

Abstract
Endothelial dysfunction and loss of nitric oxide (NO) is an integral part of the initiation and maintenance of the inflammatory process such as that occurring in traumatic shock, and is considered responsible for much of the trauma induced microvascular injury.
We investigated the effects of a vascular endothelial growth factor (VEGF) in a rat model of traumatic shock. Pentobarbital-anaesthetized rats subjected to Noble-Collip drum trauma developed a shock state characterized by marked hypotension and a 93% mortality rate with a mean survival time of 108±10 min in 14 rats. Accompanying these effects was a significant degree of endothelial dysfunction and a markedly elevated intestinal myeloperoxidase (MPO) activity.
Treatment with 125 μg kg−1 VEGF administered intravenously 18 h pre-trauma, increased survival rate to 67% (P<0.01), and prolonged survival time to 252±24 min in 12 rats (P<0.01). VEGF also significantly preserved the endothelium-dependent relaxation to ACh indicating a preservation of endothelium-derived NO.
Our results indicate that endothelial dysfunction with its accompanying loss of NO plays an important role in tissue injury associated with trauma, and that preservation of NO is beneficial in traumatic shock. The mechanisms of the protective effect of VEGF in trauma involves preservation of eNOS function and diminished neutrophil accumulation resulting in reduced neutrophil-mediated tissue injury.
Keywords: NO, superior mesenteric artery, endothelium-dependent vasorelaxation, neutrophils
Introduction
Traumatic shock is a severe condition with often a fatal outcome associated with multiple organ dysfunction. The underlying pathophysiology of traumatic shock includes diffuse endothelial damage similar to that occurring in other forms of circulatory shock including ischaemia-reperfusion (Lefer & Ma, 1992; Wang et al., 1993) and haemorrhagic shock (Scalia et al., 1996; Lefer & Lefer, 1993). Moreover, enhancement of NO production can preserve endothelial function and limit the degree of tissue injury in a variety of ischaemia-reperfusion and shock states (Gauthier et al., 1994). VEGF has been shown to upregulate eNOS production and maintain NO levels (Murohara et al., 1998). However, the effects of VEGF are not known in shock-like states including traumatic shock. We hypothesized that enhancement of eNOS with VEGF at an early step could favourably influence the course and alter the outcome of traumatic shock.
To evaluate this hypothesis, we employed the experimental animal model of Noble-Collip drum trauma which results in a severe form of traumatic shock state characterized by marked hypotension, significant splanchnic tissue injury, and a high mortality rate (Noble & Collip, 1942). Early in this trauma model there is a severe early endothelial dysfunction that is accompanied by a progressive reduction in endothelial NO release, attenuation of endothelium-dependent vasorelaxation, and increased adherence of neutrophils to the vascular endothelium leading to perturbations of vascular homeostasis (Scalia et al., 1995; 1996; Siegfried et al., 1992; Skurk et al., 1994; Christopher et al., 1994). The purpose of our study was (a) to determine the effects of VEGF, a promoter of eNOS expression, in a well established model of traumatic shock, (b) to investigate the mechanisms of any observed beneficial effects of VEGF under these conditions.
Methods
Experimental protocol
Male Sprague-Dawley rats weighing 175–225 g were anaesthetized with sodium pentobarbital (60 mg kg−1) given intraperitoneally (i.p.) prior to any experimental procedures. The trachea was then cannulated with polyethylene tubing (PE-240, Becton Dickinson, Parsippany, NJ, U.S.A.) to ensure a patent airway. Traumatic shock was induced in anaesthetized rats by whole body trauma administered in a Noble-Collip drum apparatus (Noble & Collip, 1942). The rats were subjected to a total of 525 revolutions at a rate of 60 r.p.m. Approximately 20% of the rats subjected to trauma died during the induction of trauma (i.e., in the drum) and were excluded from the study. Polyethylene catheters (PE-50) filled with heparinized 0.9% NaCl solution were inserted into the left common carotid artery to allow continuous monitoring of mean arterial blood pressure (MABP). MABP was recorded every 30 min over the entire 5 h observation period using Statham P23AC pressure transducers connected to a Grass Model 7 oscillographic recorder (Grass Instrument Co., Quincy, MA, U.S.A.). All cannulations were completed within 10 min after removal of the rat from the Noble-Collip drum apparatus.
All animals were randomly assigned to one of four experimental groups: (a) nine sham trauma rats given 125 μg kg−1 VEGF i.v., (b) 14 trauma rats given 0.9% NaCl, and, (c) 12 trauma rats given 125 μg kg−1 VEGF i.v. rhVEGF165 was obtained from N. Ferrara (Genentech, Inc., South San Francisco, CA, U.S.A.). This is a recombinant human non-glycosylated form of VEGF, which acts similarly to the glycosylated form. This dose regimen of VEGF has been shown to exert a significant endothelial protective effect in myocardial ischaemia and reperfusion (Chuhran et al., 1999). The drug or its vehicle was administered as an i.v. bolus (i.e., 0.5 ml) over a 1 min period 18 h prior to the start of the experimental protocol. Sham trauma rats were anaesthetized and subjected to the same surgical procedures as the traumatized rats, except that they were not subjected to drum trauma. Supplemental pentobarbital was administered intraperitoneally to all animals as necessary to maintain a surgical plane of anaesthesia.
The experiments were terminated 5 h post-trauma or when MABP fell below 45 mmHg. Following the termination of the study, 0.3 g segments of the ileum were removed for MPO analysis. The superior mesenteric artery was then isolated and removed for in vitro study of vascular reactivity. All rats were autopsied to determine whether gross evidence of traumatic injury occurred in the splanchnic viscera (i.e., serosanginous peritoneal fluid, bowel ischaemia, splanchnic vascular engorgement) and absence of significant vascular injury with pure peritoneal haemorrhage. Rats were excluded from the study if serosanginous fluid and splanchnic vascular engorgement did not occur, or if the animal died earlier than 30 min post-trauma from apnea or haemorrhage. Only three of the 35 rats subjected to trauma were excluded from the study for these reasons. All three excluded animals were trauma rats given vehicle.
All experiments were performed in accordance with the National Institutes of Health guidelines for the use of experimental animals and the study protocol was approved by the Institutional Animal Care and Use Committee (IACUC) of Thomas Jefferson University.
Isolated superior mesenteric artery ring studies
Superior mesenteric artery (SMA) segments were removed from rats at the end of the experiment and placed into warmed Krebs-Henselheit (K-H) buffer consisting of (in mmol/L): NaCl 118, KCl 4.75, CaCl2·2H2O 2.54, KH2PO4 1.19, MgSO4·7H2O 1.19, NaHCO3 12.5, and glucose 10.0. Isolated vessels were carefully freed of connective tissue, and cut into rings 2–3 mm in length. The rings were then mounted on stainless steel hooks, suspended in a 10 ml tissue bath (warmed to 37°C) and connected to FT-03 force displacement transducers (Grass Instrument Co., Quincy, MA, U.S.A.) in order to record changes in force on a Grass model 7 oscillographic recorder. The baths were filled with K-H buffer and aerated at 37°C with a gas mixture of 95% O2 and 5% CO2. SMA rings were initially stretched to give an optimal preload of 0.5 g of force and then equilibrated for 60–90 min. During this period, the buffer in the tissue bath was replaced every 20 min, and the resting force of the vascular rings was adjusted until 0.5 g of preload was maintained. This resting force was selected since it does not injure the endothelium or interfere with the release of NO in response to endothelium-dependent vasodilators. After equilibration, the rings were exposed to 100 nM U-46619 (9,11-epoxy-methano-PGH2, Biomol Research Laboratories, Plymouth Meeting, PA, U.S.A.), a thromboxane A2 mimetic, to generate about 0.5 g of developed force above equilibration value. Once a stable contraction was obtained, acetylcholine (ACh), an endothelium-dependent vasodilator, was added to the bath in cumulative concentrations of 0.1, 1, 10 and 100 nM. After the maximum vasorelaxant response stabilized, the rings were washed and allowed to equilibrate to baseline once more. The procedure was repeated with an endothelium-independent vasodilator, acidified NaNO2 (0.1, 1, 10 and 100 μM). NaNO2 was prepared by dissolving the compound in 0.1 N HCl and titrating it to pH 2.0. Equal volumes of K-H solution titrated to pH 2.0 relaxed 22 SMA rings 2.8±2.6%, a value that was not significantly different from zero. After additional washes and equilibration period, the procedure was again repeated with cumulative concentrations of another endothelium-dependent vasodilator, the calcium ionophore A23187, at concentrations of 1, 10, 100 and 1000 nM.
Histology
Ileal segments were used to assay the amount of infiltrated PMNs by histological staining. After the final reading of trauma, the ileum was removed and placed in 4% paraformaldehyde overnight at 4°C. The tissue was cut into sections and dehydrated using graded acetone washes at 4°C. Tissue sections were embedded in plastic (Immunobed; Polysciences Inc., Warrington, PA, U.S.A.), and 4 μm-thick sections were cut and transferred to Vectabond-coated slides (Vector Laboratories, Inc., Burlingame, CA, U.S.A.). The slides were rinsed in ethanol for 10 min to remove some of the plastic embedding and allow the tissue to stain. The tissue sections were then stained with either Haematoxylin solution Gill No. 3 for 10 min (Sigma Chemical Co.) and Giemsa stain for 3 min (Sigma Chemical Co.). The PMNs were counted using a microscope.
Statistical analysis
All values for data presented in the text and figures are means±standard errors of the mean (s.e.mean). Data from the traumatic shock model were evaluated by analysis of variance (ANOVA) and post hoc with a Fisher's correct t-test, incorporating repeated measurements. The survival rate data were assessed by Gehan's generalized Wilcoxon test (Knapp & Wise, 1985). Probabilities of 0.05 or less were considered to be statistically significant.
Results
Effects of VEGF on mean arterial blood pressure
The time course of MABP changes in the three groups of rats is illustrated in Figure 1. Administration of VEGF had no significant effect on MABP in sham-operated control rats with mean values ranging between 140–170 mmHg over the 300 min observation period. The results obtained in sham-operated control rats given VEGF are comparable to historic sham-operated controls given vehicle (i.e., 0.9% NaCl). In six such control rats, MABP was 158±13 mmHg initially, and 136±14 mmHg 5 h later. These values are not significantly different from each other. This indicates that the surgical procedures as well as administration of VEGF did not contribute to the hypotension observed in rats after the induction of trauma. The initial MABP in traumatized rats given either vehicle or VEGF ranged between 50–90 mmHg, with no significant difference between rats given vehicle or VEGF. These data suggest that the trauma protocol resulted in a severe form of circulatory shock and that both trauma groups experienced a similar degree of injury. VEGF treatment resulted in a significant normalization of MABP to values at or above 100 mmHg for virtually the entire observation period. Although administration of the vehicle resulted in slight improvements in MABP during the first 30 min post-trauma, this improvement was transient, and MABP continued to decline throughout the remainder of the survival time of these rats.
Figure 1.
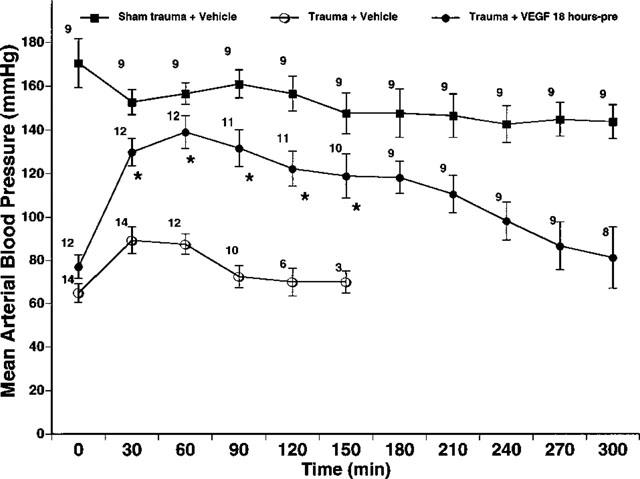
Effects of VEGF on mean arterial blood pressure (MABP). Trauma exerts profound effects on MABP. Vehicle treated trauma rats exhibited a significant decrease in MABP. VEGF (125 μg kg−1) significantly improved the MABP of the trauma rats. Data are expressed as MABP in mmHg±s.e.mean. The number to the top left of each symbol is the number of rats in each group. *P<0.01 from trauma and vehicle.
Effect of VEGF on survival time and survival rate
Figure 2 illustrates the survival times for the three experimental groups of rats. All nine sham trauma rats given VEGF survived the entire 5 h observation period (Figure 2B). In contrast, the 14 traumatized rats given only the vehicle survived for only 107±10 min, demonstrating that a lethal shock state resulted from the trauma. Nonetheless, treatment of 12 traumatized rats with VEGF resulted in a significant prolongation of survival time to 252±24 min (P<0.01).
Figure 2.
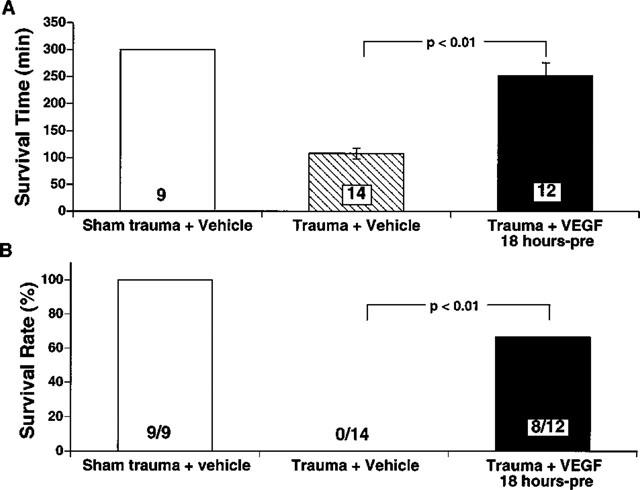
(A) Effects of VEGF on survival time following trauma. Data are expressed as mean survival time in minutes±s.e.mean. The numbers at the bottom of the bars are numbers of rats used in each group. All sham trauma rats survived the 300 min observation period. Untreated trauma rats survived only about half as long, whereas VEGF (125 μg kg−1) treated trauma rats exhibited a significantly longer survival time (P<0.01). (B) Effects of VEGF on survival rate for the 5 h observation period following trauma. VEGF (125 μg kg−1) significantly (P<0.01) prolonged survival rate compared to untreated trauma rats. Numbers at the bottom of the bars are numbers of rats survived among those used in each group.
All sham trauma rats survived for the entire 5 h observation period. Although none of 14 traumatized rats given only vehicle survived the entire observation period, 67% of the traumatized rats treated with VEGF (i.e., 8 of 12) survived the entire 5 h observation period (P<0.01) (Figure 2B). These findings support the concept of a salutary role of VEGF in shock states.
Effect of VEGF on intestinal myeloperoxidase (MPO) activity
Since endothelial dysfunction occurs in shock states and leads to increased leukocyte-endothelium interaction, we assessed intestinal neutrophil infiltration by determining MPO activity in intestinal tissue homogenates. Figure 3 illustrates MPO activity of intestinal tissue in the three experimental groups. The myeloperoxidase activity (0.1–0.7 u 100 mg−1 tissue weight) was low for all sham trauma rats, indicating that there were few resident neutrophils in normal intestinal tissue. In contrast, intestinal MPO activity in 14 traumatic shock rats given only vehicle was 18 times higher than that observed in nine sham traumatized rats (P<0.01) (i.e., 0.25±0.21 to 4.72±0.63 u g−1). Treatment with VEGF attenuated the increase in MPO activity to 2.9±0.3 u 100 mg−1 tissue weight (P<0.01) representing a one-third lower value following VEGF treatment.
Figure 3.
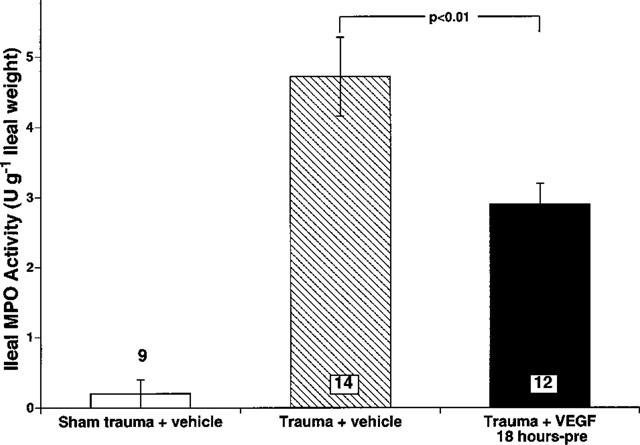
Intestinal myeloperoxidase activity (MPO) expressed as u 100 mg−1 of ileum weight. Trauma resulted in a significant increase in intestinal MPO (P<0.01) which was significantly (P<0.01) attenuated by VEGF (125 μg kg−1). Data are expressed as means±s.e.mean. The numbers at the bottom of the bars are numbers of rats used in each group.
Effect of VEGF on SMA vasorelaxation
We tested the integrity of endothelial function by comparing vasoactivity of isolated SMA rings to endothelium-dependent vasodilators as an index of the endothelial release of NO (Figure 4). The thromboxane A2 mimetic, U46619, was used to pre-contract SMA rings, and the rings were tested with endothelium-dependent (i.e., ACh and A23187) and endothelium-independent (i.e., NaNO2) vasodilators. Both ACh and A23187 fully relaxed SMA rings isolated from sham trauma rats given VEGF (i.e., 100% relaxation, Figure 4). However, the vasorelaxant responses to both ACh and A23187 in SMA rings isolated from untreated traumatized rats were significantly lower (P<0.01). VEGF treatment markedly preserved relaxation to these two endothelium-dependent vasodilators (P<0.01), indicating a significant endothelial protective effect of VEGF in this model of traumatic shock. No significant differences were observed within any of the groups regarding vasorelaxant responses to the endothelium-independent vasodilator NaNO2, indicating that the trauma did not result in a marked injury to the vascular smooth muscle of the superior mesenteric artery.
Figure 4.
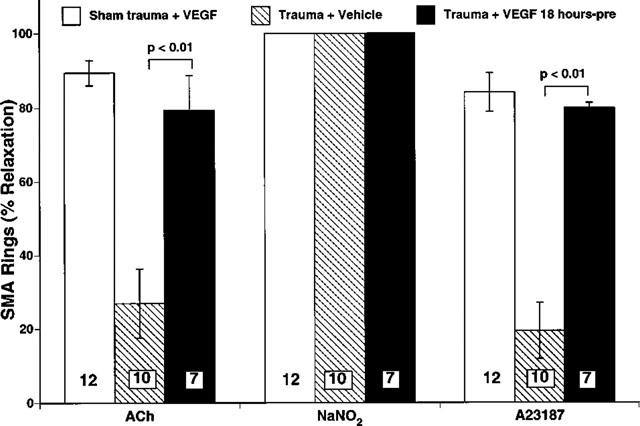
Summary of vasorelaxation responses of isolated rat superior mesenteric artery rings (SMA). Vasodilation was measured in response to the highest concentration of vasodilators: 100 nM ACh, 1 μM A23187, and 100 μM NaNO2. Trauma resulted in a significant retardation of relaxation to endothelium-dependent vasodilators (i.e., ACh and A23187) (P<0.01) but not to the endothelium-independent vasodilator (NaNO2). There was no significant dysfunction in the vascular smooth muscle of the SMA in any of the Sham trauma or trauma groups since the responses to NaNO2 were all normal. Traumatized rats treated with 125 μg kg−1 VEGF 18 h pre-trauma did not develop endothelial dysfunction. Bar heights are the means; brackets indicate ±s.e.mean. The numbers in the parenthesis indicate the number of rings prepared from each experimental group.
Effects of VEGF on leukocyte infiltration
Since endothelial dysfunction leads to increased leukocyte-endothelium interaction, we assessed the intestinal neutrophil infiltration by histological analysis. Figure 5 illustrates the degree of leukocyte infiltration into the intestinal tissue in the three experimental groups of rats. The amount of infiltrated leukocytes was low (6.1±0.81 cells mm2) for all nine sham trauma rats consistent with the finding that there was no significant neutrophil accumulation in normal intestinal tissue as measured by MPO activity. In contrast, intestinal leukocyte infiltration in 14 trauma rats given only vehicle was 34.5±1.7 cells mm2, a value almost six times higher than that observed in sham traumatized rats. VEGF treatment attenuated this increase in leukocyte infiltration by about half (P<0.01).
Figure 5.
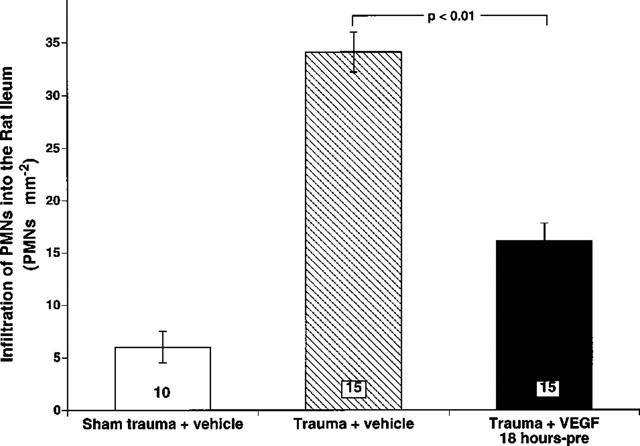
Quantification of histological staining of neutrophils (PMNs) in traumatized rats. Trauma resulted in a significant increase of infiltrated PMNs in the intestine. A significant attenuation of the infiltration was seen with the addition of 125 μg kg−1 VEGF (P<0.01). Data are expressed as means±s.e.mean. The numbers at the bottom of the bars are numbers of fields counted in each of the three groups.
Discussion
Noble-Collip drum trauma results in a very severe form of circulatory shock with a mortality rate (Noble & Collip, 1942; Araki et al., 1980) of 80–100% which is comparable to the ‘systemic inflammatory response syndrome' or SIRS observed clinically following significant blunt tissue trauma (Goris et al., 1986). This nonbacterial inflammatory reaction is induced by the initial injury and usually consists of five major pathophysiologic processes: (a) tissue ischaemia and hypoxia, (b) reperfusion with subsequent generation of oxygen free radicals, (c) stimulation of phagocytes by complement split products or other inflammatory factors, (d) activation of endothelial cells showing increased surface expression of adhesion molecules, and (e) increased leukocyte-endothelial interaction (Araki et al., 1980; Goris et al., 1986; Lefer & Lefer, 1996). A key early event in this process is vascular endothelial dysfunction characterized by a loss of nitric oxide (Scalia et al., 1996; Lefer & Lefer, 1993).
Scalia et al. (1996) studied the time course of endothelial dysfunction in murine traumatic shock and showed that significant endothelial dysfunction occurs early in the rat mesenteric vasculature (i.e., 15–30 min followed trauma). This endothelial dysfunction is characterized by impaired vaso-relaxation to endothelium-dependent dilators due to diminished endothelial release of nitric oxide. This decrease in endothelial NO release facilitates increased neutrophil adhesion and transendothelial migration through increased expression of endothelial adhesion molecules (i.e., P-selectin and ICAM-1), a process that normally is suppressed by physiologic amounts of NO (Lefer & Lefer, 1996; Davenpeck et al., 1994; Gauthier et al., 1995). The overall consequence of the endothelial dysfunction is therefore the infiltration and activation of neutrophils into the injured tissues. Activated neutrophils release cytotoxic mediators that lead to further splanchnic circulatory dysfunction, associated with fluid loss and a consistent time-dependent increase in neutrophil infiltration into the injured tissues. This process results in an irreversible shock state with marked hypotension and a fatal outcome.
Evidence is accumulating to show that decreased nitric oxide plays a major role in initiating and maintaining the inflammatory response following trauma (Gauthier et al., 1994). VEGF has been shown to maintain vascular endothelial function in murine models of limb ischaemia (Takeshita et al., 1998) and balloon-injured arteries (Asahara et al., 1995; Callow et al., 1994). VEGF also upregulates endothelium-derived nitric oxide synthase (eNOS) expression in human endothelial cells (Hood et al., 1998) and in canine coronary arteries (Ku et al., 1993). This up-regulation of eNOS leading to increased NO release has been shown to decrease endothelial cell adhesion molecule expression (Gauthier et al., 1995). This reduced adhesion molecule expression is a key component of the mechanism for the protective effects of VEGF in ischaemia/reperfusion states. This study represents the first report of VEGF used in a circulatory shock state. In particular VEGF demonstrated significant protective effects during traumatic shock in terms of endothelial preservation, attenuation of neutrophil accumulation, prolongation of survival time, and increased survival rate in this model of lethal traumatic shock. These effects cannot be attributed to differences in the severity of traumatic injury because both trauma groups were subjected to the same degree of trauma (i.e., the same number of rotations in the Noble-Collip drum), and both developed the same degree of post-trauma hypotension.
Although the precise mechanism through which VEGF attenuates traumatic shock remains to be clarified, a number of possibilities may be considered. Early endothelial dysfunction post-traumatic insult appears to be the first step in a cascade of events that occur in traumatic shock (Scalia et al., 1996). The damaged surface of the vascular endothelium then activates the complement system (Seifert et al., 1988) resulting in further endothelial injury as reflected by the progressive loss of endothelium-dependent relaxation. This may be due to a direct toxic effect of the activated complement products on the endothelium particularly the terminal membrane attack complex (C5b-9), or to the subsequent production of other inflammatory mediators (Seifert et al., 1988). The complement system stimulates neutrophil-endothelial adhesion through induction of P-selectin translocation from the Weibel-Palade bodies to the endothelial surface (Vercelotti et al., 1991; Hattori et al., 1989). Furthermore, complement induced generation of oxygen derived free radicals can be an important stimulus for expression of the endothelial adhesion glycoprotein P-selectin (Patel et al., 1991). An important stimulus seems to be a diminished basal and agonist-stimulated release of NO from the dysfunctional endothelium. Nitric oxide is known to suppress the up-regulation of P-selectin, due to quenching of oxygen derived free radicals and therefore diminution of leukocyte-endothelial interaction (Lefer & Lefer, 1996; Gauthier et al., 1995; Gaboury et al., 1993). Thus the loss of nitric oxide probably triggered increased leukocyte adhesiveness, which could in turn aggravate the existing endothelial dysfunction and promote PMN migration into injured tissues. A single bolus of VEGF was effective in attenuating this sequelae of trauma leading to enhanced leukocyte infiltration into injured tissue presumably by enhancing eNOS expression. That VEGF releases NO formed from eNOS has been clearly shown by Scalia et al. (1999) who studied the effects of VEGF on leukocyte-endothelium interaction in the mesenteric microvasculature of mice. VEGF released NO in wild type mice but not in eNOS deficient mice. Thus, it is clear that VEGF interacts primarily with eNOS to enhance NO production.
In conclusion, we have demonstrated that VEGF exerts beneficial effects in a rat model of traumatic shock. In vivo administration of VEGF prior to the induction of trauma increased the survival rate and prolonged survival time, attenuated infiltration of PMNs into intestinal tissue, and preserved vascular endothelial function. This improved endothelial function may contribute to reduced microvascular leakiness as well as increased NO synthesis and release thus maintaining circulatory function (Lefer & Lefer, 1996; Gaboury et al., 1993). Furthermore, we provided evidence that these effects may be at least partially explained by inhibition of neutrophil-endothelial interaction. Further studies are necessary to provide information on the potential significance of these findings in long-term survival studies and in other shock models.
Acknowledgments
We gratefully acknowledge Robert Craig for his excellent technical assistance in the course of this investigation.
Abbreviations
- ACh
acetylcholine
- eNOS
endothelial nitric oxide synthase
- K-H solution
Krebs-Henseleit solution
- MABP
mean arterial blood pressure
- MPO
myeloperoxidase
- NO
nitric oxide
- PMNs
polymorphonuclear leukocytes
- VEGF
vascular endothelial growth factor
References
- ARAKI H., SOLOTT S.J., LEFER A.M. Role of splanchnic visceral organs in development of tolerance to traumatic shock. J. Trauma. 1980;20:1046–1051. doi: 10.1097/00005373-198012000-00007. [DOI] [PubMed] [Google Scholar]
- ASAHARA T., BAUTERS C., PASTORE C., BUNTING S., FERRARA N., SYMES J.F., ISNER J.M. Local delivery of vascular endothelial growth factor accelerates re-endothelialization and attenuates intimal hyperplasia in balloon-injured rat carotid artery. Circulation. 1995;91:2802–2809. doi: 10.1161/01.cir.91.11.2793. [DOI] [PubMed] [Google Scholar]
- CALLOW A.D., CHOI E.T., TRACHTENBERG J.D., STEVENS S.L., CONNOLLY D.T., RODI C., RYAN U.S. Vascular permeability factor accelertes endothelial regrowth following balloon angioplasty. Growth Factors. 1994;10:223–228. doi: 10.3109/08977199409000240. [DOI] [PubMed] [Google Scholar]
- CHRISTOPHER T.A., MA X.L., LEFER A.M. Beneficial actions of S-Nitrosopenillamine, a nitric oxide donor, in murine traumatic shock. Shock. 1994;1:19–24. doi: 10.1097/00024382-199401000-00004. [DOI] [PubMed] [Google Scholar]
- CHUHRAN C.M., CAMPBELL B., LEFER A.M. Cardioprotective effects of vascular endothelial growth factor during PMN-mediated injury following ischemia/reperfusion Cardiovasc. Pathobiol. 2000(in press)
- DAVENPECK K.L., GAUTHIER T.W., LEFER A.M. Inhibition of endothelial derived nitric oxide promotes P-selectin expression and actions in the rat microcirculation. Gastroenterology. 1994;107:1050–1058. doi: 10.1016/0016-5085(94)90229-1. [DOI] [PubMed] [Google Scholar]
- GABOURY J., WOODMAN R.C., GRANGER D.N., REINHARD P., KUBES P. Nitric Oxide prevents leukocyte adherence: Role of superoxide. Am. J. Physiol. 1993;265:H862–H867. doi: 10.1152/ajpheart.1993.265.3.H862. [DOI] [PubMed] [Google Scholar]
- GAUTHIER T.W., DAVENPECK K.L., LEFER A.M. Nitric oxide attenuates leukocyte- endothelial interaction via P-selectin in splanchnic ischemia-reperfusion. Am. J. Physiol. 1994;267:G562–G568. doi: 10.1152/ajpgi.1994.267.4.G562. [DOI] [PubMed] [Google Scholar]
- GAUTHIER T.W., SCALIA R., MUROHARA T., GUO J.-P., LEFER A.M. Nitric oxide protects against leukocyte-endothelium interactions in the early stages of hypercholesterolemia. Arterio. Thromb. Vasc. Biol. 1995;15:1652–1659. doi: 10.1161/01.atv.15.10.1652. [DOI] [PubMed] [Google Scholar]
- GORIS R.J., BOEKHOLTZ W.K., VAN BEBBER I.P., NUYTINCK J.K., SCILLINGS P.M. Multiple organ failure and sepsis without bacteria: An experimental model. Arch. Surgery. 1986;121:897–901. doi: 10.1001/archsurg.1986.01400080039006. [DOI] [PubMed] [Google Scholar]
- HATTORI R., HAMILTON K.K., MCEVER R.P., SIMS P.J. Complement protein C5b-9 induces secretion of high molecular weight multimers of endothelial von Willebrand factor and translocation of granule membrane protein GMP-140 to the cell surface. J. Biol. Chem. 1989;264:9053–9060. [PubMed] [Google Scholar]
- HOOD J.D., MEININGER C.J., ZICHE M., GRANGER H.J. VEGF upregulates ecNOS message, protein and NO production in human endothelial cells. Am. J. Physiol. 1998;274:H1054–H1058. doi: 10.1152/ajpheart.1998.274.3.H1054. [DOI] [PubMed] [Google Scholar]
- KNAPP R.G., WISE W.C. A more appropriate statistical method for analyzing mortality data in shock research. Circ. Shock. 1985;16:375–381. [PubMed] [Google Scholar]
- KU D.D., ZALESKI J.K., JIU S., BROCK T.A. Vascular endothelial growth factor induces EDRF-dependent relaxation in coronary arteries. Am. J. Physiol. 1993;265:H586–H592. doi: 10.1152/ajpheart.1993.265.2.H586. [DOI] [PubMed] [Google Scholar]
- LEFER A.M., LEFER D.J. Pharmacology of the endothelium in ischemia-reperfusion and circulatory shock. Annu. Rev. Pharmacol. Toxicol. 1993;33:71–90. doi: 10.1146/annurev.pa.33.040193.000443. [DOI] [PubMed] [Google Scholar]
- LEFER A.M., LEFER D.J. The role of nitric oxide and cell adhesion molecules on the microcirculation in ischemia-reperfusion. Cardiovasc. Res. 1996;32:743–751. [PubMed] [Google Scholar]
- LEFER A.M., MA X.-L. Endothelial dysfunction as an early critical event in ischemia and shock Host Defense Dysfunction in Trauma, Shock and Sepsis 1992Springer: Heidelberg, Germany; 107–112.In: Fast, E., Meakins, J.L., Schildberg, F.W. (eds) [Google Scholar]
- MUROHARA T., ASHARA T., SILVER M., BAUTERS C., MASUDA H., KALKA C., KEARNEY, CHEN D., SYMES J.F., FISHMAN M.C., HUANG P.L., ISNER J.M. Nitric oxide synthase modulates angiogenesis in response to tissue ischemia. J. Clin. Invest. 1998;101:2567–2578. doi: 10.1172/JCI1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NOBLE R.L., COLLIP J.B. A quantitative method of production of experimental trauma without hemorrhage in unanesthetized animals. Q. J. Exp. Physiol. 1942;31:187–202. [Google Scholar]
- PATEL K.D., ZIMMERMAN G.A., PRESCOTT S.M., MCEVER R.P., MCINTYRE T.M. Oxygen radicals induce human endothelial cells to express GMP-140 and bind neutrophils. J. Cell. Biol. 1991;112:749–759. doi: 10.1083/jcb.112.4.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCALIA R., BOOTH G., LEFER D.J. Vascular endothelial growth factor attenuates leukocyte-endothelium interaction during acute endothelial dysfunction: essential role of endothelium-derived nitric oxide. FASEB J. 1999;13:1039–1046. doi: 10.1096/fasebj.13.9.1039. [DOI] [PubMed] [Google Scholar]
- SCALIA R., GAUTHIER T.W., LEFER A.M. Beneficial effects of LEX032, a novel recombinant serine protease inhibitor in murine traumatic shock. Shock. 1995;4:251–256. doi: 10.1097/00024382-199510000-00004. [DOI] [PubMed] [Google Scholar]
- SCALIA R., PEARLMAN S., CAMPBELL B., LEFER A.M. Time course of endothelial dysfunction and neutrophil adherence and infiltration during traumatic shock. Shock. 1996;6:177–182. [PubMed] [Google Scholar]
- SEIFERT P.S., CATALFAMO J.L., DODDS W.J. Complement C5a (desArg) generation in serum exposed to damaged aortic endothelium. Exp. Mol. Pathol. 1988;48:216–225. doi: 10.1016/0014-4800(88)90058-5. [DOI] [PubMed] [Google Scholar]
- SIEGFRIED M.R., MA X.-L., LEFER A.M. Splanchnic vascular endothelial dysfunction in rat endotoxemia: Role of superoxide radicals. Eur. J. Pharmacol. 1992;212:171–176. doi: 10.1016/0014-2999(92)90326-y. [DOI] [PubMed] [Google Scholar]
- SKURK C., BUERKE M., GUO J.-P., PAULSON J., LEFER A.M. Sialyl-Lewisx containing oligosaccharide exerts beneficial effects in murine traumatic shock. Am. J. Physiol. 1994;267:H2124–H2131. doi: 10.1152/ajpheart.1994.267.6.H2124. [DOI] [PubMed] [Google Scholar]
- TAKESHITA S., TAKAAKI I., MASAHIKO O., KOJI E., HIDEZO M., TANAKA E., KEIJI U., TOMOHIDE S. Endothelium-dependent relaxation of collateral microvessels after intramuscular gene transfer of vascular endothelial growth factor in a rat model of hindlimb ischemia. Circulation. 1998;98:1261–1263. doi: 10.1161/01.cir.98.13.1261. [DOI] [PubMed] [Google Scholar]
- VERCELOTTI G.M., PLATT J.L., BACH F.H., DALMASSO A.P. Neutrophil adhesion to xenogenic endothelium via iC3b. J. Immunol. 1991;146:730–734. [PubMed] [Google Scholar]
- WANG P., BA Z.F., CHAUDRY I.H. Endothelial cell dysfunction occurs very early following trauma-hemorrhage and persists despite fluid administration. Am. J. Physiol. 1993;265:H973–H979. doi: 10.1152/ajpheart.1993.265.3.H973. [DOI] [PubMed] [Google Scholar]