

Abstract
The charge density distribution of a protein has been refined experimentally. Diffraction data for a crambin crystal were measured to ultra-high resolution (0.54 Å) at low temperature by using short-wavelength synchrotron radiation. The crystal structure was refined with a model for charged, nonspherical, multipolar atoms to accurately describe the molecular electron density distribution. The refined parameters agree within 25% with our transferable electron density library derived from accurate single crystal diffraction analyses of several amino acids and small peptides. The resulting electron density maps of redistributed valence electrons (deformation maps) compare quantitatively well with a high-level quantum mechanical calculation performed on a monopeptide. This study provides validation for experimentally derived parameters and a window into charge density analysis of biological macromolecules.
The electronic charge density distribution of a molecule carries information (1) that determines its intermolecular interactions. For example, the charge distribution of an enzyme complements that of the substrate it recognizes and binds. The electrostatic potential and electric moments derivable from the charge density (1–3) provide maps that can guide the design of molecules for specified interactions. Furthermore, powerful insights into the nature and strength of hydrogen bonding and ionic interactions result from analysis of the electron density gradient and Laplacian (4–6). Extension of such analyses to proteins would permit a unique understanding of the driving forces between biomolecules as well as the subtleties of enzymatic reactions (7).
Experimental electron density distributions are obtained by analysis of single-crystal x-ray diffraction data measured to ultra-high resolution, typically to a diffraction resolution limit dmin ≈ 0.5 Å (1, 8, 9). The crystallographic studies usually map and analyze the deformation density, which is the difference between the actual electron density of the molecule and the density calculated for the promolecule, a molecular superposition of spherical, neutral, i.e., free, atoms. The deformation density thus reveals the redistribution of valence electron density caused by chemical bonding and intermolecular interactions and also is used to calibrate theoretical electron density calculations (10). However, a difficulty in crystallography is the separation of the anisotropic atomic mean-square displacements from the static molecular electron distribution (11). Proper experimental deconvolution requires very accurate diffraction data to ultra-high resolution. Thus, charge density studies have so far been limited to small-unit-cell crystals, and proteins still await study.
We have shown (12, 13) that effective thermal displacement deconvolution and meaningful deformation density distributions can be achieved for larger structures at lower resolution by transferring average experimental electron density parameters. We have built a database of such parameters derived from ultra-high resolution crystal structures of amino acids and small peptides that are transferable to polypeptides and small proteins. The limits of the transferability have been analyzed in a study of a helical octapeptide with diffraction data to dmin = 0.82-Å resolution (13). The deformation density showed well-defined bond peaks between atoms with small to moderate displacement parameters (Beq < 4 Å2) when the Fourier synthesis was performed with diffraction data complete to at least 0.9-Å resolution. The analysis also yielded partial atomic charges that compare well with the charges in the amber (14) molecular modeling dictionary.
Here we report on the crystallographic charge density refinement of a protein, crambin (46 residues), which is present in seeds of Crambe abyssinica and homologous to membrane-active plant toxins (15). The structure of the protein (Fig. 1) at a resolution of 0.83 Å already has been analyzed extensively to determine deviations from peptide backbone geometry, crystallographic water structure, and disorder correlations (17–19).
Figure 1.

Ribbon diagram (16) showing the general fold of crambin. The disulfide bridges are shown in yellow. β-sheet and extended chain are shown in green and the helices are red.
Materials and Methods
Data Collection.
Crambin was purified and crystallized as described (17). The crystals belong to the space group P21 with unit cell parameters a = 40.82; b = 18.49; c = 22.37 Å; β = 90.47°, and one molecule per asymmetric unit. X-ray diffraction data from a single crambin crystal at T = 100 K were measured to dmin = 0.54 Å by using λ = 0.54 Å synchrotron radiation and a 300-mm MarResearch imaging plate detector at the European Molecular Biology Laboratory BW7A beamline at the DORIS storage ring, Deutsches Elektronen Synchrotron, Hamburg. Using the denzo (20) and drear (21, 22) program suites, the data were reduced to a 97.6% complete data set (Table 1). The structure was refined against all of the reflections, except 5% of them used to compute the free-R crystallographic residual (23).
Table 1.
X-ray diffraction data statistics
| Number of measured reflections | 489,969 |
| Number of independent reflections | 112,293 |
| Number of reflections with I > 3σI | 79,868 |
| Rmerge(I)* (%) | 5.5 |
| Rw(F) and Rwfree† (%) | |
| Spherical atom model | 9.2 9.6 |
| Multipolar atom model | 9.0 9.4 |
Rmerge(I) = Σ (n/(n − 1))1/2|I − 〈I〉|/Σ I, where n is the number of equivalent reflections measured.
† Rw(F) = [Σ w⋅(|Fobs| − |Fcalc|)2/Σ w⋅|Fobs|2]1/2, where w = 1/σ2 (Fobs).
Electron Density Modeling.
The electron density analysis used a simplified Hansen and Coppens (24) multipolar pseudoatom model,
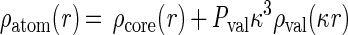 |
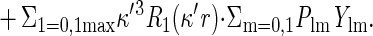 |
The first two terms on the right describe spherically symmetric core-plus-valence density, and the third term describes nonspherical, multipolar redistribution of valence-shell density caused by chemical bonding. The valence-shell electron populations Pval adjust for inter-atomic charge transfer, and the multipole populations Plm for aspherical intra-atomic redistribution of valence electron density. The Rl are Slater-type radial functions, and the Ylm± are real spherical harmonic angular functions (see ref. 24 for more details). The dilatation coefficients of the spherical (κ) and multipolar (κ′) valence electron density were not considered as variables and were set to the standard value of unity (κ = 1.16 for hydrogen atoms).
The present analysis was performed by using the mopro least-squares computer program (C.J. and C.L., unpublished work) for multipolar charged atom modeling, which is a version of molly (24) extensively modified for protein applications. The conjugate gradient method has been implemented, and stereochemical restraints, analogous to those of the shelxl-97 structure refinement program (25) have been introduced. Distance restraints were applied in the disordered parts of the structure. To facilitate the deconvolution of the deformation density from vibrational smearing, rigid bond (11) restraints were applied to the thermal displacement parameters. All of the hydrogen atoms in the ordered part of crambin were located in Fourier difference maps. For nonhydrogen atoms, the multipole expansions included the monopolar, dipolar, quadrupolar, and octupolar terms that have significantly nonzero Plm values in the amino acid and peptide database (12, 13). For hydrogen atoms, only the Pval monopole and a dipole directed along the H-X covalent bond were included.
Crystallographic Refinement.
The electron density modeling was done in three stages. In refinement I, a conventional, spherical, neutral atom model was used. Atomic positions, anisotropic displacement parameters, and site occupancies for solvent molecules and disordered protein atoms were refined by using the programs shelxl-97 (25) and mopro successively.
In refinement II, the multipolar charged atom model was applied with electron density parameters (valence population and multipoles) transferred from the database (12, 13) to the protein polypeptide main chain. For the side chains, only the multipole parameters were transferred, as the valence populations Pval show a lower degree of transferability in the database. Water molecules were considered spherical and neutral. The main-chain periodic moiety -HαCαCONH- was set to be globally electrically neutral; as the protein contains a variety of side chains, their global effect on the polypeptide main-chain charge can be expected to cancel out.
The hydrogen atoms were moved outward along the X-H bond directions to adjust the bond lengths to the values expected from neutron diffraction studies (26). The atomic positions and anisotropic thermal displacement parameters were refined (alternatively) further with fixed multipole and partial net charge parameters transferred from the database. To ensure that the structure “forgot” the spherical atom model, the temperature factors were annealed twice from random shifts of up to ± 10%. As the convergence was slow, at least 400 refinement cycles were necessary.
In refinement III, the average electron density parameters for the polypeptide main chain were allowed to vary. All of the peptide groups -Ca(-Ha)-C(=O)-N(-HN)- were constrained to be electrically neutral and equivalent. The disordered portions, accounting for 30% of the protein atoms, were modeled with fixed electron density parameters that were updated regularly during refinement III. The coordinates, the thermal displacement parameters, and the charge density parameters were refined alternatively.
Charge Refinement.
Diffraction-derived atomic charges are given by qatom = Nvalatom − Pvalatom, where Nval is the number of valence electrons in the neutral free atom and Pval is the refined valence-shell population in the bound pseudoatom. Charges derived from the Pval values might, however, be biased by effective charge transfer through the multipoles. To compensate for this bias, the Pval parameters were refitted by using a spherical atom model. In this procedure (27), only the valence population and dilatation coefficient were refined while the multipole population parameters were reset to and held fixed at Plm = 0, and the atomic positional and thermal parameters were held fixed at their refined values from refinement II or III.
Electron Density Maps.
Residual maps were computed as Fourier transforms of the structure factor differences (|Fobs| − |Fcalc|) exp (iφcalc).
The experimental static deformation density was computed from the crystallographic modeling as the atomic superposition-sum over the molecule Δρ = Σatom ρatommultipolar − ρatomspherical. This density is “static” in that it is computed for atoms at rest.
A theoretical static deformation density Δρ = ρmolecule − Σatom ρfree-atom had been computed from a wavefunction for a pseudo-monopeptide (5) from an ab initio Hartree-Fock self-consistent-field molecular orbitals calculation performed with a triplezeta-plus-(C,N,O-d and H-p)-polarization-function Gaussian basis set.
The dynamic experimental deformation electron density was obtained by Fourier transformation of the difference |Fobs|exp(iΦcalcmultipolar) − |Fcalcspherical|exp(iΦcalcspherical). This density is “dynamic” in that both the structure factor amplitudes and phases are affected by atomic thermal vibrational smearing. It is “experimental” in that the Fourier sum is truncated at the experimental diffraction resolution limit, and the |Fobs| coefficients incorporate experimental error.
Results
Residual Maps.
An important question is whether significant deviations from the spherical-atom approximation can be experimentally observed for proteins. To address this issue, the crambin structure was first refined classically, by using a spherical, neutral atom model (refinement I). The quality of the fit to this spherical scattering factor model was evaluated by residual electron density maps. These maps show systematic bonding density features (Fig. 2A), but also contained a significant amount of random noise, which was not in favor of a charge density refinement for individual atoms.
Figure 2.

Residual electron density in the peptide bond plane. (A) For the peptide Ala-9–Arg-10 when using a spherical neutral atom model, contour level 0.05 e−/Å3. (B) Averaged over the 34 nondisordered peptides in crambin when using a spherical neutral atom model. (C) When using a multipolar charged atom model transferred from the database and (D) with average valence populations and multipoles refined, contour level 0.02 e−/Å3. Positive: red lines; negative: blue lines.
Taking advantage of the repetition of the same chemical motif along the polypeptide main chain, the signal to noise ratio of the crambin residual map was increased by averaging over the 34 nondisordered peptide groups (Fig. 2B). The averaged map displayed significant positive residual density in the bonds between the nonhydrogen atoms (with a maximum Δρ = 0.12 e−/Å3 in the Cα-C bond), and negative residual density was found in the Ha and HN hydrogen regions, indicating unmodeled electron depletion on the hydrogen atoms. These features in the residual map clearly demonstrate that the spherical-atom model does not provide an adequate fit to the experimental diffraction data (13, 28).
After transfer of the statistically significant multipoles from the database (Table 2), the residual features were greatly reduced in the peptide-averaged residual electron density (Fig. 2C). In fact, residual bond densities were even negative, indicating that the deformation density transferred from the database (12) needs further adjustments (V.P.-P., C.J., C.L., and B. Guillot, unpublished work).
Table 2.
Average valence and multipole populations for the atoms of the polypeptide main chain
| Atom | Local O-x axis ∖y | Valence population | Dipoles | Quadruples | Octupoles | ||
|---|---|---|---|---|---|---|---|
| Cα | Cα-C | Pval | Oxz2 | Oyz2 | Ox3 | Oz3 | |
| ∖N | 4.04 (7) | −0.177 (16) | −0.276 (25) | 0.269 (32) | 0.069 (12) | ||
| 3.96 (4) | −0.092 (11) | −0.246 (11) | 0.184 (10) | 0.049 (10) | |||
| C | C-O | Dx | Qz2 | Qx2-y2 | Ox3 | ||
| ∖N | 3.98 (4) | 0.106 (10) | −0.308 (15) | 0.107 (14) | 0.424 (19) | ||
| 4.32 (4) | 0.068 (9) | −0.268 (11) | 0.093 (10) | 0.342 (11) | |||
| O | O-C | Dx | Qz2 | Qx2-y2 | |||
| ∖N | 6.29 (2) | −0.076 (10) | −0.074 (7) | −0.073(7) | |||
| 5.96 (3) | Fixed | Fixed | Fixed | ||||
| N | N-C | Dy | Qz2 | Ox3 | |||
| ∖Cα | 5.31 (3) | −0.077 (12) | −0.0522 (13) | 0.271 (9) | |||
| 4.95 (4) | −0.011 (10) | 0.030 (10) | 0.150 (8) | ||||
| HN | HN-N | Dx | |||||
| ∖C | 0.61 (3) | 0.177 (16) | |||||
| 0.88 (2) | 0.229 (12) | ||||||
| Hα | Hα-Cα | Dx | |||||
| ∖N | 0.77 (2) | 0.151 (11) | |||||
| 0.89 (2) | 0.194 (11) | ||||||
The upper value is the average charge density parameter in the database, with the SD in the sample given in parenthesis. The lower value and its uncertainty is obtained from the crambin multipole refinement. In the local axis system of an atom, the Ox direction is defined by the first two atoms, and the Oxy plane is defined by the third atom.
The well-defined peaks on the covalent bonds as well as the low noise level in the averaged residual maps (Fig. 2 B and C) encouraged us to obtain chemically meaningful deformation densities for the main-chain peptide moiety. The average statistically significant multipole and partial net charge parameters of the peptide main-chain atoms thus were allowed to vary in the refinement. The resultant peptide-averaged residual density map (Fig. 2D) showed substantially less negative residual density along the peptide bonds with |ΔΦ| < 0.06 e−/Å3. The progressive flattening of residual density features through the three stages of refinement (Fig. 2 B–D) is convincing physical evidence of real improvement in the modeling. The remaining global negative residual density in the peptide bond plane (Fig. 2D) may be caused by the constrains applied: first, the peptide moiety was constrained to be neutral; second, in this version of the database, only the multipoles that are significantly different from zero were considered and refined, and third, the dilatation coefficients of the valence density were fixed. Also the scale factor had decreased by 0.3% from the spherical atom model to the final refinement, which also might account for a global negative residual density.
Crystallographic Statistics.
Although the spherical and multipolar atom models led to significantly different (averaged) residual densities, they yielded only slightly different values (Table 1) of their crystallographic amplitude agreement indices and their phase agreement statistics from free-R likelihood estimates computed with the rflexpl program (29). The unweighted average phase error is 6.3° and 6.0° when using the spherical and multipolar atom model, respectively. The effect of the atomic model on the calculated phase also has been analyzed. The average phase difference between the spherical and multipolar refinements is <|ΔΦ|> = 3.8°, which is smaller than the estimated phase error itself. This difference is presumably small because the anisotropic thermal displacement parameters in a spherical atoms model can accommodate much of the valence density deformation (13, 30) that should be properly described by a multipolar model. The importance of model testing against residual maps also must be emphasized.
Electron Density Parameters.
The charge density refinement of the average peptide led to a realistic static deformation density, except for the peptide oxygen atom lone pairs. The charge density parameters of the oxygen atom therefore were readjusted to their database values. The oxygen lone pair density had indeed showed up less clearly than the bonding deformation density in Fig. 2B. Proper refinement of the charge density parameters describing the oxygen lone pairs would require even higher-resolution diffraction data and a lower thermal motion as these features are only 0.3 Å distant from the oxygen nucleus.
As expected from the residual density in Fig. 2C, the multipole populations on the peptide nonhydrogen atoms generally decreased from their database values in refinement III (Table 2), whereas for the peptide hydrogen atoms Ha and HN the dipole populations increased by about 25%. This electron transfer in the H→X direction was compensated by a corresponding increase of the electronic populations Pval on the hydrogen atoms (Table 2).
Deformation Maps.
The static deformation density maps after transfer and refinement of the statistically significant multipoles (refinements II and III) are shown in Fig. 3 A and B. The two densities display a correlation coefficient of 0.89 in the peptide bond plane displayed in Fig. 3. As expected from Fig. 2C, there is a decrease of the rms deformation density from 0.24 to 0.18 e−/Å3 in the peptide bond plane. The database deformation density is clearly overestimated, as the rms value computed on a sample of 13 experimental deformation maps of peptide bonds present in the database is 0.194 ± 0.005. A preliminary analysis of the current version of the database seems to indicate that the overestimation of the deformation density is caused by the dilatation coefficients κ and κ′, which were fixed to unity for the non-H atoms. When average contraction/expansion coefficients are used, the database deformation density has a rms value of 0.18, and the correlation coefficient with the crambin-derived map reaches 92%.
Figure 3.

Deformation map in peptide bond plane. (A) Static map obtained from the database parameters. (B) Experimental static map obtained by using atomic charges and multipole parameters refined against the crambin diffraction data. (C) Theoretical static map computed for the pseudo-monopeptide (Z)-N-acetyl-α,β-dehydrophenylalanine methylamide (5). (D) Experimental dynamic map for the peptide Ala-9–Arg-10 of crambin. The contour level is 0.05 e−/Å3. Positive: red lines; negative: blue lines.
To further validate the results, the crambin experimental deformation density maps were compared with corresponding theoretical maps obtained from quantum mechanical calculations on the monopeptide (Z)-N-acetyl-α, β-dehydrophenylalanine methylamide (5) (Fig. 3C). The bond peak heights display an almost quantitative agreement in the two static maps (Fig. 3 B and C). The experimental and theoretical deformation densities have rms values of 0.18 and 0.20 e−/Å3, respectively and give a correlation coefficient of 0.88 (if the regions within 0.15-Å radius around the atomic nuclei are ignored). The excellent consistency between the experimental results of this charge density analysis of a protein and theoretical results for a high-level calculation on a small peptide is remarkable.
According to the rms deformation densities, the 6–311G++ calculations on the monopeptide do not agree with the database transferred map, which again seems overestimated. The theoretical map has a rms deformation density (0.20) that is in good agreement with the experimental values found on 13 peptide bonds (0.194 ± 0.005) present in the database; it is, however, slightly higher than the crambin refined value (0.18).
The slightly lower deformation density features of the crambin refined map might be caused by the less than small molecule quality of the diffraction data and by the higher thermal motion. This attenuation effect has been observed even more dramatically in the refinement of atomic charges (see next paragraph).
A usual X-X refinement (24), where the deformation density is in principle deconvoluted from the thermal motion, was not attempted, as the electron density residual in Fig. 2B was weaker than what is usually observed for small molecules. Also, such a refinement is possible only for atoms with a sufficiently low thermal motion that have thus a contribution to the diffraction that extends to the subatomic resolution level (0.5 Å). The charge density parameters refinement turned out to have a slow convergence; it was thus performed only after the database transfer, which is a point close to the real structure and therefore yields the best coordinates and thermal displacement parameters (12).
To see the charge density on individual atoms, the (unaveraged) dynamic deformation map after refinement III has been computed. This density incorporates the thermal motion of the atoms and the errors of the diffraction data. The dynamic deformation density is shown in Fig. 3D for the peptide bond Ala-9–Arg-10. The density peaks are generally visible on the covalent bonds; however, they are strongly affected by the vibrationnal smearing and presumably also by the noise in the diffraction data. As expected, lone-pair peaks on the oxygen atom are attenuated because of thermal-motion smearing.
Atomic Charges Refinement.
An important longer-range aim of our studies is to determine experimental electrostatic parameters for biological macromolecules and to calibrate the theoretically derived electrostatic parameters used in biomolecular modeling calculations (31). Net atomic charges can, for instance, be derived experimentally from x-ray diffraction data via pseudoatom modeling (3, 27, 31). In analyses of small-molecule structures, spherical-atom charges refitted after multipolar refinements have been shown to yield molecular electric moments that agree well with independently measured experimental values (3, 27).
The refitted charges of the crambin peptide atoms after refinement II were: Ca +0.06, C +0.32, O −0.18, N −0.44, Ha +0.08, and HN +0.16 electrons. These are charges for volume-occupying spherical atoms, and they are chemically sensible, although generally smaller than the nuclear-centered effective point-charges in the amber molecular modeling dictionary (14). The refitted charges after refinement III were further attenuated to unrealistically small values, despite the apparent reasonableness of the refined bonding deformation density. Presumably the refinement of atomic charges is more sensitive to the quality of the diffraction data, the scale factor calculation, and the level of thermal motion, and it requires more accurate data to even higher resolution.
Discussion
The resolution (0.54 Å) and the quality of the crambin diffraction data presented in this study permit the refinement of the average multipole parameters for the polypeptide backbone, but not of the individual atoms as suggested by the noise level in the nonaveraged maps (Figs. 2A and 3D). With the availability of intense third-generation synchrotron sources, crystallographic data could be collected with high signal/noise ratios to even higher resolution for the current crambin crystals. This experiment might allow meaningful, nonaveraged, individual-atom charge density analysis for the inner region of the protein that has a low thermal motion. The high diffraction power of the crambin crystals with respect to protein crystallography is correlated with the low thermal displacement parameters of the protein atoms (13); the median value is only Beq = 2.5 Å2.
This moderate thermal motion may be attributed to the three disulfide bridges present in the crambin structure, the small unit cell, and the tight crystal packing with only 30% (vol/vol) solvent, nearly all of which is ordered. Low thermal vibration (Beq < 4 Å2) of the molecular structure is a prerequisite for measuring diffraction intensities to ultra-high resolution (13). The thermal motion level in crambin is, however, high compared with usual values found in small molecules crystallography (B ≈1–2 Å2).
The work described here opens the way to numerous electron density studies for proteins, because atomic-resolution protein diffraction is becoming more and more accessible and accurate (32). Depositions at the Protein Data Bank of crystal structures of small and even medium size proteins at resolutions better than 1.2 Å are becoming more frequent (33). For instance, from crystals of human aldose reductase (34), a 36-kDa protein, a diffraction data set to 0.65-Å resolution recently has been collected and the multipolar refinement is underway.
Further enhancements in protein crystal diffraction to the ultra-high resolution level will extend the number of proteins that can be studied with electron density methods. Research in the manipulation of crystal growth conditions and the use of cryo-crystallography will be crucial. These techniques can reduce the crystal mosaicity and/or lower the attenuating contribution of the atomic displacement parameters to diffraction intensities. This study shows that, at least for small proteins, the collection of diffraction data to the subatomic level has become feasible because of the remarkable technological advances in synchrotron beamline facilities and area detectors.
The determination of the charges and the electronic distribution for the atoms in the active site of enzymes will provide new information and enable a better understanding of their function (7). Another important application of charge density to structural biology would be the determination of electronic properties and oxidation states of reactive metallic centers in redox and electron transfer metalloproteins. Ultra-high resolution crystallographic studies performed in parallel on metalloproteins and biomimetic compounds (35, 36) in combination with quantum mechanical calculations will yield new insights into redox processes in biology. With the enhancement of charge density modeling, the development of the database of transferable parameters and the continued technological advances of experimental crystallography, analyzing the electronic structure of macrobiomolecules has considerable unexplored potential (8).
Acknowledgments
We thank Dr. Marie-Madeleine Rohmer (Strasbourg) for her help in the computation of Fig. 3C, the two referees for their comments on the database parameters, and the European Union for financial support for part of this work through Framework IV Biotechnology Contract BIO4-CT96–1809. R.H.B. is grateful for research support from National Institutes of Health Grant GM56829.
Footnotes
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID code 1ejg).
References
- 1.Coppens P. X-Ray Charge Densities and Chemical Bonding. Oxford: Oxford Univ. Press; 1997. pp. 233–286. [Google Scholar]
- 2.Stewart R F, Craven B M. Biophys J. 1993;65:998–1005. doi: 10.1016/S0006-3495(93)81142-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spackman M A, Byrom P G. Acta Crystallogr B. 1996;52:1023–1035. [Google Scholar]
- 4.Bader R W F. In: Atoms in Molecules: A Quantum Theory. Helpern J, Green M L H, editors. Oxford: Oxford Science; 1990. pp. 288–315. [Google Scholar]
- 5.Souhassou M, Blessing R H. J Appl Crystallogr. 1999;32:210–217. [Google Scholar]
- 6.Espinosa E, Molins E, Lecomte C. Chem Phys Lett. 1999;300:745–748. [Google Scholar]
- 7.Mesecar A D, Stoddard B L, Koshland D E. Science. 1997;277:202–206. doi: 10.1126/science.277.5323.202. [DOI] [PubMed] [Google Scholar]
- 8.Coppens P. Acta Crystallogr A. 1998;54:779–788. [Google Scholar]
- 9.Lecomte C. In: Advances in Molecular Structure Research. Hargittai I, Hargittai M, editors. Greenwich, CT: JAI Press; 1995. pp. 261–302. [Google Scholar]
- 10.Souhassou M, Lecomte C, Ghermani N E, Rohmer M M, Wiest R, Bénard M, Blessing R H. J Am Chem Soc. 1992;114:2371–2382. [Google Scholar]
- 11.Hirshfeld F L. Acta Crystallogr A. 1976;32:239–244. [Google Scholar]
- 12.Pichon-Pesme V, Lecomte C, Lachekar H. J Phys Chem. 1995;99:6242–6250. [Google Scholar]
- 13.Jelsch C, Pichon-Pesme V, Lecomte C, Aubry A. Acta Crystallogr D. 1998;54:1306–1318. doi: 10.1107/s0907444998004466. [DOI] [PubMed] [Google Scholar]
- 14.Bayly C I, Cieplak P, Cornell W D, Kollman P A. J Phys Chem. 1993;97:10269–10280. [Google Scholar]
- 15.Rao U, Stec B, Teeter M M. Acta Crystallogr D. 1995;51:914–924. doi: 10.1107/S0907444995002976. [DOI] [PubMed] [Google Scholar]
- 16.Kraulis P G. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 17.Teeter M M, Roe S M, Heo N H. J Mol Biol. 1993;230:292–311. doi: 10.1006/jmbi.1993.1143. [DOI] [PubMed] [Google Scholar]
- 18.Stec B, Zhou R, Teeter M M. Acta Crystallogr D. 1995;51:663–681. doi: 10.1107/S0907444994014484. [DOI] [PubMed] [Google Scholar]
- 19.Yamano A, Heo N H, Teeter M M. J Biol Chem. 1997;272:9597–9600. doi: 10.1074/jbc.272.15.9597. [DOI] [PubMed] [Google Scholar]
- 20.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 21.Blessing R H. J Appl Crystallogr. 1997;30:421–426. [Google Scholar]
- 22.Blessing R H, Guo D Y, Langs D A. In: Direct Methods for Solving Macromolecular Structures, NATO ASI Series Volume, Series C: Mathematical and Physical Sciences. Fortier S, editor. Vol. 507. Dordrecht, The Netherlands: Kluwer; 1998. pp. 47–71. [Google Scholar]
- 23.Brünger A T. Nature (London) 1992;355:472–474. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- 24.Hansen N K, Coppens P. Acta Crystallogr A. 1978;34:909–921. [Google Scholar]
- 25.Sheldrick G M, Schneider T. Methods Enzymol. 1997;276:319–343. [PubMed] [Google Scholar]
- 26.Allen F H. Acta Crystallogr B. 1986;42:515–522. [Google Scholar]
- 27.Coppens P, Guru Row T N, Leung P, Stevens E D, Becker P J, Yang Y W. Acta Crystallogr A. 1979;35:63–72. [Google Scholar]
- 28.Lamzin V S, Morris R J, Dauter Z, Wilson K S, Teeter M M. J Biol Chem. 1999;274:20753–20755. doi: 10.1074/jbc.274.30.20753. [DOI] [PubMed] [Google Scholar]
- 29.Urzhumtsev A G, Skovoroda T P, Lunin V Y. J Appl Crystallogr. 1996;29:741–744. [Google Scholar]
- 30.Coppens P. Science. 1967;158:1577–1579. doi: 10.1126/science.158.3808.1577. [DOI] [PubMed] [Google Scholar]
- 31.Bouhmaida N, Ghermani N-E, Lecomte C, Thalal A. Acta Crystallogr. 1998;53:556–563. doi: 10.1107/s0108767399000689. [DOI] [PubMed] [Google Scholar]
- 32.Dauter Z, Lamzin V S, Wilson K S. Curr Opin Struct Biol. 1997;7:681–688. doi: 10.1016/s0959-440x(97)80078-4. [DOI] [PubMed] [Google Scholar]
- 33.Longhi S, Czjzek M, Cambillau C. Curr Opin Struct Biol. 1998;8:730–737. doi: 10.1016/s0959-440x(98)80093-6. [DOI] [PubMed] [Google Scholar]
- 34.Lamour V, Barth P, Rogniaux H, Poterszman A, Howard E, Mitschler A, Van Dorsselaer A, Podjarny A, Moras D. Acta Crystallogr D. 1999;55:721–723. doi: 10.1107/s0907444998013365. [DOI] [PubMed] [Google Scholar]
- 35.Lecomte C, Blessing R H, Coppens P, Tabard A. J Am Chem Soc. 1986;108:6942–6949. [Google Scholar]
- 36.Lecomte C, Rohmer M M, Bénard M. In: in The Porphyrin Handbook: Theoretical and Physical Characterization. Kadish K M, Smith K, Guilard R, editors. Vol. 7. New York: Academic; 1999. pp. 39–79. [Google Scholar]