

Abstract
The main purpose of the study was to clarify to which extent nitric oxide (NO) contributes to acetylcholine (ACh) induced relaxation of human subcutaneous small arteries.
Arterial segments were mounted in myographs for recording of isometric tension, NO concentration and smooth muscle membrane potential.
In noradrenaline-contracted arteries, ACh induced endothelium-dependent relaxations. The NO synthase inhibitor, NG-nitro-L-arginine (L-NOARG) had a small significant effect on the concentration-response curves for ACh, and in the presence of L-NOARG, indomethacin only caused a small additional rightward shift in the ACh relaxation.
The NO scavenger, oxyhaemoglobin attenuated relaxations for ACh and for the NO donor S-nitroso-N-acetylpenicillamine (SNAP). Inhibition of guanylyl cyclase with 1H-[1,2,4]oxadiazolo[4,3-a]quinoxaline-1-one (ODQ), and inhibition of protein kinase G with β-phenyl-1, N2-etheno-8-bromoguanosine- 3′, 5′- cyclic monophosphorothioate, Rp-isomer, slightly attenuated ACh relaxation, but abolished SNAP induced relaxation.
ACh induced relaxation without increases in the free NO concentration. In contrast, for equivalent relaxation, SNAP increased the NO concentration 32±8 nM.
ACh hyperpolarized the arterial smooth muscle cells with 11.4±1.3 mV and 10.5±1.3 mV in the absence and presence of L-NOARG, respectively. SNAP only elicited a hyperpolarization of 1.6±0.9 mV.
In the presence of indomethacin and L-NOARG, ACh relaxation was almost unaffected by lipoxygenase inhibition with nordihydroguaiaretic acid, or cytochrome P450 inhibition with 17-octadecynoic acid or econazole. ACh relaxation was strongly reduced by the combination of charybdotoxin and apamin, but small increments in the extracellular potassium concentration induced no relaxations.
The study demonstrates that the NO/L-arginine pathway is present in human subcutaneous small arteries and to a limited extent is involved in ACh induced relaxation. The study also suggests a small contribution of arachidonic acid metabolites. However, ACh relaxation is mainly dependent on a non-NO, non-prostanoid endothelium dependent hyperpolarization.
Keywords: Acetylcholine, endothelium, human artery, nitric oxide, membrane potential, NO sensitive microelectrode
Introduction
The vascular endothelium plays an important role in the regulation of vascular tone through the release of a number of smooth muscle relaxing factors such as nitric oxide (NO) and prostaglandins. Acetylcholine (ACh) is the most widely used drug for assessment of the endothelial L-arginine/NO pathway of the arterial circulation in man and isolated arteries under pathophysiological conditions (Taddei et al., 1998; Buus et al., 1999). However, in vivo inhibition of the endothelial cell production of NO by L-arginine analogues only attenuates the ACh induced increase in blood flow to a limited extent (Chowienczyk et al., 1993; Lefroy et al., 1993; Bruning et al., 1996). Also inhibition of prostanoid production has either a very small or no effect on ACh induced vasodilatation (Duffy et al., 1998). These in vivo findings may suggest that factors other than NO or prostanoids mediate ACh-induced relaxation in the human circulation.
Investigations of isolated animal arteries have also suggested that the endothelium releases a non-NO, non-prostanoid factor (Cohen & Vanhoutte, 1995). The nature of this factor has been intensively investigated and epoxyeicosatrienoic acids (EET's) and potassium are considered as likely candidates (Harder et al., 1997; Edwards et al., 1998; Vanhoutte, 1999). Indeed, in the human coronary circulation, arachidonic acid derivatives formed through the cytochrome P450 system were suggested to be of importance (Campbell et al., 1996; Miura & Gutterman, 1998), but this was not the case in human mesenteric small arteries (Urakami Harasawa et al., 1997). Thus, the nature of the non-NO, non-prostanoid relaxing factor remains to be clarified in human small arteries.
In animal arteries, ACh relaxation is associated with hyperpolarization of the smooth muscle cells which persists in the presence of inhibitors of NO synthase and cyclo-oxygenase (Mombouli & Vanhoutte, 1997; Vanheel & Van de Voorde, 1998). Recent studies, based on the use of high potassium concentrations and potassium channel blockers, suggest that potassium channels are responsible for the relaxation caused by endothelium dependent vasodilators such as ACh, bradykinin and substance P also in isolated human arteries (Deng et al., 1995; Petersson, et al. 1995; Wallerstedt & Bodelsson, 1997), but there are few studies in human small arteries where changes in membrane potential have actually been measured, and then only by applying bradykinin or arachidonic acid (Urakami Harasawa et al., 1997; Miura & Gutterman, 1998). Therefore, there is a lack of information concerning the effect of acetylcholine on membrane potential in human systemic small arteries.
There is evidence suggesting that NO cannot be excluded as a candidate for the endothelium derived hyperpolarizing factor (EDHF). NO and nitrosothiols can cause hyperpolarization in different animal arteries (Myers et al., 1990; Parkington et al., 1995). Moreover, the role for NO in the vasodilating response to ACh is often based on the effect of inhibitors of NO production. However, it is possible that inhibitors of NO synthase, like NG-nitro-L-arginine (L-NOARG) and NG-nitro-L-arginine methyl ester, only partially inhibit ACh induced NO release (Cohen et al., 1997; Simonsen et al., 1999). Therefore, it is not possible to clarify only by the use of inhibitors of NO synthase whether NO or a nitrosothiol is mediating the endothelium-dependent relaxations resistant to NO synthase and cyclo-oxygenase inhibition.
The main purpose of the present study was to clarify to which extent NO and/or a nitrosothiol contribute to ACh induced vasorelaxation in human subcutaneous small arteries. Three different approaches were applied. First, the effect of other more selective inhibitors such as oxyhaemoglobin and ODQ of the L-arginine/NO pathway on the relaxations to ACh and the nitrosothiol, S-nitroso-N-acetylpenicillamine (SNAP) was examined. Second, simultaneous measurements of relaxation and intraluminal NO concentration were performed. Third, the effect on the membrane potential of ACh and SNAP was compared. Finally, we also addressed whether a cytochrome P450 derivative or potassium mediate the endothelium dependent relaxations induced by ACh.
Methods
Materials
Subcutaneous tissue was obtained from patients undergoing thoracic surgery. A total of 128 arteries from 69 patients was studied. Patients had thoracic surgery for lung cancer (60%), oesophageal cancer (10%), valve replacement (10%), or coronary bypass (20%). At the time of biopsy approximately 15% were treated with cardiovascular drugs like ACE inhibitors, statins or nitrates. Immediately after the biopsy was excised, it was placed in an ice-cold physiological salt solution (PSS, for composition see below). Mean standardized luminal diameter of the arteries was 267 μm (range 124–484 μm). The study was approved by the Local Ethics Committee and participants gave informed consent.
In a few control experiments either the superior mesenteric or small mesenteric artery from 12 week old male Wistar rats was used.
Small artery preparation and wall tension measurement
After excision of the biopsy, 1-4 small arteries were carefully dissected from the subcutaneous fat specimen. Approximately 2 mm long vessel segments were mounted as ring preparations in an isometric small vessel myograph (Mulvany & Halpern, 1977). The vessels were threaded on two stainless steel wires, 40 μm in diameter, attached to a force transducer and a micrometer, respectively. In this way the myograph permitted direct measurement of vessel wall tension while the internal diameter was controlled. After equilibration in oxygenated (5% CO2 in O2) PSS, pH 7.4, at 37°C for 30 min, a standardized normalization procedure was performed (Mulvany & Halpern, 1977). This defines the lumen diameter that the artery would have had in vivo, when relaxed and under a transmural pressure of 100 mmHg using the Laplace law: P=2T/d, where P is transmural pressure, T is wall tension, and d is the lumen diameter. The arteries were then set to the lumen diameter d1=0.9×d100 where active force development is maximal. To test tissue viability, all vessels were subjected to 5 μM noradrenaline for 2 min. If active force exceeded 10 kPa, the vessel was considered viable and used for further experiments.
ACh relaxation of noradrenaline constricted arteries
Arteries were contracted with noradrenaline (0.5–2 μM) resulting in a contraction level of 80–100% of the initial response to 5 μM noradrenaline, and concentration-response curves for ACh (1 nM–10 μM) and the NO donor SNAP (10 nM–10 μM) were constructed. The reproducibility of the relaxation to ACh and SNAP was tested by obtaining a second concentration-response curve under similar conditions.
To assess whether the relaxations induced by ACh were dependent on the endothelium, the vessels were contracted with noradrenaline and a first cumulative concentration-response curve for ACh was obtained. The endothelial cells in one vessel were removed by introducing into the lumen a human scalp hair and rubbing back and forth several times. From the same patient another vessel which served as control was left endothelium-intact. Then a second concentration-response curve for ACh and also for SNAP was obtained. Endothelial cell removal was verified by subsequent histological examination of formaldehyde fixed and eosin stained longitudinal sections of the arteries.
The effect of L-NOARG on ACh relaxation was examined. A first concentration-response curve for ACh was constructed in noradrenaline activated arteries, and a second after incubation with either 10 μM, 100 μM or 1 mM L-NOARG for at least 20 min. Each artery was only subjected to one concentration of L-NOARG. Similar levels of constriction with noradrenaline were achieved in untreated arteries and arteries treated with L-NOARG. The effect of indomethacin (3 μM) on ACh relaxation was tested in arteries already incubated with L-NOARG (100 μM). In other experiments the effect of the NO scavenger oxyhaemoglobin (10 μM), the guanylyl cyclase inhibitor 1H-[1,2,4]oxadiazolo[4,3-a]quinoxaline-1-one (ODQ, 3 μM) and the protein kinase G inhibitor β-phenyl-1, N2-etheno-8-bromoguanosine- 3′, 5′- cyclic monophosphorothioate, Rp-isomer (Rp-8-Br-PET-cGMPS, 30 μM) was tested in indomethacin pretreated arteries. The effect of oxyhaemoglobin, ODQ and Rp-8-Br-PET-cGMPS on the concentration response curves for SNAP were also examined.
To study whether arachidonic acid metabolites play a role in ACh relaxation, concentration-response curves for arachidonic acid (100 nM–100 μM) were obtained in noradrenaline-contracted endothelium-intact vessels and in vessels in which the endothelium was removed with a hair. Removal of the endothelium in these vessels was verified by the absence of ACh relaxation. Secondly, in arteries pretreated with L-NOARG (100 μM) and indomethacin (3 μM), the non-NO, non-prostanoid part of the ACh relaxation was investigated by evaluating the effects of the cytochrome P450 enzyme inhibitors econazole (5 μM) and 17-octadecynoic acid (17-ODYA, 3 μM), and the lipoxygenase inhibitor nordihydroguaiaretic acid (NDGA, 5 μM). To examine whether econazole had an effect on potassium channels, concentration-response curves for the potassium channel opener pinacidil (10 nM–10 μM) were obtained in the absence and presence of econazole.
To address whether potassium is mediating the endothelium dependent relaxations, a first control concentration-response curve was constructed for ACh and a second in the presence of the combination of the potassium channel blockers charybdotoxin (100 nM) and apamin (100 nM). Similar experiments were performed in rat mesenteric small arteries of the same diameter. Moreover, to investigate whether small increases in the extracellular potassium concentration relaxes noradrenaline-contracted segments, the potassium concentration was increased to 7, 10, 15 and 20 mM by adding PSS containing 125 mM KCl to the vessel chamber. Similar experiments were performed in rat mesenteric small arteries.
Simultaneous measurements of NO concentration and vasorelaxation
For simultaneous measurements of force and NO concentrations, an NO sensitive microelectrode with a diameter of 30–50 μm was introduced into the lumen of the artery as previously described (Simonsen et al., 1999). The arteries used for these experiments were mounted as described above except that one of the two 40-μm wires was exchanged with a 100-μm wire to obtain a slightly larger distance between the two opposing walls of the vessel in order to insert the electrode. The microelectrode (ISONOP30, World Precision Instruments, Stevenage, U.K.) used for these experiments is constructed from a carbon fibre with a polymer coating giving selectivity for NO. The electrode has a high selectivity for NO and a detection sensitivity around 1 nM (Simonsen et al., 1999). Selectivity was tested by adding nitrite (1 and 10 μM) or, during the experiment, by adding noradrenaline as earlier described (Simonsen et al., 1999). Before each experiment, calibration of the electrode with an NO solution prepared from deoxygenated distilled water was carried out at 37°C in a glass vial with stirring. After calibration, the electrode was introduced into the vessel lumen by means of a micromanipulator through a hole drilled in one side of the myograph. In that way, the tip of the electrode was placed at a distance from the endothelial surface of 10–20 μm. The NO electrode was connected to an amplifier (NO-meter, WPI) and the amplified signal was registered on a recorder allowing simultaneous measurements of changes in NO concentrations and force. Measurements of NO concentrations were performed in noradrenaline constricted arteries in the presence of indomethacin (3 μM) during stimulation with ACh (10 μM) or SNAP (10 μM). Furthermore, oxyhaemoglobin (10 μM) was added to test the effect of NO scavenging on the NO concentration measured. Similar experiments were also performed in the rat superior mesenteric artery.
Measurement of smooth muscle membrane potential
The arteries were mounted on two 40-μm wires in a myograph as described above. A hydraulic micromanipulator (Narishige MW3) was attached to the myograph stage, allowing intracellular potential recordings of smooth muscle cells to be made with glass electrodes inserted from the adventitial side of the artery. The electrode was connected to an amplifier (M-707, WPI). Electrodes were prepared on a horizontal puller (Sutter P-87, WPI) from aluminosilicate glass and had resistances of 80–100 MΩ when filled with 3 M KCl. Criteria for acceptable measurements were as described previously (Mulvany et al., 1982). Drugs were dissolved in PSS and added to the artery by superfusion from a reservoir bubbled with 5% CO2 in O2. All measurements were performed in the presence of indomethacin (3 μM). Prior to measuring membrane potential, endothelial function was tested by adding ACh (10 μM) to noradrenaline-contracted arteries. Membrane potential was measured under resting conditions, in arteries stimulated with ACh (10 μM), ACh in the presence of L-NOARG (100 μM), and in arteries stimulated with SNAP (10 μM).
Solutions and drugs
Arteries were dissected, mounted, and held in a physiological salt solution (PSS) of the following composition (mM): NaCl 119, NaHCO3 25, KCl 4.7, KH2PO4 1.18, MgSO4 1.17, CaCl2 2.5, EDTA 0.026, and glucose 5.5. Acetylcholine hydrochloride, noradrenaline-HCl, NG-nitro-L-arginine (L-NOARG), S-nitroso-N-acetylpenicillamine (SNAP), indomethacin, 17-octadecynoic acid (17-ODYA), econazole, nordihydroguaiaretic acid (NDGA), charybdotoxin and apamin were from Sigma Chemical Inc. (St. Louis, MO, U.S.A.). 1H-[1,2,4]oxadiazolo[4,3-a]quinoxaline-1-one (ODQ) were obtained from Tocris Cookson (Langford Bristol, U.K.). β-Phenyl-1, N2-etheno-8-bromoguanosine- 3′, 5′- cyclic monophosphorothioate, Rp-isomer (Rp-8-Br-PET-cGMPS) was from Life Science Institute (Bremen, Germany). Pinacidil was a gift from Løvens Kemiske Fabrik (Copenhagen, Denmark). Drugs were dissolved in distilled water except L-NOARG which was dissolved in calcium free PSS and econazole and ODQ which were diluted in dimethyl sulphoxide. Oxyhaemoglobin was prepared from a 1 mM solution of bovine haemoglobin (Sigma) by addition of sodium dithionite (10 mM). The reducing agent converting methaemoglobin to oxyhaemoglobin was removed by dialysis in distilled water and gassed with N2 at 4°C. The purity of oxyhaemoglobin was determined spectrophotometrically giving a final concentration of around 10 mM in the stock solution.
Calculations and statistical evaluation
All data are expressed as mean±s.e.mean, where n equals the number of patients. Relaxations are expressed as percentage of the noradrenaline induced constriction level. Concentration-response curves were analysed by iterative nonlinear regression analysis (GraphPad, San Diego, CA, U.S.A.). Each regression line was fitted to a sigmoid equation: R/Rmax=Ac/(Ac+(EC50)c) where Rmax is the maximal relaxation caused by the agonist, A is the concentration of agonist, EC50 is the concentration required for half maximum relaxation and c is a constant. Sensitivities are expressed as pD2 equal to −log (EC50 (M)). Differences between means were analysed with Student's paired or unpaired two-tailed t-test, as appropriate, or by ANOVA. The level of significance in all tests was set at P<0.05.
Results
The human small vessels did not show any resting tension. The initial stimulation with noradrenaline (5 μM) resulted in an active tension development of 2.80±0.14 Nm−1 (n=128).
Role of the L-arginine/NO pathway in ACh relaxation
In noradrenaline-contracted arteries, ACh induced reproducible relaxations with pD2 values and maximal relaxations of 7.43±0.10 and 90±3%, and 7.42±0.14 and 92±2% (n=7) in a first and second concentration response curve, respectively. In contrast to ACh, relaxation to SNAP was not reproducible, as the second concentration-response curve was shifted to the right compared to a first control curve (n=3). Thus, only one concentration-response curve for SNAP was performed in each artery. In arteries where the endothelial layer was removed by intimal rubbing, ACh relaxation totally disappeared, whereas SNAP relaxation was preserved (Figure 1, n=3). Figure 2A–C and Table 1 show the effect of increasing concentrations of L-NOARG on ACh relaxation. L-NOARG did not increase resting tension. For the lowest concentration of L-NOARG (10 μM) there was no effect on the ACh relaxation. The higher concentrations significantly attenuated ACh relaxation, but only to a small extent. In arteries incubated with L-NOARG, indomethacin induced small additional rightward shifts in the concentration-response curves for ACh (Table 1).
Figure 1.
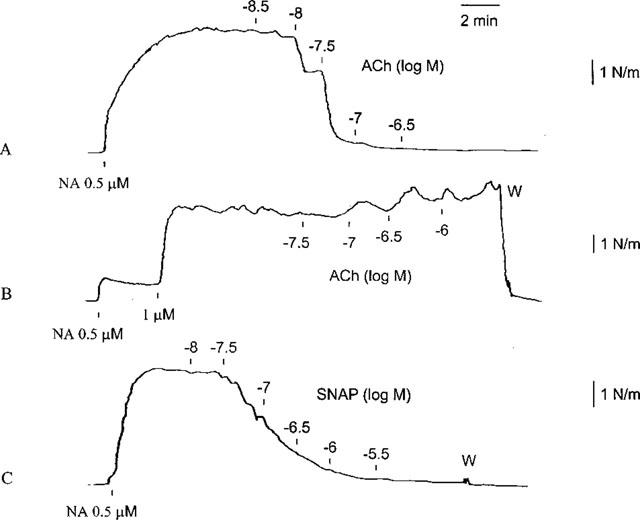
Traces demonstrating the effect of acetylcholine (ACh) in a human small artery contracted with noradrenaline (NA) before (A) and after (B) removal of the endothelium. After removal of the endothelium SNAP still caused relaxation of the artery (C). W, Wash out.
Figure 2.
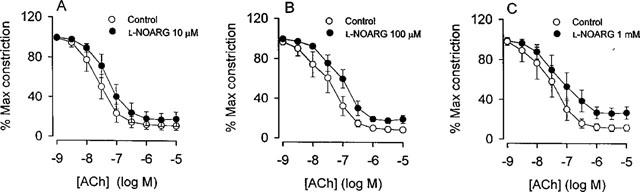
ACh relaxation of human small arteries in the absence and presence of three different concentrations of NG-nitro-L-arginine (L-NOARG): (A) 10 μM (n=7), (B) 100 μM (n=8) and (C) 1 mM (n=6). The arteries were constricted with noradrenaline and relaxations are expressed as percentage of the initial constriction level. Data are means±s.e.mean.
Table 1.
Effect of L-NOARG, indomethacin, oxyhaemoglobin, ODQ and Rp-8-Br-PET-cGMPS on ACh relaxation of subcutaneous small arteries
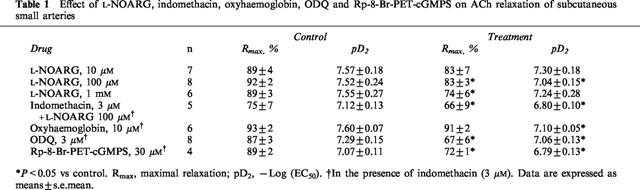
In arteries incubated with indomethacin, the NO scavenger, oxyhaemoglobin, caused significant rightward shifts in the concentration-response curves for ACh and SNAP, and reduced also the maximal relaxations induced by SNAP (Figure 3A–B and Table 1). Oxyhaemoglobin had no effect on resting tension. The inhibitor of soluble guanylyl cyclase, ODQ, induced small rightward shifts in the concentration-response curves and inhibited the maximal relaxations induced by ACh (Figure 3C and Table 1). In contrast, ODQ almost completely inhibited the relaxing effect of SNAP (Figure 3D). The inhibitor of protein kinase G, Rp-8-Br-PET-cGMPS, only caused a modest attenuation of ACh relaxation (Figure 3E and Table 1), while SNAP relaxation was abolished (Figure 3F).
Figure 3.
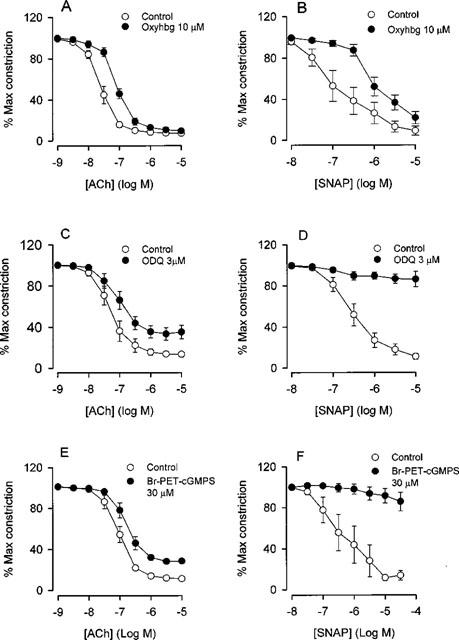
(A) Relaxation with ACh (n=6) and (B) relaxation with the NO donor SNAP (n=5) in the absence and presence of oxyhaemoglobin (Oxyhgb). (C) ACh relaxation (n=8) and (D) SNAP relaxation (n=5) in the absence and presence of ODQ. (E) ACh relaxation (n=4) and (F) SNAP relaxation (n=3) in the absence and presence of Rp-8-Br-PET-cGMPS (Br-PET-cGMPS). The human small arteries were all pretreated with indomethacin (3 μM). Data are means±s.e.mean.
Simultaneous measurements of NO concentration and relaxation
In the rat superior mesenteric artery, we have previously found that ACh increases the NO concentration simultaneously with the relaxation (Simonsen et al., 1999). Stimulation with ACh in the human small arteries resulted in a mean relaxation of 64±7% (n=4). However, in contrast to the rat superior mesenteric artery, there was no detectable increase in the NO concentration (Figure 4B). Addition of SNAP resulted in relaxations of 82±8% and an increase in the NO concentration of 32±8 nM (n=5, Figure 4B). Addition of oxyhaemoglobin reversed the increase in NO concentration induced by SNAP and caused a small reversal in the relaxation to 53±13% (n=4, Figure 4B).
Figure 4.
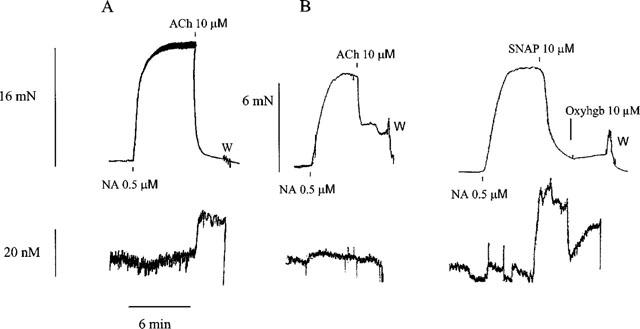
Traces illustrating simultaneous measurements of force (upper traces) and changes in NO concentrations (lower traces) in (A) the rat superior mesenteric artery relaxed with ACh and (B) a small human artery relaxed with ACh and SNAP. In this artery oxyhaemoglobin (Oxyhbg) partly reversed the increase in NO concentration, with only a small change in force. NA, noradrenaline; W, wash out.
Smooth muscle membrane potential
Smooth muscle resting membrane potential in arteries incubated with indomethacin averaged 54.8±1.4 mV (n=6). Addition of ACh induced hyperpolarizations in all arteries as illustrated on the trace in Figure 5A. In most arteries, the hyperpolarizations were transient and membrane potential returned to the initial resting level within 2–5 min. In arteries incubated with both L-NOARG and indomethacin, membrane potential was 59.6±2.4 mV (n=7); thus L-NOARG did not significantly change resting membrane potential. In these arteries, ACh also induced transient hyperpolarizations with a duration of 2–5 min (Figure 5B). Resting membrane potential in SNAP stimulated arteries was 56.5±2.3 mV (n=6). The hyperpolarization induced by SNAP was significantly smaller than the hyperpolarization induced by ACh (Figure 5C). Average values of maximal hyperpolarizations are shown in Figure 5D.
Figure 5.
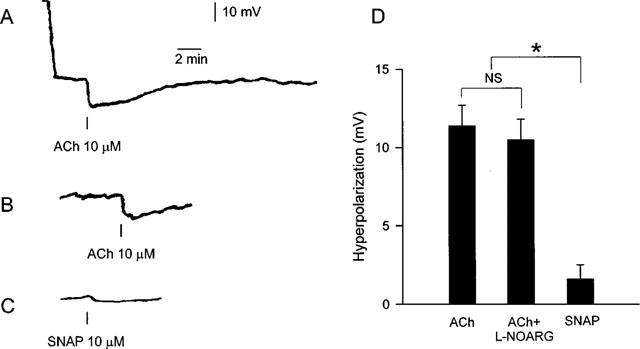
Traces illustrating measurements of smooth muscle membrane potential in human small arteries pretreated with indomethacin and stimulated with (A) ACh, where the initial part of the trace shows the change in potential upon impalement with the electrode, (B) ACh in the presence of L-NOARG (100 μM) and (C) SNAP. (D) Average changes in smooth muscle membrane potential of indomethacin treated arteries during stimulation with ACh (10 μM, n=6), ACh (10 μM) in the presence of L-NOARG (100 μM, n=7) and SNAP (10 μM, n=6). NS, nonsignificant; *P<0.01 compared to ACh and ACh+L-NOARG. Data are means±s.e.mean.
ACh relaxation resistant to L-NOARG and indomethacin
Arachidonic acid induced similar maximal relaxations of 62±11% (n=5) and 60±11% (n=5) in noradrenaline-contracted endothelium-intact and -denuded vessels, respectively. Neither 17-ODYA nor NDGA changed sensitivity or maximal relaxation to ACh (Table 2). Econazole caused a small rightward shift in the concentration-response curves for ACh, without any change in the maximal response (Table 2). Econazole did not change relaxation to pinacidil. Thus, relaxations obtained to pinacidil (10 μM) were 83±3% and 69±9% (P=NS, n=7), respectively, in the absence and presence of econazole.
Table 2.
Effect of 17-octadecynoic acid (17-ODYA), nordihydroguaiaretic acid (NDGA), econazole and the combination of charybdotoxin (ChTX) and apamin on ACh relaxation in small arteries

In the presence of L-NOARG and indomethacin, ACh relaxation was strongly attenuated by the combination of charybdotoxin and apamin in the human arteries (Figure 6, Table 2). Similar results were obtained in rat mesenteric small arteries, where the maximal relaxation induced by ACh was reduced from 94±1% to 6±5% (n=3, data not shown). Increasing the potassium concentration in rat mesenteric small arteries elicited a transient relaxation (n=3, Figure 7A), whereas increasing the extracellular potassium concentration in human small arteries did not result in relaxation (n=4, Figure 7B).
Figure 6.
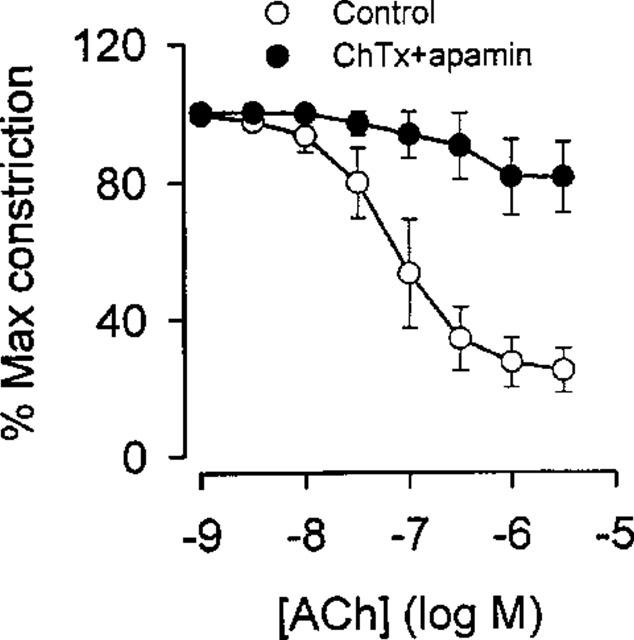
ACh relaxation of noradrenaline constricted human small arteries (n=4) in the absence and presence of the combination of the potassium channel blockers charybdotoxin (ChTx, 100 nM) and apamin (100 nM). The arteries were treated with L-NOARG (100 μM) and indomethacin (3 μM). Data are means±s.e.mean.
Figure 7.
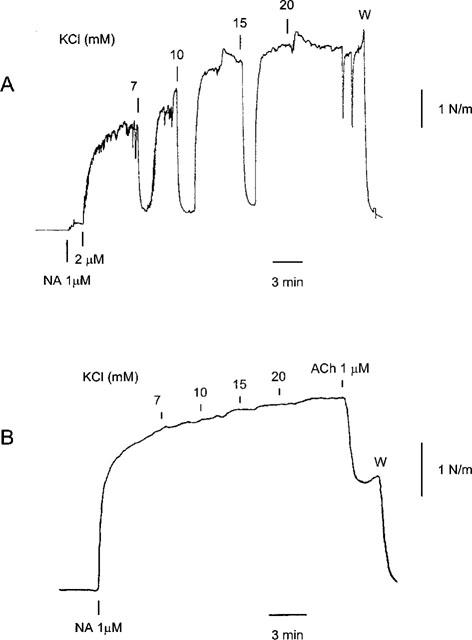
Traces illustrating the effect of increasing extracellular potassium concentration in a rat mesenteric small artery (A) and a human small artery (B). The arteries were treated with L-NOARG (100 μM) and indomethacin (3 μM) and contracted with noradrenaline (NA). Note that in the human artery ACh caused relaxation.
Discussion
The main finding of this study is that ACh relaxation of noradrenaline contracted human small subcutaneous arteries is dependent mainly on factors other than endothelial cell release of NO or NO containing factors. However, the results do show that the NO/L-arginine pathway is present in these vessels, and suggest a small and not negligible role for its products. Similarly, a small role for arachidonic acid derivatives has been demonstrated. These conclusions are strongly supported by our finding that ACh, but not SNAP, causes smooth muscle hyperpolarization.
Role of the L-arginine/NO pathway in ACh relaxation
Our finding that ACh relaxation is only minimally affected by NO synthase inhibition is in accordance with previous in vitro data on human small arteries (Woolfson & Poston, 1990; Deng et al., 1995; Pascoal & Umans, 1996; Miller Jr. et al., 1998). Similar results have been obtained with other endothelium dependent agonists such as bradykinin and substance P in human arteries from various organs (Petersson et al., 1995; Pascoal & Umans, 1996; Kemp & Cocks, 1997; Urakama-Harasawa et al., 1997; Wallerstedt & Bodelsson, 1997). These in vitro findings are similar to in vivo experiments in the forearm, where NO synthase inhibition only results in a minor rightward shift in the vasodilating response to ACh (Bruning et al., 1996; Meredith et al., 1996). In the present study these findings are expanded to a range of concentrations of the NO synthase inhibitor L-NOARG and demonstrate that inhibition is not markedly different at concentrations of 10 μM and 1 mM. These results underline that the lack of inhibition of the ACh relaxation is not related to application of an insufficient concentration of the NO synthase inhibitor.
The small displacement to the right of the ACh relaxation curve caused by interference with the L-arginine/NO pathway can be difficult to interpret in terms of the amount of NO released by the endothelium. In the rat superior mesenteric artery, we have observed that L-NOARG does not completely inhibit NO release (Simonsen et al., 1999). It was for these reasons we used the NO sensitive microelectrode to assess directly the concentration of NO. The finding that relaxation induced by ACh was not associated with a detectable increase in NO concentration raises the question whether the sensitivity of the NO electrode is sufficient. In a previous study, using exactly the same setup and experimental protocol, we found that ACh induced relaxation in the rat superior mesenteric artery resulted in an average increase in the NO concentration of 33 nM, and that SNAP relaxation was associated with an increase in NO concentration of 40 nM (Simonsen et al., 1999). In the human arteries, we measured similar increases in NO concentrations during SNAP relaxation. These results therefore demonstrate that electrode sensitivity is similar during NO measurements in the rat superior mesenteric artery and the human arteries. Furthermore, as the rat superior artery and the human small arteries were equally sensitive to SNAP it is reasonable to assume that NO sensitivity is not markedly different in these preparations and therefore the large difference seen in NO concentrations during ACh stimulation is likely to reflect a true difference in endothelial NO release.
The lack of increase in free extracellular NO concentration during ACh stimulation does not by itself exclude a role for NO or NO containing endothelium derived mediators in human subcutaneous arteries. Thus, it can not be excluded that small amounts of NO, under the detection limit for the electrode, are released. Furthermore, the NO electrode does not measure abluminally released NO. Finally the electrode only measures free NO, and therefore these experiments do not exclude the release of endothelial derived nitrosothiols. However, our findings with the NO sensitive microelectrode are in agreement with the pharmacological results that inhibition of NO synthase in the rat superior mesenteric artery almost abolishes ACh relaxation (Simonsen et al., 1999), whereas in the human small arteries the effect of L-NOARG is small. Thus, the direct measurements of NO concentrations support the pharmacological findings that the relative contribution of NO to ACh induced relaxation is less in human small arteries.
The relatively modest effect of the NO scavenger oxyhaemoglobin on SNAP relaxation has also been observed by others (Kemp & Cocks, 1997), and may be related to possible intracellular release of NO. SNAP is a nitrosothiol and both in vitro and in vivo studies suggest that these substances could play a role as endothelium derived mediators (Myers et al., 1990; Creager et al., 1997). However, the inhibitors of soluble guanylyl cyclase and protein kinase G abolished SNAP induced relaxations, while they hardly changed the ACh relaxation. Thus, blockade of smooth muscle intracellular signalling pathways for NO supports the results obtained with L-NOARG and oxyhaemoglobin that NO and NO containing factors, such as nitrosothiols, only to a limited extent are involved in ACh relaxation of human subcutaneous arteries.
Although the effects of oxyhaemoglobin on ACh induced relaxation were modest, with no effect on maximal relaxation, oxyhaemoglobin did cause a small right-shift in the ACh concentration response curve. This suggests (see Figure 3) that at low concentrations of ACh, NO mediated relaxation may play a larger relative role than at higher concentrations. Importantly, however, the oxyhaemoglobin results indicate the presence of the NO/L-arginine pathway in these arteries.
The L-arginine pathway and changes in membrane potential
The maximal hyperpolarization induced by ACh was around 11 mV, which is similar to observations by Urakami-Harasawa et al. (1997) in gastric small arteries stimulated with bradykinin. The magnitude of ACh induced maximal hyperpolarization was not significantly changed by NO synthase inhibition suggesting that NO does not contribute to the hyperpolarization. This is further supported by the finding that SNAP in a concentration, resulting in much higher concentrations of NO, hyperpolarized the smooth muscle cells by less than 2 mV. Previous studies have suggested that NO contributes to the hyperpolarization in some arteries (Tare et al., 1990; Parkington et al., 1995; Cohen et al., 1997). However, using injections of exogenous NO into the organ bath gives concentrations of NO considerably exceeding the concentrations obtained with ACh induced relaxation (Simonsen et al., 1999). In our experiments on human small arteries, SNAP resulted in 80–90% vasorelaxation and a NO concentration similar to that induced by ACh in the rat superior mesenteric artery. As this was only associated with a very small change in membrane potential, we suggest that NO in these human small arteries does not substantially contribute to hyperpolarization. Although membrane potential measurements were only performed in resting arteries, these results indicate, together with the relaxation experiments and the measurements of NO concentrations, that ACh induced relaxations in human arteries only to a limited extent is dependent on the release of NO or NO containing factors. In addition these results show that an EDHF is involved in the ACh response.
Role for arachidonic acid metabolites in ACh relaxation
The mechanical experiments and measurements of the membrane potential in the present study suggest that a non-NO factor mediates ACh relaxation and hyperpolarization. Arachidonic acid metabolites such as prostaglandins and leukotrienes and recently also EET's were found to be involved in endothelium-dependent relaxations and hyperpolarization (Harder et al., 1997). However, in the human subcutaneous small arteries arachidonic acid only induced endothelium-independent relaxations. Moreover, the inhibitors of cyclo-oxygenase, indomethacin, and lipoxygenase, NDGA, had only modest or no effect, suggesting that prostaglandins only play a minor role in ACh relaxation of these arteries.
In the present study, inhibition of the cytochrome P450 system, and thereby the formation of EET's, with 17-ODYA and econazole, either had no or a very small effect on ACh relaxation. This is in accordance with previous findings on rat mesenteric small arteries (Zygmunt et al., 1996; Vanheel & Van de Voorde, 1998). In human subcutaneous arteries, the cytochrome P450 inhibitor SKF 525a did not block the bradykinin induced relaxation (Urakama-Harasawa et al., 1997). However, this is in contrast to findings from the human (Miura & Gutterman, 1998) and porcine (Popp et al., 1996) coronary microcirculation, where P450 metabolites seem to play an important role for endothelium dependent relaxation. These observations emphasize the diversity in endothelium dependent relaxation mechanisms between different vascular beds. In the human subcutaneous small arteries our results argue against any major role for arachidonic acid metabolites in the ACh relaxation, although the lack of positive controls in this preparation for the inhibitors we have used limits the strength of this conclusion.
Role for K+ in ACh relaxation
Recent studies have proposed that potassium released from the endothelial cells may act as an EDHF, as increases in the extracellular potassium concentration causes vasorelaxation and hyperpolarization (Knot et al., 1996; Edwards et al., 1998). In these studies the combination of the blockers of Ca2+-activated K+ channels, charybdotoxin and apamin, was found to block the release of K+ from the endothelial cells (Edwards et al., 1998). In the present study both in the human small arteries and rat mesenteric small arteries the non-NO, non-prostanoid ACh relaxation was also strongly attenuated by the combination of charybdotoxin and apamin. However, despite our confirmation of the results by Edwards et al. (1998) of potassium induced relaxation in rat small arteries, we did not observe vasorelaxation in the human small arteries by increasing the extracellular potassium concentration. This is in agreement with recent results in guinea-pig arteries where the combination of charybdotoxin and apamin also inhibited ACh relaxation, but low K+ concentrations did not cause relaxation (Quignard et al., 1999). Thus, our results suggest that changes in the extracellular potassium concentration are not involved in the ACh relaxation of human subcutaneous arteries.
Possible limitations
Given the large number of arteries used in this study, it was not practical to obtain them from healthy volunteers. Thus, the conditions of the patients involved, or their pre- and perioperative drug treatment, might have influenced endothelial cell function. However, the NO and prostanoid independent relaxation was observed in all arteries we studied. Furthermore, subcutaneous small arteries from healthy volunteers show ACh induced relaxation similar to that observed previously by us (Buus et al., 1999), as well as NO independent ACh relaxation (Deng et al., 1995). Thus, the findings are unlikely to be a disease related phenomenon.
Concluding remarks
This paper demonstrates that the L-arginine/NO pathway is present in human small subcutaneous arteries, but is only to a limited extent involved in ACh relaxation. Our study also suggests only a small role for arachidonic acid metabolites. Furthermore, we have shown that ACh, in contrast to NO, causes hyperpolarization of the smooth muscle cells. Thus, apart from a diffusible EDHF, the relaxation of underlying smooth muscle cells could occur by gap junctions coupled to endothelial cells, as demonstrated in rabbit mesenteric small arteries (Hutcheson et al., 1999). A role for possible endothelial-smooth muscle gap junctions in human small vessels has not, however, been investigated.
In conclusion, the results suggest that in these vessels ACh induced relaxation is closely and primarily associated with a non-NO, non-prostanoid mechanism.
Acknowledgments
Laboratory technicians Lotte Paaby and Jørgen Andresen are thanked for their technical assistance. This work was supported by Aarhus Universitets Forskningsfond and Fonden til Lægevidenskabens Fremme. M.J. Mulvany is supported by the Danish Medical Research Council, the Danish Heart Foundation and the European Union.
Abbreviations
- ACh
acetylcholine
- EDHF
endothelium derived hyperpolarizing factor
- EET
epoxyeicosatrienoic acid
- L-NOARG
NG-nitro-L-arginine
- NDGA
nordihydroguaiaretic acid
- NO
nitric oxide
- ODQ
1H-[1,2,4]oxadiazolo[4,3-a]quinoxaline-1-one
- PSS
physiological salt solution
- Rp-8-Br-PET-cGMPS
β-Phenyl-1, N2-etheno-8-bromoguanosine- 3′, 5′- cyclic monophosphorothioate, Rp-isomer
- SNAP
S-nitroso-N-acetylpenicillamine
- 17-ODYA
17-octadecynoic acid
References
- BRUNING T.A., CHANG P.C., KEMME M.J., VERMEIJ P., PFAFFENDORF M., VAN ZWIETEN P.A. Comparison of cholinergic vasodilator responses to acetylcholine and methacholine in the human forearm. Blood Press. 1996;5:333–341. doi: 10.3109/08037059609078071. [DOI] [PubMed] [Google Scholar]
- BUUS N.H., BØTTCHER M., BØTKER H.E., SØRENSEN K.E., NIELSEN T.T., MULVANY M.J. Reduced vasodilator capacity in syndrome X related to structure and function of resistance arteries. Am. J. Cardiol. 1999;83:149–154. doi: 10.1016/s0002-9149(98)00815-7. [DOI] [PubMed] [Google Scholar]
- CAMPBELL W.B., GEBREMEDHIN D., PRATT P.F., HARDER D.R. Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circ. Res. 1996;78:415–423. doi: 10.1161/01.res.78.3.415. [DOI] [PubMed] [Google Scholar]
- CHOWIENCZYK P.J., COCKCROFT J.R., RITTER J.M. Differential inhibition by NG-monomethyl-L-arginine of vasodilator effects of acetylcholine and methacholine in human forearm vasculature. Br. J. Pharmacol. 1993;110:736–738. doi: 10.1111/j.1476-5381.1993.tb13873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COHEN R.A., PLANE F., NAJIBI S., HUK I., MALINSKI T., GARLAND C.J. Nitric oxide is the mediator of both endothelium-dependent relaxation and hyperpolarization of the rabbit carotid artery. Proc. Natl. Acad. Sci. U.S.A. 1997;94:4193–4198. doi: 10.1073/pnas.94.8.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COHEN R.A., VANHOUTTE P.M. Endothelium-dependent hyperpolarization. Beyond nitric oxide and cyclic GMP. Circulation. 1995;92:3337–3349. doi: 10.1161/01.cir.92.11.3337. [DOI] [PubMed] [Google Scholar]
- CREAGER M.A., RODDY M.A., BOLES K., STAMLER J.S. Nacetylcysteine does not influence the activity of endotheliumderived relaxing factor in vivo. Hypertension. 1997;29:668–672. doi: 10.1161/01.hyp.29.2.668. [DOI] [PubMed] [Google Scholar]
- DENG L.Y., LI J.S., SCHIFFRIN E.L. Endothelium-dependent relaxation of small arteries from essential hypertensive patients: mechanisms and comparison with normotensive subjects and with responses of vessels from spontaneously hypertensive rats. Clin. Sci. Colch. 1995;88:611–622. doi: 10.1042/cs0880611. [DOI] [PubMed] [Google Scholar]
- DUFFY S.J., TRAN B.T., NEW G., TUDBALL R.N., ESLER M.D., HARPER R.W., MEREDITH I.T. Continuous release of vasodilator prostanoids contributes to regulation of resting forearm blood flow in humans. Am. J. Physiol. 1998;274:H283–H289. doi: 10.1152/ajpheart.1998.274.4.H1174. [DOI] [PubMed] [Google Scholar]
- EDWARDS G., DORA K.A., GARDENER M.J., GARLAND C.J., WESTON A.H. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 1998;396:269–272. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- HARDER D.R., LANGE A.R., GEBREMEDHIN D., BIRKS E.K., ROMAN R.J. Cytochrome P450 metabolites of arachidonic acid as intracellular signaling molecules in vascular tissue. J. Vasc. Res. 1997;34:237–243. doi: 10.1159/000159228. [DOI] [PubMed] [Google Scholar]
- HUTCHESON I.R., CHAYTOR A.T., EVANS W.H., GRIFFITH T.M. Nitric oxide-independent relaxations to acetylcholine and A23187 involve different routes of heterocellular communication. Role of Gap junctions and phospholipase A2. Circ. Res. 1999;84:53–63. doi: 10.1161/01.res.84.1.53. [DOI] [PubMed] [Google Scholar]
- KEMP B.K., COCKS T.M. Evidence that mechanisms dependent and independent of nitric oxide mediate endothelium-dependent relaxation to bradykinin in human small resistance-like coronary arteries. Br. J. Pharmacol. 1997;120:757–762. doi: 10.1038/sj.bjp.0700928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KNOT H.J., ZIMMERMANN P.A., NELSON M.T. Extracellular K(+)-induced hyperpolarizations and dilatations of rat coronary and cerebral arteries involve inward rectifier K(+) channels. J. Physiol. 1996;492:419–430. doi: 10.1113/jphysiol.1996.sp021318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEFROY D.C., CRAKE T., UREN N.G., DAVIES G.J., MASERI A. Effect of inhibition of nitric oxide synthesis on epicardial coronary artery caliber and coronary blood flow in humans. Circulation. 1993;88:43–54. doi: 10.1161/01.cir.88.1.43. [DOI] [PubMed] [Google Scholar]
- MEREDITH I.T., CURRIE K.E., ANDERSON T.J., RODDY M.A., GANZ P., CREAGER M.A. Postischemic vasodilation in human forearm is dependent on endothelium-derived nitric oxide. Am. J. Physiol. 1996;270:H1435–1440. doi: 10.1152/ajpheart.1996.270.4.H1435. [DOI] [PubMed] [Google Scholar]
- MILLER F.J., JR, DELLSPERGER K.C., GUTTERMAN D.D. Pharmacologic activation of the human coronary microcirculation in vitro: endothelium-dependent dilation and differential responses to acetylcholine. Cardiovasc. Res. 1998;38:744–750. doi: 10.1016/s0008-6363(98)00035-2. [DOI] [PubMed] [Google Scholar]
- MIURA H., GUTTERMAN D.D. Human coronary arteriolar dilation to arachidonic acid depends on cytochrome P450 monooxygenase and Ca2+-activated K+ channels. Circ. Res. 1998;83:501–507. doi: 10.1161/01.res.83.5.501. [DOI] [PubMed] [Google Scholar]
- MOMBOULI J.V., VANHOUTTE P.M. Endothelium-derived hyperpolarizing factor(s): updating the unknown. Trends. Pharmacol. Sci. 1997;18:252–256. [PubMed] [Google Scholar]
- MULVANY M.J. , HALPERN W. Contractile properties of small arterial resistance vessels in spontaneously hypertensive and normotensive rats. Circ. Res. 1977;41:19–26. doi: 10.1161/01.res.41.1.19. [DOI] [PubMed] [Google Scholar]
- MULVANY M.J., NILSSON H. , FLATMAN J.A. Role of membrane potential in the response of rat small mesenteric arteries to exogenous noradrenaline stimulation. J. Physiol. 1982;332:363–373. doi: 10.1113/jphysiol.1982.sp014418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MYERS P.R., MINOR R.L., JR, GUERRA R., JR, BATES J.N., HARRISON D.G. Vasorelaxant properties of the endothelium-derived relaxing factor more closely resemble S-nitrosocysteine than nitric oxide. Nature. 1990;345:161–163. doi: 10.1038/345161a0. [DOI] [PubMed] [Google Scholar]
- PARKINGTON H.C., TONTA M.A., COLEMAN H.A., TARE M. Role of membrane potential in endothelium-dependent relaxation of guinea-pig coronary arterial smooth muscle. J. Physiol. 1995;484:469–480. doi: 10.1113/jphysiol.1995.sp020679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PASCOAL I.F. and , UMANS J.G. Effect of pregnancy on mechanisms of relaxation in human omental microvessels. Hypertension. 1996;28:183–187. doi: 10.1161/01.hyp.28.2.183. [DOI] [PubMed] [Google Scholar]
- PETERSSON J., ZYGMUNT P.M., BRANDT L., HÖGESTATT E.D. Substance P-induced relaxation and hyperpolarization in human cerebral arteries. Br. J. Pharmacol. 1995;115:889–894. doi: 10.1111/j.1476-5381.1995.tb15893.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POPP R., BAUERSACHS J., HECKER M., FLEMING I., BUSSE R. A transferable, beta-naphthoflavone-inducible, hyperpolarizing factor is synthesized by native and cultured porcine coronary endothelial cells. J. Physiol. 1996;497:699–709. doi: 10.1113/jphysiol.1996.sp021801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QUIGNARD J.F., FELETOU M., THOLLON C., VILAINE J.P., DUHAULT J., VANHOUTTE P.M. Potassium ions and endothelium-derived hyperpolarizing factor in guinea-pig carotid and porcine coronary arteries. Br. J. Pharmacol. 1999;127:27–34. doi: 10.1038/sj.bjp.0702493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SIMONSEN U., WADSWORTH R.M., BUUS N.H., MULVANY M.J. Simultaneous nitric oxide release and relaxation of the rat superior mesenteric artery. J. Physiol. 1999;516:271–282. doi: 10.1111/j.1469-7793.1999.271aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TADDEI S., VIRDIS A., GHIADONI L., MATTEI P., SALVETTI A. Effects of angiotensin converting enzyme inhibition on endothelium- dependent vasodilatation in essential hypertensive patients. J. Hypertens. 1998;16:447–456. doi: 10.1097/00004872-199816040-00006. [DOI] [PubMed] [Google Scholar]
- TARE M., PARKINGTON H.C., COLEMAN H.A., NEILD T.O., DUSTING G.J. Hyperpolarization and relaxation of arterial smooth muscle caused by nitric oxide derived from the endothelium. Nature. 1990;346:69–71. doi: 10.1038/346069a0. [DOI] [PubMed] [Google Scholar]
- URAKAMA-HARASAWA L., SHIMOKAWA H., NAKASHIMA M., EGASHIRA K., TAKESHITA A. Importance of endothelium-derived hyperpolarizing factor in human arteries. J. Clin. Invest. 1997;100:2793–2799. doi: 10.1172/JCI119826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VANHEEL B., VAN DE VOORDE J. Evidence against the involvement of cytochrome P450 metabolites in endothelium-dependent hyperpolarization of the rat main mesenteric artery. J. Physiol. 1998;501:331–341. doi: 10.1111/j.1469-7793.1997.331bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VANHOUTTE P.M. Old-timer makes a comeback. Nature. 1999;396:213–216. doi: 10.1038/24261. [DOI] [PubMed] [Google Scholar]
- WALLERSTEDT S.M., BODELSSON M. Endothelium-dependent relaxation by substance P in human isolated omental arteries and veins: relative contribution of prostanoids, nitric oxide and hyperpolarization. Br. J. Pharmacol. 1997;120:25–30. doi: 10.1038/sj.bjp.0700879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WOOLFSON R.G., POSTON L. Effect of NG-monomethyl-L-arginine on endothelium-dependent relaxation of human subcutaneous resistance arteries. Clin. Sci. Colch. 1990;79:273–278. doi: 10.1042/cs0790273. [DOI] [PubMed] [Google Scholar]
- ZYGMUNT P.M., EDWARDS G., WESTON A.H., DAVIS S.C., HÖGESTATT E.D. Effects of cytochrome P450 inhibitors on EDHF-mediated relaxation in the rat hepatic artery. Br. J. Pharmacol. 1996;118:1147–1152. doi: 10.1111/j.1476-5381.1996.tb15517.x. [DOI] [PMC free article] [PubMed] [Google Scholar]