortex [53], assessed substrate specificity, inhibitor sensitivity, and energetics in more depth: We provided additional evidence that of the substrates for the various citric acid cycle carriers, only dicarboxylates specifically interacted with GSH for transport across the mitochondrial inner membrane. Although l-glutamate was inhibitory, the interaction is not due to the two compounds being transported by the same carrier. Rather, the glutamyl residue of the GSH molecule appears to play a role in binding to the carrier(s). Whereas l-glutamate inhibited GSH uptake, GSH did not affect rates of l-glutamate uptake, which were approximately 5-fold higher than those for GSH. Mitochondrial uptake of GSH was also found to be significantly diminished in a nominally phosphate-free or low (≤1 mM) phosphate-containing buffer, consistent with the function of the DIC in GSH uptake.
Function of the DIC and OGC in GSH uptake were further demonstrated in studies where the inner membrane carriers from mitochondria of rabbit kidney cortex were enriched and reconstituted into proteoliposomes [57], using reconstitution methods previously developed for study of the DIC and OGC [58–61]. This approach has the advantage of eliminating the potentially confounding effects of matrix metabolism or volume change or induction of the MPT. Kinetics, substrate specificity, and inhibitor sensitivity were similar to those properties observed in intact mitochondria from rat renal cortex. Based on effects of butylmalonate (selective DIC inhibitor) and phenylsuccinate (selective OGC inhibitor), both singly and in combination, we estimated that of the total amount of GSH transport in renal cortical mitochondria, ~60% is mediated by the DIC and ~40% is mediated by the OGC. At least 80% of the total observable GSH transport across the mitochondrial inner membrane of kidney mitochondria could be attributed to function of the DIC and OGC [53,57]. A basic scheme illustrating the function of the two carriers in renal mitochondrial GSH transport and their relationship to both the citric acid and GSH redox cycles, is shown (Fig. 4). It should be emphasized that although the DIC and OGC account for most of the observable transport of GSH in mitochondria from renal cortex, other thus far unidentified carriers may also contribute and that the contribution of these two carriers in mitochondria from other tissues may be less than that in kidney.
Fig. 4.
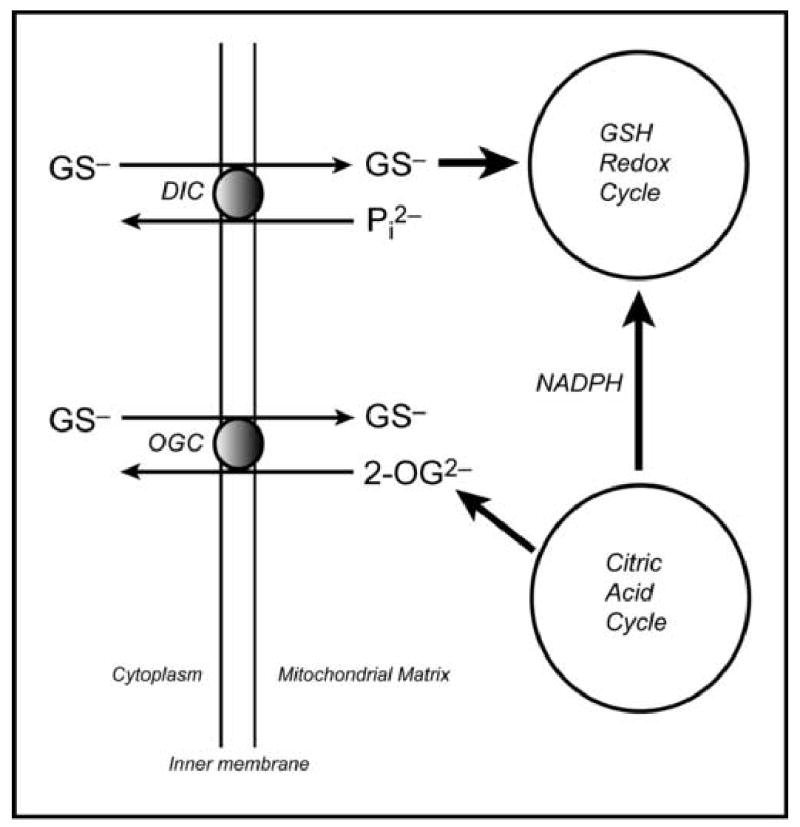
Mitochondrial transport of GSH. Generalized summary scheme, simplified from [56], illustrating the basic function of the DIC and OGC in GSH transport and their relationships with the citric acid and GSH redox cycles.
Kaplowitz and colleagues [62] identified decreased mRNA expression and transport activity of the OGC in rat liver mitochondria as being linked to the decreased mitochondrial GSH content characteristic of alcoholic liver disease, confirming the role of this carrier in mitochondrial GSH transport. More detailed mechanistic studies of the kinetics and properties of GSH transport in liver mitochondria, however, are not available. As discussed below, we have unpublished data suggesting that tissue-specific differences exist in the function of the various carriers for mitochondrial GSH transport. Fernandez-Checa and Kaplowitz [63] recently reviewed the role of hepatic mitochondrial GSH transport in disease and chemically induced toxicity. As briefly discussed below (see section 4), studies from these investigators highlight the human health and therapeutic significance of mitochondrial GSH transport.
3.2.3. Structural and functional studies of the DIC and OGC
As noted above, both the DIC and OGC appear to belong to a “superfamily” of mitochondrial inner membrane transporters with similar three-dimensional structure, as predicted by hydropathy analysis [64]. The TMpred program predicts three-dimensional structure and transmembrane-spanning domains (TMDs) using amino acid polarity and charge. Based on this program, both the DIC and OGC, as well as other carriers in the superfamily (e.g., the MCC, CIC, PiC), are predicted to have three TMDs. Each TMD is comprised of two hydrophobic stretches that span the membrane presumably as α-helices, each separated by hydrophilic loops. Both the N- and C-terminal ends of each monomer are on the cytoplasmic side of the inner membrane. The hydropathy plots can then be used to predict which amino acid residues may be essential for proper three-dimensional structure and insertion into the membrane. Both carriers are similar in size, with the DIC containing 286 or 287 amino acids, depending on species, and a molecular mass of approximately 31 kDa, and the OGC containing 314 to 322 amino acids, depending on species, and a molecular mass of 34–37 kDa. All the members of the Slc25 transporter family are believed to exist as homodimers.
Various approaches have been used to identify essential residues for proper carrier function, including inhibition by amino acid-specific reagents, site-directed and cysteine-scanning mutagenesis, and spin labeling. For example, Palmieri’s group used Arg-specific reagents [65] and different mutagenesis and spin-labeling techniques [66,67] to demonstrate the function of an Arg residue near the substrate-binding site and two other Arg residues in TMD1 and TMD2 of the OGC. Cys residues are often critical to protein function. Although the DIC and OGC exhibit some homology, their content of Cys residues and the apparent role of these residues in carrier function appear to differ. Thus, the DIC from rat or bovine liver contains five Cys residues at positions 17, 21, 22, 211, and 216. Conversion of any one of these residues in the bacterially expressed DIC from rat kidney mitochondria to Ser or Ala by site-directed mutagenesis, however, failed to significantly alter transport activity (J. Wang, F. Xu, D.A. Putt, L.H. Matherly, and L.H. Lash, unpublished observations). In contrast, the OGC contains only three Cys residues at positions 184, 221, and 224, with the latter two forming an intramolecular disulfide bond [68–71]. Conversion of C221 and C224 to Ser results in a marked reduction in transport activity, indicating that formation of the intramolecular disulfide bridge is critical for function [71].
For the OGC, therefore, the Cys residues appear to be critical for proper three-dimensional structure and function. For the DIC, however, the functional significance of the cysteine residues is unclear. In studies to purify and reconstitute the DIC [58,60], it was noted that in marked contrast with the OGC, the DIC is relatively unstable when isolated, undergoing relatively facile oxidative inactivation. Thus, although replacement of any of the five Cys residues of the DIC with a Ser or Ala does not markedly affect activity, oxidation of the thiol groups in the native protein inhibits activity.
3.2.4. Species and tissue specificity of transport
The DIC appears to be invariant across tissues and exhibits a high degree of amino acid sequence homology across species. Protein sequences from the liver mitochondrial carriers are known for three species: mouse, rat, and human. Whereas mouse and human DIC have 287 amino acid residues, rat DIC has 286. The extent of sequence homology between mouse and rat is 96% whereas that between mouse and human is 89%. The OGC, in contrast, exhibits much broader cDNA and amino acid sequence differences across both species and tissues. Using the cDNA sequence from brain as the basis for comparison, rat and mouse exhibit the highest degree of homology (95%; 919/964), with those from rat and bovine and rat and human exhibiting somewhat lower degrees of homology (rat vs. bovine: 90% or 866/964; rat vs. human: 89% or 862/964). Comparison of deduced amino acid sequences, however, shows greater species differences, with that from the mouse differing the greatest amounts from those from rat, human, or bovine. Thus, whereas the OGC protein from bovine, rat, and human contain 314 amino acid residues, that from mouse contains 322 amino acid residues. Moreover, whereas amino acid sequence homologies between the OGC from rat, bovine, and human are 95% to 96%, those between mouse and the other species is only 34%. The functional implications for these differences are unknown.
cDNA sequences for the OGC from rat that are published in GenBankTM are only available from brain and heart mitochondria. Although Coll et al. [62] quantified OGC expression in rat liver, the published cDNA sequence from rat brain was used as the template for their PCR primer design. In our study of OGC from rat kidney [71], we used total rat kidney RNA as a template. The sequence of our PCR product, which was repeated numerous times with identical results, exhibited significant differences from that of the mitochondrial OGC from rat heart and brain. Nucleotide differences in the cDNA sequences were observed for 6 bp or 10 bp between kidney and heart or kidney and brain, respectively. Amino acid differences for the deduced sequences did not appear to be significant between kidney and heart (2 amino acid residues) but were significant between kidney and brain (6 amino acid residues), with 2 differences involving changes in charge or polarity and 3 differences involving residues in or near TMDs. The nature of these differences suggests that the OGC proteins in rat liver and kidney mitochondria exhibit differences in structure that may translate into functional differences.
While extensive studies of transport kinetics and substrate specificity for the OGC have been conducted in rat kidney mitochondria [53], no such studies have as yet been published for rat liver mitochondria. We have, however, conducted preliminary studies in isolated rat liver mitochondria (Q. Zhong, L.H. Lash, unpublished observations), and one clear difference between GSH transport in mitochondria from the two tissues is that whereas function of the DIC and OGC can account for at least 80% of the total transport activity in kidney mitochondria, these two carriers account for at most 50% of total transport activity in liver mitochondria. This suggests that at least one other carrier besides the DIC and OGC plays a quantitatively significant role in GSH transport into rat liver mitochondria. Additional studies are needed to clarify the function of various carriers for GSH in rat liver mitochondria.
Besides the potential function of tissue-specific carriers, differences also exist in the level of expression and activity of carriers in various tissues. It is known, for example, that deficiencies exist in certain mitochondrial enzymes or transmembrane carriers, resulting in so-called mitochondriocytopathies [72]. Further, these disorders most severely affect those tissues that are most dependent on mitochondrial energy production, such as skeletal muscle and heart. Huizing et al. [72] determined the human tissue distribution of several key carriers involved in either oxidative phosphorylation (i.e., the AAC, PiC, and voltage-dependent anion channel) or metabolite transport (i.e., OGC, carnitine-acylcarnitine carrier, CIC). Levels of mRNA expression in various tissues generally correlated with tissue dependence on mitochondrial energy, with skeletal muscle and heart exhibiting much higher levels of mRNA for several carriers than other tissues, including brain, pancreas, lung, liver, placenta, and kidney. Of interest for GSH transport, OGC mRNA expression was by far the highest in skeletal muscle and heart, but was also relatively high in brain, liver, and kidney. Pancreas and placenta exhibited very low levels of OGC mRNA expression and that in lung was barely detectable.
3.2.5. Potential role of other carriers
As indicated above, less specific, mechanistic information on mitochondrial GSH transport is available in liver than in kidney. Although rat liver OGC may have a high degree of homology with rat brain OGC [62], its sequence has not been reported. A thorough search of the GenBankTM database and the published literature failed to find any cDNA or amino acid sequences for the OGC from rat liver mitochondria [71]. Fiermonte et al. [73], however, reported cloning and expression of a rat liver oxodicarboxylate (oxoadipate) carrier (ODC; Slc25a21) that transports 2-OG and other C5–C7 dicarboxylates. They isolated a 1456-bp cDNA with a 99-bp 5’-untranslated region, an open reading frame of 897 bp, and a 460-bp 3’-untranslated region. The cDNA encodes a polypeptide of 298 amino acids with a molecular mass of 33,276, which contrasts with the OGC from rat kidney, heart, and brain mitochondria, which are all 314 amino acids in length. The relationship between the rat liver ODC and OGC is unclear as is the potential for the ODC to transport GSH. Clearly, further investigation is needed to establish the roles of the OGC versus ODC in rat liver, to determine the significance of the OGC sequence variants in relation to kidney and liver GSH transport, and to assess the functional implications of these sequence differences for mitochondrial GSH transport.
3.2.6. Regulation of mitochondrial GSH transport
The identification of the DIC and OGC as the primary membrane carriers responsible for transport of GSH into renal mitochondria suggests that mitochondrial GSH status is closely regulated by mitochondrial energetics. Certainly, the fact that GSH is transported by the same carriers as and, therefore, competes with, dicarboxylates, suggests that nutritional status can directly influence GSH transport ability. Indeed, as described above, some of the earliest studies in liver and kidney mitochondria found marked differences in transport activity dependent on respiratory state [44,45,47]. Indeed, the principal function of the DIC is to transport dicarboxylates from the cytoplasm into the mitochondria, thereby supplying substrates for the citric acid cycle [48,49]. Interestingly, the DIC also transports thiosulfate into mitochondria, delivering it to rhodanese and thiosulfate reductase. Although the OGC can also deliver substrates to the enzymes of the citric acid cycle, it is primarily viewed as functioning in the malate-aspartate and 2-OG-isocitrate shuttles, nitrogen metabolism, and gluconeogenesis from lactate.
Correlation between mitochondrial GSH status and energetics makes sense because the mitochondria are the primary sites within the cell for oxygen consumption and, hence, for endogenous generation of ROS. One would expect, therefore, that higher concentrations of GSH in the matrix would be advantageous in rapidly respiring mitochondria to minimize the potential release of toxic ROS generated during electron transport. Although some investigation of the relationship between rates of GSH transport and respiratory state has been done, the hypothesis or expectation stated above has not been tested directly.
There is, therefore, some expectation that ROS or RNS may directly affect activity of GSH carriers by interaction with key Cys residues. For the DIC, while the five Cys residues do not seem to be essential for activity (as replacement with Ser did not affect activity), they may serve as regulatory sites or sensors of oxidative and/or nitrosative stress. Our hypothesis is that relatively low levels of ROS or RNS can up-regulate expression of one or more of the mitochondrial GSH carriers. Higher levels of these species, however, may lead to in activation of the carriers. The relatively facile autoxidation and inactivation of the DIC upon purification and isolation [58,60] supports this notion. For the OGC, its three Cys residues appear to function much differently than those of the DIC. Whereas C221 and C224 are important for intramolecular disulfide bond formation, C184 may function similarly to the Cys residues of the DIC as a redox sensor, although the relative stability of the purified OGC to autoxidation and inactivation could be interpreted as arguing against this hypothesis. Additional studies are needed to directly assess the influence of ROS and RNS on GSH carrier expression and function.
4. Mitochondrial GSH status and disease or pathological states
An increasing number of toxic or pathological states are being recognized for being associated with marked depletion and/or oxidation of the mitochondrial GSH pool. These observations highlight the importance of this pool. Additionally, our increasing knowledge about the carriers that are responsible for GSH transport into mitochondria provides us with therapeutic targets. For example, hydrogen peroxide generation in liver mitochondria, which leads to oxidative injury, is stimulated only when mitochondrial GSH is depleted below a critical level of approximately 40% of normal [74,75]. Chronic ethanol ingestion and alcoholic liver disease are associated with marked decreases in liver mitochondrial GSH content [76–81]. One consequence of this ethanol-induced decrease is an increase in susceptibility to certain toxicants, such as acetaminophen [82,83], and to toxic cytokines such as TNFα [84–86]. Cirrhosis and other forms of biliary obstruction are also characterized by mitochondrial dysfunction and depletion of the matrix GSH pool [87,88]. Type II diabetes has been reported to be associated with mitochondrial oxidative stress and a depleted and/or oxidized state of mitochondrial GSH in rat heart, brain, and kidney [89–91].
As noted above, Kaplowitz and colleagues [62] provided direct evidence that the altered state of mitochondrial GSH in alcoholic liver disease is due, at least in part, to decreased expression and activity of the OGC. This was the first evidence of this type for any disease or pathological state that is characterized by mitochondrial damage or oxidative stress. The specific involvement of defects in or decreased expression of GSH carriers in other diseases has not been established. As discussed above, Fernandez-Checa and Kaplowitz [63] recently reviewed the role of alterations in hepatic mitochondrial GSH in various pathological and toxic states.
5. Manipulation of mitochondrial GSH
Numerous approaches can and have been used to alter concentrations of GSH in cells. For therapeutic objectives, an increase in either GSH content or GSH/GSSG redox state would typically be desired. Due to feedback inhibition of γ-glutamylcysteine synthetase (GCS) by GSH, however, achievable levels of GSH inside cells have an upper limit. The most common approach to increasing cellular GSH concentrations to bypass this limit is to incubate cells with either GSH, GSH ethyl ester, or amino acid precursors, depending on whether or not intact GSH is transported across the plasma membrane [56]. Another approach would involve induction or overexpression of GCS. If one wants to specifically increase GSH content in a particular subcellular organelle (e.g., the mitochondria), then simply increasing GCS activity or expression or providing the extracellular medium with GSH or GSH precursors will not be very effective. Rather, because the mitochondrial GSH pool appears to be largely, if not entirely, determined by transport from the cytoplasm, the most logical method is to alter expression of the DIC or OGC to effect the desired changes in mitochondrial GSH status [71,92].
In our initial study [92], we transiently transfected NRK-52E cells, an immortalized cell line derived from rat proximal tubules that exhibits many properties that are favorable for study of mitochondrial GSH transport [93], with the cDNA for the rat DIC. Besides exhibiting many transport and metabolic functions that are normally found in the proximal tubular cell, NRK-52E cells exhibit relatively high activities of GSH synthesis and other GSH-dependent metabolic reactions with the exception of GGT. As is typical with immortalized epithelial cell lines, the brush-border membrane is largely lost so that activities of enzymes normally found there will be low. For our purposes, this is an advantage as it largely removes the possibly confounding effect of degradation during measurement of transport.
Three clones that transiently overexpressed the DIC exhibited 3- to 11-fold increases (mean = 5.5-fold) in rates of GSH uptake into mitochondria (Fig. 5A). Similarly, NRK-52E cells that stably expressed the OGC exhibited 6.1-fold increases in mitochondrial GSH uptake as compared to the wild-type cells. Mutation of the two Cys residues of the OGC involved in an intramolecular disulfide bond to Ser (OGC-C221,224S) resulted in a modified carrier protein that exhibited a marked reduction in GSH transport so that mitochondrial accumulation of GSH in these cells was actually slightly less than that in non-transfected, wild-type cells.
Fig. 5.
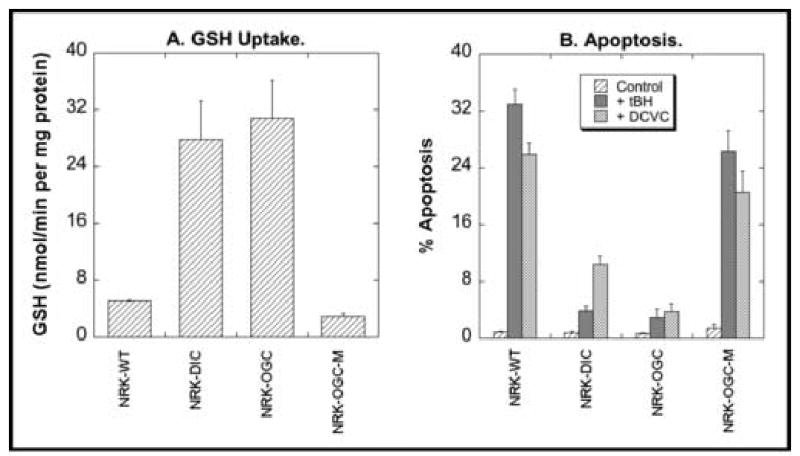
Genetic modulation of mitochondrial GSH transport in NRK-52E cells and susceptibility to chemically induced apoptosis. The cDNA for the DIC was transiently overexpressed in NRK-52E cells (NRK-DIC) and the cDNA for either the OGC or a double-cysteine mutant of the OGC (NRK-OGC or NRK-OGC-M, respectively) was stably overexpressed in NRK-52E cells. A. Uptake rates for GSH into mitochondria from different genetically modified NRK-52E cell populations. Mitochondria from each cell population were incubated with [3H]-GSH (final concentration = 5 mM). Data are expressed as uptake rates and are means ± SEM of measurements from 3–5 separate experiments. B. Fraction of apoptotic cells. Each cell population was incubated for 4 hr with either medium (= Control), 10 μM tert-butyl hydroperoxide (tBH), or 50 μM S-(1,2-dichlorovinyl)-L-cysteine (DCVC). The fraction of cells undergoing apoptosis was estimated by propidium iodide staining, flow cytometry and FACS analysis. Results are means ± SEM of 4–5 separate experiments. These data were derived from studies originally presented in refs. 71 and 92, and were combined to illustrate toxicologic effects of genetic manipulation of mitochondrial GSH carriers.
To investigate the toxicological consequences of altering mitochondrial GSH transport capability, we incubated the different populations of NRK-52E cells with either of two well-characterized mitochondrial toxicants, tert-butyl hydroperoxide (tBH) or S-(1,2-dichlorovinyl)-L-cysteine (DCVC), and determined the fraction of cells undergoing apoptosis (Fig. 5B). Dramatic differences were observed in the sensitivity of the different cell populations to the two agents, with the cells overexpressing either the DIC or OGC exhibiting resistance to cell injury. In contrast, cells overexpressing the double-cysteine mutant of the OGC exhibited similar sensitivity to tBH or DCVC as the wild-type cells. These results support the use of approaches to enhance mitochondrial GSH to protect cells from oxidants and other cytotoxic chemicals. The absence of protection with cells overexpressing the double-cysteine mutant of the OGC suggests that a threshold level of mitochondrial GSH exists for optimal mitochondrial function and resistance to chemically induced toxicity.
6. Summary and conclusions
Studies using a variety of experimental models, including isolated mitochondria, purified and reconstituted carrier proteins, bacterial-expressed and reconstituted carrier proteins, and cell lines overexpressing specific carrier cDNAs, have established that transport of GSH from the cytoplasm into mitochondria in rat kidney proximal tubule is primarily mediated by the DIC and OGC. Some tissue-specific differences have been identified, so that additional carriers may be more important in some tissues. Genetic manipulation of carrier activity and expression has been demonstrated to be an effective means of producing severalfold increases in mitochondrial contents of GSH, thereby protecting cells from oxidants and other mitochondrial toxicants. The existence of several diseases that are characterized by oxidation or depletion of the mitochondrial GSH pool in specific tissues illustrates the potential human health significance of approaches that enhance mitochondrial GSH transport.
Acknowledgments
Research from the author’s laboratory was supported by NIDDK Grant R01-DK40725 and the NIEHS Center for Molecular Toxicology with Human Applicants (Grant P30-ES06639) at Wayne State University.
References
- 1.Griffith OW, Meister A. Origin and turnover of mitochondrial glutathione. Proc Natl Acad Sci USA. 1985;82:4668–4672. doi: 10.1073/pnas.82.14.4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McKernan TM, Woods EB, Lash LH. Uptake of glutathione by renal cortical mitochondria. Arch Biochem Biophys. 1991;288:653–663. doi: 10.1016/0003-9861(91)90248-h. [DOI] [PubMed] [Google Scholar]
- 3.Hinchman CA, Ballatori N. Glutathione-degrading capacities of liver and kidney in different species. Biochem Pharmacol. 1990;40:1131–1135. doi: 10.1016/0006-2952(90)90503-d. [DOI] [PubMed] [Google Scholar]
- 4.D. P. Jones, L. H. Lash. Introduction: Criteria for assessing normal and abnormal mitochondrial function, in: L.H. Lash, D.P. Jones (Eds.), Mitochondrial Dysfunction, Methods in Toxicology, vol. 2, Academic Press, San Diego, 1993, pp. 1–7.
- 5.Hunter FE, Jr, Scott A, Hoffsten PE, Gebicki JM, Weinstein J, Schneider A. Studies on the mechanism of swelling, lysis, and disintegration of isolated liver mitochondria exposed to mixtures of oxidized and reduced glutathione. J Biol Chem. 1964;239:614–621. [PubMed] [Google Scholar]
- 6.Jocelyn PC. Some properties of mitochondrial glutathione. Biochim Biophys Acta. 1975;369:427–436. doi: 10.1016/0005-2728(75)90148-6. [DOI] [PubMed] [Google Scholar]
- 7.Jocelyn PC, Dickson J. Glutathione and the mitochondrial reduction of hydroperoxides. Biochim Biophys Acta. 1980;590:1–12. doi: 10.1016/0005-2728(80)90141-3. [DOI] [PubMed] [Google Scholar]
- 8.Jocelyn PC, Kamminga A. The non-protein thiol of rat liver mitochondria. Biochim Biophys Acta. 1974;343:356–362. doi: 10.1016/0304-4165(74)90099-3. [DOI] [PubMed] [Google Scholar]
- 9.Brodie AE, Reed DJ. Glutathione disulfide reduction in tumor mitochondria after t-butyl hydroperoxide treatment. Chem Biol Interact. 1992;84:125–132. doi: 10.1016/0009-2797(92)90073-t. [DOI] [PubMed] [Google Scholar]
- 10.Maddaiah VT. Glutathione correlates with lipid peroxidation in liver mitochondria of triiodothyronine-injected hypophysectomized rats. FASEB J. 1990;4:1513–1518. doi: 10.1096/fasebj.4.5.2307329. [DOI] [PubMed] [Google Scholar]
- 11.Meredith MJ, Reed DJ. Depletion in vitro of mitochondrial glutathione in rat hepatocytes and enhancement of lipid peroxidation by adriamycin and 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU) Biochem Pharmacol. 1983;32:1383–1388. doi: 10.1016/0006-2952(83)90451-3. [DOI] [PubMed] [Google Scholar]
- 12.Olafsdottir K, Reed DJ. Retention of oxidized glutathione by isolated rat liver mitochondria during hydroperoxide treatment. Biochim Biophys Acta. 1988;964:377–382. doi: 10.1016/0304-4165(88)90038-4. [DOI] [PubMed] [Google Scholar]
- 13.Ravindranath V, Reed DJ. Glutathione depletion and formation of glutathione-protein mixed disulfide following exposure of brain mitochondria to oxidative stress. Biochem Biophys Res Commun. 1990;169:1075–1079. doi: 10.1016/0006-291x(90)92004-j. [DOI] [PubMed] [Google Scholar]
- 14.Shertzer HG, Bannenberg GL, Zhu H, Liu RM, Moldéus P. The role of thiols in mitochondrial susceptibility to iron and tert-butyl hydroperoxide-mediated toxicity in cultured mouse hepatocytes. Chem Res Toxicol. 1994;7:358–366. doi: 10.1021/tx00039a013. [DOI] [PubMed] [Google Scholar]
- 15.Sies H, Moss KM. A role of mitochondrial glutathione peroxidase in modulating mitochondrial oxidations in liver. Eur J Biochem. 1978;84:377–383. doi: 10.1111/j.1432-1033.1978.tb12178.x. [DOI] [PubMed] [Google Scholar]
- 16.Beatrice MC, Stiers DL, Pfeiffer DR. The role of glutathione in the retention of Ca2+ by liver mitochondria. J Biol Chem. 1984;259:1279–1287. [PubMed] [Google Scholar]
- 17.Lotscher HR, Winterhalter KH, Carafoli E, Richter C. Hydroperoxides can modulate the redox state of pyridine nucleotides and the calcium balance in rat liver mitochondria. Proc Natl Acad Sci USA. 1979;76:4340–4344. doi: 10.1073/pnas.76.9.4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moore GA, Jewell SA, Bellomo G, Orrenius S. On the relationship between Ca2+ efflux and membrane damage during t-butyl hydroperoxide metabolism by liver mitochondria. FEBS Lett. 1983;153:289–292. doi: 10.1016/0014-5793(83)80626-7. [DOI] [PubMed] [Google Scholar]
- 19.Olafsdottir K, Pascoe GA, Reed DJ. Mitochondrial glutathione status during Ca2+ ionophore-induced injury to isolated hepatocytes. Arch Biochem Biophys. 1988;263:226–235. doi: 10.1016/0003-9861(88)90631-5. [DOI] [PubMed] [Google Scholar]
- 20.Savage MK, Jones DP, Reed DJ. Calcium- and phosphate-dependent release and loading of glutathione by liver mitochondria. Arch Biochem Biophys. 1991;290:51–56. doi: 10.1016/0003-9861(91)90590-f. [DOI] [PubMed] [Google Scholar]
- 21.Savage MK, Reed DJ. Release of mitochondrial glutathione and calcium by a cyclosporin A-sensitive mechanism occurs without large amplitude swelling. Arch Biochem Biophys. 1994;315:142–152. doi: 10.1006/abbi.1994.1483. [DOI] [PubMed] [Google Scholar]
- 22.de la Asuncion JG, Millan A, Pla R, Bruseghini L, Esteras A, Pallardo FV, Sastre J, Vina J. Mitochondrial glutathione oxidation correlates with age-associated damage to mitochondrial DNA. FASEB J. 1996;10:333–338. doi: 10.1096/fasebj.10.2.8641567. [DOI] [PubMed] [Google Scholar]
- 23.Esteve JM, Mompo J, Garcia de la Asuncion J, Sastre J, Asensi M, Boix J, Vina JR, Pallardo FV. Oxidative damage to mitochondrial DNA and glutathione oxidation in apoptosis: studies in vivo and in vitro. FASEB J. 1999;13:1055–1064. doi: 10.1096/fasebj.13.9.1055. [DOI] [PubMed] [Google Scholar]
- 24.Chernyak BV, Bernardi P. The mitochondrial permeability transition pore is modulated by oxidative agents through both pyridine nucleotides and glutathione at two separate sites. Eur J Biochem. 1996;238:623–630. doi: 10.1111/j.1432-1033.1996.0623w.x. [DOI] [PubMed] [Google Scholar]
- 25.Costantini P, Chernyak BV, Petronilli V, Bernardi P. Modulation of the mitochondrial permeability transition pore by pyridine nucleotides and dithiol oxidation at two separate sites. J Biol Chem. 1996;271:6746–6751. doi: 10.1074/jbc.271.12.6746. [DOI] [PubMed] [Google Scholar]
- 26.Halestrap AP, Woodfield KY, Connern CP. Oxidative stress, thiol reagents, and membrane potential modulate the mitochondrial permeability transition by affecting nucleotide binding to the adenine nucleotide translocase. J Biol Chem. 1997;272:3346–3354. doi: 10.1074/jbc.272.6.3346. [DOI] [PubMed] [Google Scholar]
- 27.Masini A, Ceccarelli D, Trenti T, Gallesi D, Muscatello U. Mitochondrial inner membrane permeability changes induced by octadecadienoic acid hydroperoxide: Role of mitochondrial GSH pool. Biochim Biophys Acta. 1992;1101:84–89. doi: 10.1016/0167-4838(92)90471-o. [DOI] [PubMed] [Google Scholar]
- 28.Reed DJ, Savage MK. Influence of metabolic inhibitors on mitochondrial permeability transition and glutathione status. Biochim Biophys Acta. 1995;1271:43–50. doi: 10.1016/0925-4439(95)00008-r. [DOI] [PubMed] [Google Scholar]
- 29.Scarlett JL, Packer MA, Porteus CM, Murphy MP. Alterations to glutathione and nicotinamide nucleotides during the mitochondrial permeability transition induced by peroxynitrite. Biochem Pharmacol. 1996;52:1047–1055. doi: 10.1016/0006-2952(96)99426-5. [DOI] [PubMed] [Google Scholar]
- 30.Davis W, Jr, Ronai Z, Tew KD. Cellular thiols and reactive oxygen species in drug-induced apoptosis. J Pharmacol Exp Ther. 2001;296:1–6. [PubMed] [Google Scholar]
- 31.Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: A primary site for bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- 32.Marchetti P, Decaudin D, Macho A, Zamzami N, Hirsch T, Susin SA, Kroemer G. Redox regulation of apoptosis: Impact of thiol oxidation status on mitochondrial function. Eur J Immunol. 1997;27:289–296. doi: 10.1002/eji.1830270142. [DOI] [PubMed] [Google Scholar]
- 33.Petit PX, Susin SA, Zamzami N, Mignotte B, Kroemer G. Mitochondria and programmed cell death: Back to the future. FEBS Lett. 1996;396:7–13. doi: 10.1016/0014-5793(96)00988-x. [DOI] [PubMed] [Google Scholar]
- 34.Richter C, Schweizer M, Cossarizza A, Franceschi C. Control of apoptosis by the cellular ATP level. FEBS Lett. 1996;378:107–110. doi: 10.1016/0014-5793(95)01431-4. [DOI] [PubMed] [Google Scholar]
- 35.Skulachev VP. Why are mitochondria involved in apoptosis? Permeability transition pores and apoptosis as selective mechanisms to eliminate superoxide-producing mitochondria and cells. FEBS Lett. 1996;397:7–10. doi: 10.1016/0014-5793(96)00989-1. [DOI] [PubMed] [Google Scholar]
- 36.Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP, Wang X. Prevention of apoptosis by Bcl-2: Release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- 37.Zamzami N, Susin SA, Marchetti P, Hirsch T, Gomez-Monterrey I, Castedo M, Kroemer G. Mitochondrial control of nuclear apoptosis. J Exp Med. 1996;183:1533–1544. doi: 10.1084/jem.183.4.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Filomeni G, Rotilio G, Ciriolo MR. Cell signaling and the glutathione redox system. Biochem Pharmacol. 2002;64:1057–1064. doi: 10.1016/s0006-2952(02)01176-0. [DOI] [PubMed] [Google Scholar]
- 39.Hammond CL, Lee TK, Ballatori N. Novel roles for glutathione in gene expression, cell death, and membrane transport of organic solutes. J Hepatology. 2001;34:946–954. doi: 10.1016/s0168-8278(01)00037-x. [DOI] [PubMed] [Google Scholar]
- 40.Wong PSY, Fukuto JM. Reaction of organic nitrate esters and S-nitrosothiols with reduced flavins: A possible mechanism of bioactivation. Drug Metab Dispos. 1999;27:502–509. [PubMed] [Google Scholar]
- 41.Ji Y, Akerboom TP, Sies H, Thomas JA. S-Nitrosylation and S-glutathionylation of protein sulfhydryls by S-nitrosoglutathione. Arch Biochem Biophys. 1999;362:67–78. doi: 10.1006/abbi.1998.1013. [DOI] [PubMed] [Google Scholar]
- 42.Sandau KB, Callsen D, Brüne B. Protection against nitric oxide-induced apoptosis in rat mesangial cells demands mitogen-activated protein kinases and reduced glutathione. Mol Pharmacol. 1999;56:744–751. [PubMed] [Google Scholar]
- 43.Sarkela TM, Berthiaume J, Elfering S, Gybina AA, Giulivi C. The modulation of oxygen radical production by nitric oxide in mitochondria. J Biol Chem. 2001;276:6945–6949. doi: 10.1074/jbc.M007625200. [DOI] [PubMed] [Google Scholar]
- 44.Kurosawa K, Hayashi N, Sato N, Kamada T, Tagawa K. Transport of glutathione across the mitochondrial membranes. Biochem Biophys Res Commun. 1990;167:367–372. doi: 10.1016/0006-291x(90)91774-m. [DOI] [PubMed] [Google Scholar]
- 45.Martensson J, Lai JCK, Meister A. High-affinity transport of glutathione is part of a multicomponent system essential for mitochondrial function. Proc Natl Acad Sci USA. 1990;87:7185–7189. doi: 10.1073/pnas.87.18.7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Garcia-Ruiz C, Morales A, Cole A, Rodes J, Kaplowitz N, Fernandez-Checa JC. Evidence that the rat hepatic mitochondrial carrier is distinct from the sinusoidal and canalicular transporters for reduced glutathione. J Biol Chem. 1995;270:15946–15949. doi: 10.1074/jbc.270.27.15946. [DOI] [PubMed] [Google Scholar]
- 47.Schnellmann RG. Renal mitochondrial glutathione transport. Life Sci. 1991;49:393–398. doi: 10.1016/0024-3205(91)90447-j. [DOI] [PubMed] [Google Scholar]
- 48.Klingenberg M. Overview on mitochondrial metabolite transport systems. Methods Enzymol. 1979;56:245–252. doi: 10.1016/0076-6879(79)56027-3. [DOI] [PubMed] [Google Scholar]
- 49.Palmieri F. The mitochondrial transporter family (SLC25): physiological and pathological implications. Pflügers Arch. 2004;447:689–709. doi: 10.1007/s00424-003-1099-7. [DOI] [PubMed] [Google Scholar]
- 50.Palmieri F, Bisaccia F, Capobianco L, Dolce V, Fiermonte G, Iacobazzi V, Indiveri C, Palmieri L. Mitochondrial metabolite transporters. Biochim Biophys Acta. 1996;1275:127–132. doi: 10.1016/0005-2728(96)00062-x. [DOI] [PubMed] [Google Scholar]
- 51.L. H. Lash, J.M. Sall. Mitochondrial isolation from liver and kidney: Strategy, techniques, and criteria for purity, in: L.H. Lash, D.P. Jones (Eds.), Mitochondrial Dysfunction, Methods in Toxicology, vol. 2, Academic Press, San Diego, 1993, pp. 8–28.
- 52.Lash LH. Intracellular distribution of thiols and disulfides: Assay of mitochondrial GSH transport. Methods Enzymol. 1995;252:14–26. doi: 10.1016/0076-6879(95)52004-x. [DOI] [PubMed] [Google Scholar]
- 53.Chen Z, Lash LH. Evidence for mitochondrial uptake of glutathione by dicarboxylate and 2-oxoglutarate carriers. J Pharmacol Exp Ther. 1998;285:608–618. [PubMed] [Google Scholar]
- 54.Griner RD, Aleo MD, Schnellmann RG. The role of short chain fatty acid substrates in aerobic and glycolytic metabolism in primary cultures of renal proximal tubule cells. In Vitro Cell Dev Biol. 1993;29A:649–655. doi: 10.1007/BF02634554. [DOI] [PubMed] [Google Scholar]
- 55.Griner RD, Schnellmann RG. Decreasing glycolysis increases sensitivity to mitochondrial inhibition in primary cultures of renal proximal tubule cells. In Vitro Cell Dev Biol. 1994;30A:30–34. doi: 10.1007/BF02631415. [DOI] [PubMed] [Google Scholar]
- 56.Lash LH. Role of glutathione transport processes in kidney function. Toxicol Appl Pharmacol. 2005;204:329–342. doi: 10.1016/j.taap.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 57.Chen Z, Putt DA, Lash LH. Enrichment and functional reconstitution of glutathione transport activity from rabbit kidney mitochondria: Further evidence for the role of the dicarboxylate and 2-oxoglutarate carriers in mitochondrial glutathione transport. Arch Biochem Biophys. 2000;373:193–202. doi: 10.1006/abbi.1999.1527. [DOI] [PubMed] [Google Scholar]
- 58.Kaplan RS, Pedersen PL. Isolation and reconstitution of the n-butylmalonate-sensitive dicarboxylate transporter from rat liver mitochondria. J Biol Chem. 1985;260:10293–10298. [PubMed] [Google Scholar]
- 59.Bisaccia F, Indiveri C, Palmieri F. Purification of reconstitutively active α-oxoglutarate carrier from pig heart mitochondria. Biochim Biophys Acta. 1985;810:362–369. doi: 10.1016/0005-2728(85)90222-1. [DOI] [PubMed] [Google Scholar]
- 60.Bisaccia F, Indiveri C, Palmieri F. Purification and reconstitution of two anion carriers from rat liver mitochondria: the dicarboxylate and the 2-oxoglutarate carrier. Biochim Biophys Acta. 1988;933:229–240. doi: 10.1016/0005-2728(88)90030-8. [DOI] [PubMed] [Google Scholar]
- 61.Lançar-Benba J, Foucher B, Saint-Macary M. Purification of the rat-liver mitochondrial dicarboxylate carrier by affinity chromatography on immobilized malate dehydrogenase. Biochim Biophys Acta. 1994;1190:213–216. doi: 10.1016/0005-2736(94)90076-0. [DOI] [PubMed] [Google Scholar]
- 62.Coll O, Colell A, Garcia-Ruiz C, Kaplowitz N, Fernandez-Checa JC. Sensitivity of the 2-oxoglutarate carrier to alcohol intake contributes to mitochondrial glutathione depletion. Hepatology. 2003;38:692–702. doi: 10.1053/jhep.2003.50351. [DOI] [PubMed] [Google Scholar]
- 63.Fernandez-Checa JC, Kaplowitz N. Hepatic mitochondrial glutathione: transport and role in disease and toxicity. Toxicol Appl Pharmacol. 2005;204:263–273. doi: 10.1016/j.taap.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 64.Hofmann K, Stoffel W. TMbase – A database of membrane-spanning proteins segments. Biol Chem Hoppe-Seyler. 1993;347:166. [Google Scholar]
- 65.Stipani I, Mangiullo G, Stipani V, Daddabbo L, Natuzzi D, Palmieri F. Inhibition of the reconstituted mitochondrial oxoglutarate carrier by arginine-specific reagents. Arch Biochem Biophys. 1996;331:48–54. doi: 10.1006/abbi.1996.0281. [DOI] [PubMed] [Google Scholar]
- 66.Stipani V, Cappello AR, Daddabbo L, Natuzzi D, Miniero DV, Stipani I, Palmieri F. The mitochondrial oxoglutarate carrier: cysteine-scanning mutagenesis of transmembrane domain IV and sensitivity of Cys mutants to sulphydryl reagents. Biochemistry. 2001;40:15805–15810. doi: 10.1021/bi011616j. [DOI] [PubMed] [Google Scholar]
- 67.Morozzo della Rocca B, Lauria G, Venerini F, Palmieri L, Polizio F, Capobianco L, Stipani V, Pedersen J, Cappello AR, Desideri A, Palmieri F. The mitochondrial oxoglutarate carrier: structural and dynamic properties of transmembrane segment IV studied by site-directed spin labeling. Biochemistry. 2003;42:5493–5499. doi: 10.1021/bi027025q. [DOI] [PubMed] [Google Scholar]
- 68.Capobianco L, Bisaccia F, Mazzeo M, Palmieri F. The mitochondrial oxoglutarate carrier: Sulfhydryl reagents bind to cysteine-184, and this interaction is enhanced by substrate binding. Biochemistry. 1996;35:8974–8980. doi: 10.1021/bi960258v. [DOI] [PubMed] [Google Scholar]
- 69.Bisaccia F, Capobianco L, Mazzeo M, Palmieri F. The mitochondrial oxoglutarate carrier protein contains a disulfide bridge between intramembranous cysteines 221 and 224. FEBS Lett. 1996;392:54–58. doi: 10.1016/0014-5793(96)00784-3. [DOI] [PubMed] [Google Scholar]
- 70.Bisaccia F, Zara V, Capobianco L, Iacobazzi V, Mazzeo M, Palmieri F. The formation of a disulfide cross-link between the two subunits demonstrates the dimeric structure of the mitochondrial oxoglutarate carrier. Biochim Biophys Acta. 1996;1292:281–288. doi: 10.1016/0167-4838(95)00215-4. [DOI] [PubMed] [Google Scholar]
- 71.Xu F, Putt DA, Matherly LH, Lash LH. Modulation of expression of rat mitochondrial 2-oxoglutarate carrier in NRK-52E cells alters mitochondrial transport and accumulation of glutathione and susceptibility to chemically induced apoptosis. J Pharmacol Exp Ther. 2006;316:1175–1186. doi: 10.1124/jpet.105.094599. [DOI] [PubMed] [Google Scholar]
- 72.Huizing M, Ruitenbeek W, van den Heuvel LP, Dolce V, Iacobazzi V, Smeitink JAM, Palmieri F, Trijbels JMF. Human mitochondrial transmembrane metabolite carriers: Tissue distribution and its implication for mitochondrial disorders. J Bioenerg Biomembr. 1998;30:277–284. doi: 10.1023/a:1020501021222. [DOI] [PubMed] [Google Scholar]
- 73.Fiermonte G, Dolce V, Palmieri L, Ventura M, Runswick MJ, Palmieri F, Walker JE. Identification of the human mitochondrial oxodicarboxylate carrier: Bacterial expression, reconstitution, functional characterization, tissue distribution, and chromosomal location. J Biol Chem. 2001;276:8225–8230. doi: 10.1074/jbc.M009607200. [DOI] [PubMed] [Google Scholar]
- 74.Garcia-Ruiz C, Colell A, Morales A, Kaplowitz N, Fernandez-Checa JC. Role of oxidative stress generated from the mitochondrial electron transport chain and mitochondrial glutathione status in loss of mitochondrial function and activation of transcription factor nuclear factor-kappa B: studies with isolated mitochondria and rat hepatocytes. Mol Pharmacol. 1995;48:825–834. [PubMed] [Google Scholar]
- 75.Han D, Canali R, Rettori D, Kaplowitz N. Effect of glutathione depletion on sites and topology of superoxide and hydrogen peroxide production in mitochondria. Mol Pharmacol. 2003;64:1136–1144. doi: 10.1124/mol.64.5.1136. [DOI] [PubMed] [Google Scholar]
- 76.Garcia-Ruiz C, Morales A, Ballesta A, Rodes J, Kaplowitz N, Fernandez-Checa JC. Effect of chronic ethanol feeding on glutathione and functional integrity of mitochondria in periportal and perivenous rat hepatocytes. J Clin Invest. 1994;94:193–201. doi: 10.1172/JCI117306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fernandez-Checa JC, Ookhtens M, Kaplowitz N. Effect of chronic ethanol feeding on rat hepatocyte glutathione. Compartmentation, efflux, and response to incubation with ethanol. J Clin Invest. 1987;80:57–62. doi: 10.1172/JCI113063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fernandez-Checa JC, Garcia-Ruiz C, Ookhtens M, Kaplowitz N. Impaired uptake of glutathione by hepatic mitochondria from chronic ethanol-fed rats. Tracer kinetic studies in vitro and in vivo and susceptibility to oxidant stress. J Clin Invest. 1991;87:397–405. doi: 10.1172/JCI115010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fernandez-Checa JC, Hirano T, Tsukamoto H, Kaplowitz N. Mitochondrial glutathione depletion in alcoholic liver disease. Alcohol. 1993;10:469–475. doi: 10.1016/0741-8329(93)90067-x. [DOI] [PubMed] [Google Scholar]
- 80.Fernandez-Checa JC, Colell A, Garcia-Ruiz C. S-Adenosyl-l-methionine and mitochondrial reduced glutathione depletion in alcoholic liver disease. Alcohol. 2002;27:179–183. doi: 10.1016/s0741-8329(02)00229-x. [DOI] [PubMed] [Google Scholar]
- 81.Garcia-Ruiz C, Morales A, Colell A, Ballesta A, Rodés J, Kaplowitz N, Fernandez-Checa JC. Feeding S-adenosyl-L-methionine attenuates both ethanol-induced depletion of mitochondrial glutathione and mitochondrial dysfunction in periportal and perivenous rat hepatocytes. Hepatology. 1995;21:207–214. doi: 10.1002/hep.1840210133. [DOI] [PubMed] [Google Scholar]
- 82.Zhao P, Slattery JT. Effects of ethanol dose and ethanol withdrawal on rat liver mitochondrial glutathione: Implication of potentiated acetaminophen toxicity in alcoholics. Drug Metab Dispos. 2002;30:1413–1417. doi: 10.1124/dmd.30.12.1413. [DOI] [PubMed] [Google Scholar]
- 83.Zhao P, Kahorn TF, Slattery JT. Selective mitochondrial glutathione depletion by ethanol enhances acetaminophen toxicity in rat liver. Hepatology. 2002;36:326–335. doi: 10.1053/jhep.2002.34943. [DOI] [PubMed] [Google Scholar]
- 84.Colell A, Garcia-Ruiz C, Miranda M, Ardite E, Mari M, Morales A, Corrales F, Kaplowitz N, Fernandez-Checa JC. Selective glutathione depletion of mitochondria by ethanol sensitizes hepatocytes to tumor necrosis factor. Gastroenterology. 1998;115:1541–1551. doi: 10.1016/s0016-5085(98)70034-4. [DOI] [PubMed] [Google Scholar]
- 85.Pastorino JG, Hoek JW. Ethanol potentiates tumor necrosis factor cytotoxicity in hepatoma cells and primary rat hepatocytes by promoting induction of the mitochondrial permeability transition. Hepatology. 2000;31:1141–1152. doi: 10.1053/he.2000.7013. [DOI] [PubMed] [Google Scholar]
- 86.Minagawa M, Deng Q, Liu ZX, Tsukamoto H, Dennert G. Activated natural killer T cells induce liver injury by Fas and tumor necrosis factor-alpha during alcohol consumption. Gastroenterology. 2004;126:1387–1399. doi: 10.1053/j.gastro.2004.01.022. [DOI] [PubMed] [Google Scholar]
- 87.Krahenbuhl S, Stucki J, Reichen J. Reduced activity of the electron transport chain in liver mitochondria isolated from rats with secondary biliary cirrhosis. Hepatology. 1992;15:1160–1166. doi: 10.1002/hep.1840150630. [DOI] [PubMed] [Google Scholar]
- 88.Krahenbuhl S, Talos C, Lauterburg BH, Reichen J. Reduced antioxidative capacity in liver mitochondria from bile duct ligated rats. Hepatology. 1995;22:607–612. doi: 10.1002/hep.1840220234. [DOI] [PubMed] [Google Scholar]
- 89.Santos DL, Palmeira CM, Seiça R, Dias J, Mesquita J, Moreno AJ, Santos MS. Diabetes and mitochondrial oxidative stress: A study using heart mitochondria from the diabetic Goto-Kakizaki rat. Mol Cell Biochem. 2003;246:163–170. [PubMed] [Google Scholar]
- 90.Mastrocola R, Restivo F, Vercellinatto I, Danni O, Brignardello E, Aragno M, Boccuzzi G. Oxidative and nitrosative stress in brain mitochondria of diabetic rats. J Endocrinol. 2005;187:37–44. doi: 10.1677/joe.1.06269. [DOI] [PubMed] [Google Scholar]
- 91.Rosca MG, Mustata TG, Kinter MT, Ozdemir AM, Kern TS, Szweda LI, Brownlee M, Monnier VM, Weiss MF. Glycation of mitochondrial proteins from diabetic rat kidney is associated with excess superoxide formation. Am J Physiol. 2005;289:F420–F430. doi: 10.1152/ajprenal.00415.2004. [DOI] [PubMed] [Google Scholar]
- 92.Lash LH, Putt DA, Matherly LH. Protection of NRK-52E cells, a rat renal proximal tubular cell line, from chemical-induced apoptosis by overexpression of a mitochondrial glutathione transporter. J Pharmacol Exp Ther. 2002;303:476–486. doi: 10.1124/jpet.102.040220. [DOI] [PubMed] [Google Scholar]
- 93.Lash LH, Putt DA, Hueni SE, Cao W, Xu F, Kulidjian SJ, Horwitz JP. Cellular energetics and glutathione status in NRK-52E cells: toxicological implications. Biochem Pharmacol. 2002;64:1533–1546. doi: 10.1016/s0006-2952(02)01360-6. [DOI] [PubMed] [Google Scholar]