

Abstract
The effect of several K+ channel blockers such as glibenclamide, tolbutamide, charybdotoxin (ChTX), apamin, tetraethylammonium (TEA), 4-aminopyridine (4-AP) and cesium on the peripheral antinociceptive effect of morphine was evaluated by the paw pressure test in Wistar rats.
The intraplantar administration of a carrageenan suspension (250 μg) resulted in an acute inflammatory response and a decreased threshold to noxious pressure. Morphine administered locally into the paw (25, 50, 100 and 200 μg) elicited a dose-dependent antinociceptive effect which was demonstrated to be mediated by a peripheral site up to the 100 μg dose.
The selective blockers of ATP-sensitive K+ channels glibenclamide (20, 40 and 80 μg paw−1) and tolbutamide (40, 80 and 160 μg paw−1) antagonized the peripheral antinociception induced by morphine (100 μg paw−1).
This effect was unaffected by ChTX (0.5, 1.0 and 2.0 μg paw−1), a large conductance Ca2+-activated K+ channel blocker, or by apamin (2.5, 5.0 and 10.0 μg paw−1), a selective blocker of a small conductance Ca2+-activated K+ channel.
Intraplantar administration of the non-specific K+ channel blockers TEA (160, 320 and 640 μg), 4-AP (10, 50 and 100 μg) and cesium (125, 250 and 500 μg) also did not modify the peripheral antinociceptive effect of morphine.
These results suggest that the peripheral antinociceptive effect of morphine may result from activation of ATP-sensitive K+ channels, which may cause a hyperpolarization of peripheral terminals of primary afferents, leading to a decrease in action potential generation. In contrast, large conductance Ca2+-activated K+ channels, small conductance Ca2+-activated K+ channels as well as voltage-dependent K+ channels appear not to be involved in this transduction pathway.
Keywords: Analgesia, morphine, K+ channel blockers, μ-opioid receptor agonists, glibenclamide, tolbutamide, apamin, charybdotoxin
Introduction
Experiments at the cellular level have shown that agonists at the μ opioid receptor increase potassium conductance (North, 1989). In the central nervous system, the opening of K+ channels seems to play a role in opioid-mediated antinociception, since the ATP-sensitive K+ channel blockers (sulphonylureas) antagonize the antinociceptive effect of opioids (Ocaña et al., 1990; 1995; Wild et al., 1991; Roane & Boyd, 1993). The antinociceptive effect of opioid agonists was also demonstrated to be enhanced by ATP-sensitive K+ channel openers such as pinacidil (Vergoni et al., 1992) and cromakalim (Ocaña et al., 1996). Evidence that some opioids induce opening of calcium-activated K+ channels has also been obtained (Stretton et al., 1992). In addition, the diversity of K+ channels (Halliwell, 1990) with different electrophysiological and pharmacological characteristics described in neurones, suggests that other types of K+ channels may be involved in this effect.
Opioids can produce analgesia by inhibiting nociceptive input at supraspinal and spinal sites (Herz & Teschemacher, 1971; Yaksh & Rudy, 1976). Several studies have also indicated that exogenous as well as endogenous opioids can act on peripheral nociceptors to produce an antinociceptive effect against the hyperalgesia induced by local inflammation (Ferreira & Nakamura, 1979; Bentley et al., 1981; Stein et al., 1990). Stein et al. (1990) demonstrated the presence of opioid receptors on the peripheral terminals of primary afferents, however, the mechanism by which opioid agonists induce peripheral antinociception is unclear. It was previously shown that opioid receptors at peripheral sites are coupled with inhibitory G proteins since pertussis toxin inhibits the peripheral antinociception induced by morphine (Levine & Taiwo, 1989) and are also coupled with L-arginine-NO-cGMP pathway (Ferreira et al., 1991; Duarte et al., 1992).
The present study was undertaken to determine whether specific and non-specific K+ channel blockers have any effect on the peripheral antinociception induced by morphine. For this purpose we tested the effects of glibenclamide and tolbutamide, sulphonylureas that specifically block ATP-sensitive K+ channels (Edwards & Weston, 1993), apamin, a selective blocker of small conductance Ca2+-activated K+ channels (Romey et al., 1984), charybdotoxin (ChTX), a blocker of large conductance Ca2+-activated K+ channels (Miller et al., 1985), and non-selective K+ channel blockers, 4-aminopyridine (4-AP), tetraethylammonium (TEA) and cesium (Cook & Quast, 1990).
Methods
Animals
The experiments were performed on 180–250 g male Wistar rats (from CEBIO-UFMG). The animals were housed in a temperature-controlled room (23±1°C) on an automatic 12 h light/dark cycle (06.00–18.00 h). All testing was conducted during the light phase (12.00–17.00 h). Food and water were freely available until the beginning of the experiments. Naïve animals were used throughout.
Measurement of hyperalgesia
Hyperalgesia was induced in the hindpaw by intraplantar administration of carrageenan suspension (250 μg) and measured according to the paw pressure test described by Randall & Selitto (1957). An analgesy-meter (Ugo-Basile, Italy) with a cone-shaped paw-presser with a rounded tip, which applies a linearly increasing force to the plantar surface of the paw, was used. The weight in gram (g) required eliciting nociceptive responses such as paw flexion or struggling was defined as the nociceptive threshold. A cut-off value of 300 g was used to prevent damage to the paws. The nociceptive threshold was always (except when indicated) measured in the right hindpaw and determined by the average of three consecutive trials recorded before and 3 h after carrageenan injection. The results were calculated by the difference between these two averages (Δ of nociceptive threshold) and expressed as grams.
Experimental protocol
Morphine was administered subcutaneously into the right hindpaw 2 h after local injection of the carrageenan suspension. In the protocol used to determine whether morphine was acting at central sites carrageenan was injected into both hindpaws while morphine was administered 2 h later into the left or right paw. All the K+ channel blockers were injected subcutaneously into the right hindpaw. The sulphonylureas (glibenclamide and tolbutamide) were administered 5 min before morphine while all the other K+ channel blockers were injected 45 min after morphine (Wild et al., 1991; Ocaña & Baeyens, 1993; Yonehara & Takiuchi, 1997).
Chemicals
The drug used as hyperalgesic agent was lambda carrageenan (Sigma) and the μ-opioid receptor agonist was morphine hydrochloride (Merck). The K+ channel blockers and their suppliers were: glibenclamide (Sigma), tolbutamide (ICN Biomedicals), charybdotoxin (Sigma), apamin (Sigma), tetraethylammonium chloride (Sigma), 4-aminopyridine (Sigma) and cesium (Mitsuwa's Pure Chemicals). Morphine and carrageenan were dissolved in isotonic saline and injected in a volume of 100 μl per paw. The K+ channel blockers were dissolved in demineralized water with exception of sulphonylureas that were dissolved in saline and tween (2%) vehicle, immediately before use and injected in a volume of 50 μl per paw. For acidic or alkaline solutions the pH was adjusted closer to 7.
Statistical analysis
The statistical analyses were carried out by one-way analysis of variance (ANOVA) followed by Bonferroni's test for multiple comparisons. Probabilities less than 5% (P<0.05) were considered to be statistically significant.
Results
Antinociceptive effect of morphine
The administration of morphine (25, 50, 100, 200 μg) into the right hind paw produced an antinociceptive response against the hyperalgesia induced by prior local injection of carrageenan (Figure 1). Morphine at the dose of 100 μg, when administered into the left paw, did not produce an antinociceptive effect in the right paw, whereas morphine at the dose of 200 μg when injected into the left paw induced a potent antinociceptive effect in the contra-lateral paw (Figure 2).
Figure 1.
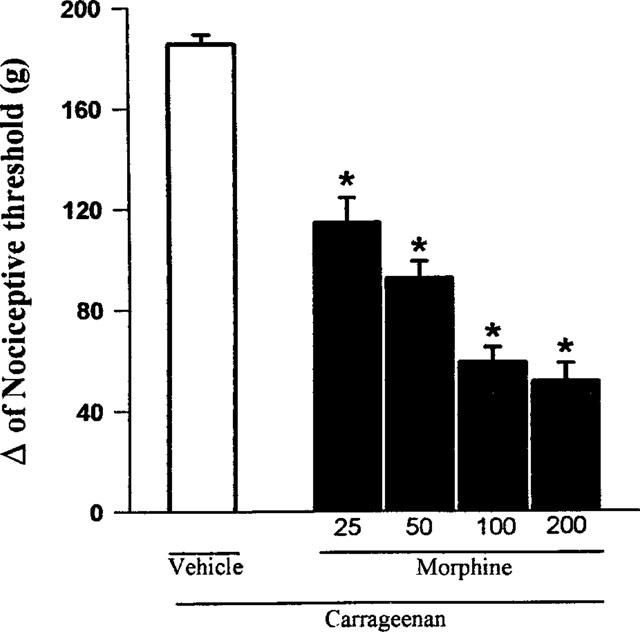
Effect of morphine on the nociceptive threshold in carrageenan-induced hyperalgesia in rats. Morphine (μg) was administered intraplantarly 2 h after the local administration of 100 μl of a carrageenan suspension (250 μg). Each column represents the mean±s.e.mean (n=10). *P<0.01 vs carrageenan+vehicle-injected control (Bonferroni's test).
Figure 2.
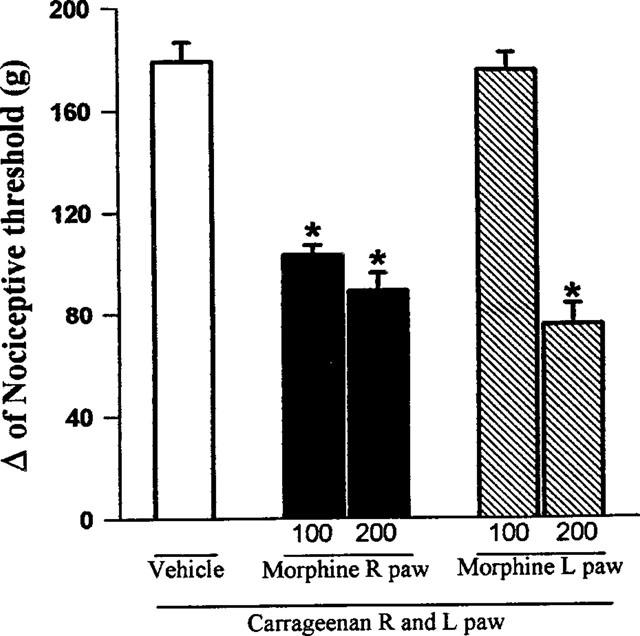
Exclusion of a central antinociceptive effect of morphine. Morphine (μg) was administered into the right (R) or left (L) paw 2 h after carrageenan administration into both hind paws. Each column represents the mean±s.e.mean (n=10). *P<0.01 vs carrageenan+ vehicle-injected control (Bonferroni's test).
Antagonism of morphine-induced antinociception by glibenclamide and tolbutamide
Glibenclamide (20, 40, 80 μg paw−1) significantly reduced the morphine-induced antinociception (100 μg paw−1) in a dose-dependent manner (Figure 3). As shown in Figure 4, the other sulphonylurea tested, tolbutamide (40, 80 and 160 μg paw−1) also significantly inhibited the morphine-induced antinociceptive effect. None of the sulphonylureas tested significantly modified the nociceptive threshold in control animals (data not shown), or induced any overt behavioural effect at the doses used. Furthermore, the maximum dose of glibenclamide (80 μg), by the same route, did not alter significantly the plasma glucose level (results not shown).
Figure 3.
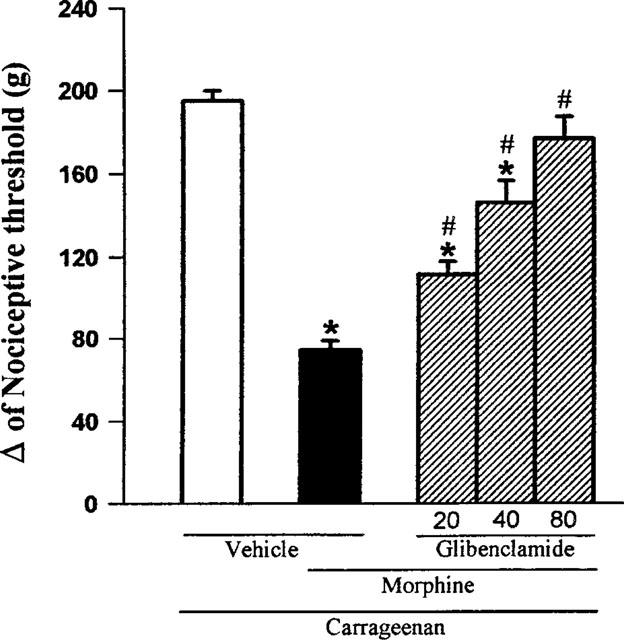
Antagonism induced by intraplantar administration of glibenclamide of the peripheral antinociception produced by morphine in hyperalgesic paws. Glibenclamide (μg) was administered 5 min before morphine (100 μg). Each column represents the mean±s.e.mean (n=5). * and # P<0.01 as compared to (Carrageenan+vehicles) and (Carrageenan+morphine+vehicle)- injected controls, respectively (Bonferroni's test).
Figure 4.
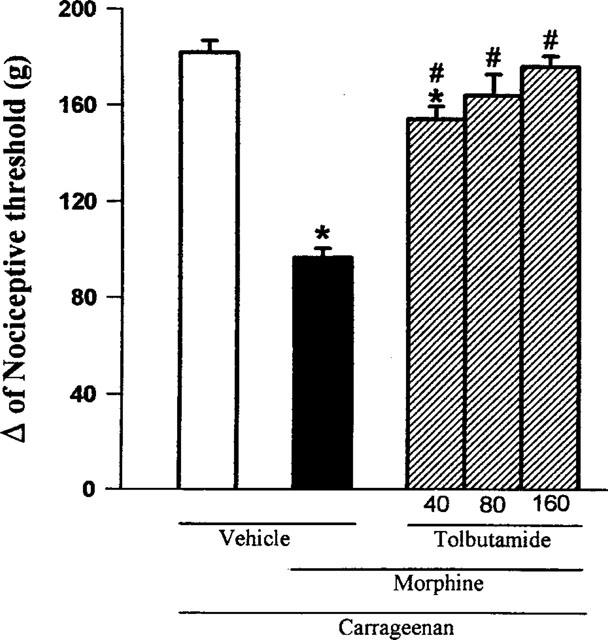
Antagonism induced by intraplantar administration of tolbutamide of the peripheral antinociception produced by morphine in hyperalgesic paws. Tolbutamide (μg) was administered 5 min before morphine (100 μg). Each column represents the mean±s.e.mean (n=5). * and # P<0.01 as compared to (Carrageenan+vehicles) and (Carrageenan+morphine+vehicle)-injected controls, respectively (Bonferroni's test).
Effect of apamin and ChTX on morphine-induced antinociception
Intraplantar injection of apamin (2.5, 5.0 and 10 μg) had no significant effect on morphine-induced antinociception, even at the highest dose tested. Charybdotoxin (0.5, 1.0 and 2.0 μg paw−1) also failed to significantly counteract the antinociception induced by morphine (Figure 5).
Figure 5.
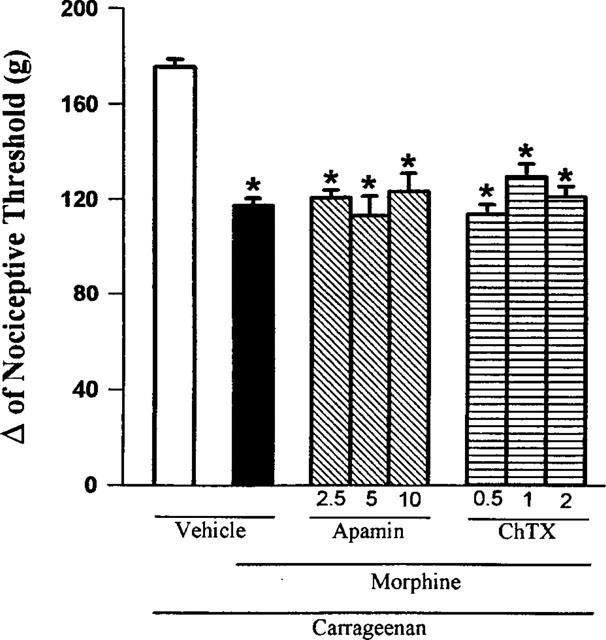
Effect of intraplantar administration of apamin and ChTX on the peripheral antinociception induced by morphine in hyperalgesic paws. Apamin and charybdotoxin (μg) were administered 45 min after morphine (100 μg). Each column represents the mean±s.e.mean (n=5). No statistically significant differences were detected between the groups treated with morphine+vehicle and morphine+apamin or ChTX in any case. *P<0.01 vs carrageenan+vehicle-injected control (Bonferroni's test).
Effect of 4-AP, TEA and cesium on morphine-induced antinociception
As shown in Figure 6, 4-AP (10, 50 and 100 μg paw−1), TEA (160, 320 and 640 μg paw−1) and Cesium (125, 250 and 500 μg paw−1) did not significantly modify the antinociception induced by morphine.
Figure 6.
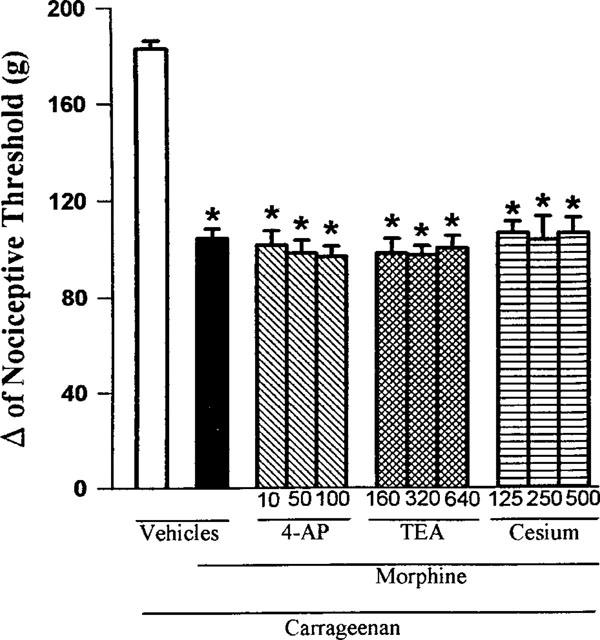
Effect of intraplantar administration of 4-AP, TEA and Cesium on the peripheral antinociception induced by morphine in hyperalgesic paws. 4-AP, TEA and Cesium (μg paw−1) were administered 45 min after morphine (100 μg paw−1). Each column represents the mean±s.e.mean (n=5). No statistically significant differences between the groups treated with carrageenan+morphine+vehicle and carrageenan+morphine+4-AP, TEA or Cesium were found in any case. *P<0.01 vs carrageenan+vehicle-injected control (Bonferroni's test).
Discussion
The present study demonstrated that the sulphonylureas glibenclamide and tolbutamide could reverse the peripheral antinociceptive effect induced by intraplantar administration of morphine in rats. Other K+ channel blockers such as apamin, ChTX, 4-AP, TEA, and Cesium did not exhibit any inhibitory effect. The doses of the ineffective blockers are compatible with those used to examine the involvement of potassium channels in the inhibitory prejunctional effect of morphine on peripheral sensory nerves in vivo (Yonehara & Takiuchi, 1997).
A growing number of both experimental and clinical studies demonstrated that locally administered opioids can produce pronounced analgesic effects by interacting with peripheral opioid receptors (Ferreira & Nakamura, 1979; Bentley, 1981; Smith, 1982; Stein et al., 1990). According to Stein (1993), μ opioid agonists are more potent than δ or κ agonists in inducing peripheral antinociceptive effects. Thus, we used morphine because it has been described as an agonist of μ opioid receptors (Zimmerman et al., 1987; Satoh & Minami, 1995). To exclude central effects of opioids many strategies can be used (Stein, 1993). In the present study, we used the strategy of evaluating the efficacy of ipsi- versus contralateral paw administration because the route and site of administration would be the same. Morphine at a dose of 100 μg was ineffective when administered into the contralateral paw, suggesting that at this dose morphine has a peripheral site of action in inflamed tissue. This effect seems to be specific and receptor mediated, since 50 μg of naloxone (when injected into the right paw, but not into the left), totally blocked the antinociceptive effect of morphine (result not shown).
Patch-clamp studies have shown that the sulphonylureas are selective inhibitors of ATP-sensitive K+ channels in pancreatic β-cells, cardiac myocytes and skeletal muscle cells (Edward & Weston, 1993). Indeed, the sensitivity to sulphonylureas, especially the potent glibenclamide, is commonly used to characterize the KATP channel (Babenko et al., 1998). However, glibenclamide also blocks an ATP-independent K+ current in a human neuroblastoma cell line (Reeve et al., 1992) and a delayed rectifier K+ current in neural and cardiac cells (Rosati et al., 1998). Blockade of these currents might mimic the effects expected from KATP blockade, thus potentially confusing the interpretation of the results. Delayed rectifying K+ channels are blocked by TEA, 4-AP and cesium (Hille, 1992) and if morphine was acting through the activation of these channels both sulphonylureas and these other blockers would revert this effect.
Raffa & Codd (1994) demonstrated that glibenclamide cannot bind directly to μ, δ or κ opioid receptors because this drug cannot alter the binding of specific agonists of these receptors. The effect of sulphonylureas against morphine-induced antinociception should not be interpreted as a counteraction by a possible increased excitability induced by the blockers, since these drugs do not cause any hyperalgesic effect when alone. Our results agree with those obtained by Nichols & Lederer (1991) who described glibenclamide as more potent in blocking ATP-sensitive K+ channels than tolbutamide in pancreatic β-cells and in smooth and cardiac muscle. In the present study, the maximum dose of glibenclamide (80 μg), by the same route, did not alter significantly the plasma glucose level (results not shown). Furthermore, all sulphonylureas tested to date, when administered by the intracerebroventricular or intrathecal route, dose-dependently antagonized the antinociception induced by systemic administration of morphine (Ocaña et al., 1990; 1995; Wild et al., 1991), suggesting that opening of ATP-sensitive K+ channels in neurones of the central nervous system underlies the antinociceptive effect of morphine.
In the present study apamin, a protein extracted from bee venom and a selective blocker of small conductance Ca2+-activated K+ channels (Romey et al., 1984), and ChTX, a toxin that blocks large conductance calcium-activated K+ channels (Miller et al., 1985), failed to antagonize the peripheral antinociceptive effect induced by morphine. Stretton et al. (1992) demonstrated that activation of charybdotoxin-sensitive, but not apamin-sensitive K+ channels may be the mechanism for the prejunctional modulation of sensory nerves in guinea-pig airways by μ-opioid agonists and suggest that the same type of K+ channel activation could be involved in the modulation of pain sensation by opiates. Our results disagree with this hypothesis and exclude the involvement of both types of Ca2+-activated K+ channels in the peripheral antinociception induced by morphine. Also, according to Garcia et al. (1997), ChTX is not specific for the large conductance Ca2+-activated K+ channels, blocking a number of other K+ channels.
Our results show that 4-AP, TEA, and Cesium administered intraplantarly had no significant effect on the peripheral antinociception induced by morphine.
These drugs block different types of K+ channels, including calcium-activated and voltage-dependent K+ channels, although they are not specific for any of them in particular (Cook & Quast, 1990). Ocaña et al. (1995) showed that 4-AP and TEA have no effect on the central antinociception induced by μ-opioid receptor agonists. Finally, North & Williams (1985) demonstrated that the K+ channels activated by μ-opioid agonists are not sensitive to 4-AP or TEA.
In conclusion, we have found that two different sulphonylureas, glibenclamide and tolbutamide, antagonized the peripheral antinociceptive effect induced by morphine in rats, suggesting that ATP-sensitive K+ channels play an important role in this effect. It is important to consider that other potassium channels might be involved, such as G-protein coupled channels. Since other K+ channel blockers failed to reverse this effect it may be inferred that other types of K+ channels such as large conductance Ca2+-activated, small conductance Ca2+-activated and voltage-dependent K+ channels appear not to be involved in the peripheral antinociceptive effect induced by morphine.
Acknowledgments
Research supported by Conselho Nacional de Pesquisa (CNPq, grants 000521719/95) and Fundação de Amparo à Pesquisa do estado de Minas Gerais (FAPEMIG, CBS-1800/95).
Abbreviations
- 4-AP
4-aminopyridine
- ATP
adenosine 5′-triphosphate
- ChTX
Charybdotoxin
- KATP
ATP-sensitive K+ channel
- L paw
left paw
- R paw
right paw
- TEA
Tetraethylammonium
References
- BABENKO A.P., AGUILAR-BRYAN L., BRYAN J. A view of Sur/KIR 6.X, KATP channels. Annu. Rev. Physiol. 1998;60:667–687. doi: 10.1146/annurev.physiol.60.1.667. [DOI] [PubMed] [Google Scholar]
- BENTLEY G.A., NEWTON S.H., STARR J. Evidence for an action of morphine and the enkephalins on sensory nerve endings in the mouse peritoneum. Br. J. Pharmacol. 1981;73:325–332. doi: 10.1111/j.1476-5381.1981.tb10425.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COOK N.S., QUAST U. Potassium channel pharmacology Potassium channels: Structure, classification, function, and therapeutic potential 1990Chichester: Ellis Horwood Limited; 181–255.ed. Cook. N.S. pp [Google Scholar]
- DUARTE I.D.G., DOSSANTOS I.R., LORENZETTI B.B., FERREIRA S.H. Analgesia by direct antagonism of nociceptor sensitisation involves the arginine-nitric oxide-cGMP pathway. Eur. J. Pharmacol. 1992;217:225–227. doi: 10.1016/0014-2999(92)90881-4. [DOI] [PubMed] [Google Scholar]
- EDWARDS G., WESTON A.H. The pharmacology of ATP-sensitive potassium channels. Annu. Rev. Pharmacol. Toxicol. 1993;33:597–637. doi: 10.1146/annurev.pa.33.040193.003121. [DOI] [PubMed] [Google Scholar]
- FERREIRA S.H., DUARTE I.D.G., LORENZETTI B.B. The molecular mechanism of action of peripheral morphine analgesia: stimulation of the cGMP system via nitric oxide release. Eur. J. Pharmacol. 1991;201:121–122. doi: 10.1016/0014-2999(91)90333-l. [DOI] [PubMed] [Google Scholar]
- FERREIRA S.H., NAKAMURA M. Prostaglandin hyperalgesia: The peripheral analgesic activity of morphine, enkephalins and opioid antagonists. Prostaglandins. 1979;18:191–200. doi: 10.1016/0090-6980(79)90104-7. [DOI] [PubMed] [Google Scholar]
- GARCIA M.L., HANNER M., KNAUS H.G., KOCH R., SCHMALHOFER W., SLAUGHTER R.S., KACZOROWSKI G.J. Pharmacology of potassium channels. Advances Pharmacol. 1997;39:425–471. doi: 10.1016/s1054-3589(08)60078-2. [DOI] [PubMed] [Google Scholar]
- HALLIWELL J.V. K+ channels in the central nervous system Potassium channels: Structure, classification, function, and therapeutic potential 1990Chichester: Ellis Horwood Limited; 348–381.ed. Cook. N.S. pp [Google Scholar]
- HERZ A., TESCHEMACHER H.J. Activities and sites of antinociceptive action of morphine-like analgesics and kinetics of distribution following intravenous, intracerebral, and intraventricular application. Adv. Drug. Res. 1971;6:79–119. [Google Scholar]
- HILLE B. Ionic channels of excitable membranes. Sinauer Associates Inc; 1992. Potassium channels and chloride channels; pp. 115–139. [Google Scholar]
- LEVINE J.D., TAIWO Y.O. Involvement of the mu-opiate receptor in peripheral analgesia. Neuroscience. 1989;32:571–575. doi: 10.1016/0306-4522(89)90279-0. [DOI] [PubMed] [Google Scholar]
- MILLER C., MOCZYDLOWSKI E., LATORRE R., PHILLIPS M. Charybdotoxin, a protein inhibitor of single Ca2+-activated K+ channels from mammalian skeletal muscle. Nature. 1985;313:316–318. doi: 10.1038/313316a0. [DOI] [PubMed] [Google Scholar]
- NICHOLS C.G., LEDERER W.J. Adenosine triphosphate-sensitive potassium channels in the cardiovascular system. Am. J. Physiol. 1991;261:H1675–H1686. doi: 10.1152/ajpheart.1991.261.6.H1675. [DOI] [PubMed] [Google Scholar]
- NORTH R.A. Drug receptors and the inhibition of nerve cells. Br. J. Pharmacol. 1989;98:13–28. doi: 10.1111/j.1476-5381.1989.tb16855.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NORTH R.A., WILLIAMS J.T. On the potassium conductance increased by opioids in rat locus coeruleus neurones. J. Physiol. 1985;364:265–280. doi: 10.1113/jphysiol.1985.sp015743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OCAÑA M., BAEYENS J.M. Differential effects of K+ channel blockers on antinociception induced by α2-adrenoceptor, GABAB and κ-opioid receptor agonists. Br. J. Pharmacol. 1993;110:1049–1054. doi: 10.1111/j.1476-5381.1993.tb13919.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OCAÑA M., BARRIOS M., BAEYENS J.M. Cromakalim differentially enhances antinociception induced by agonists of alpha2 adrenoceptors, γ-aminobutyric acid, mu and kappa opioid receptors. J. Pharmacol. Exp. Ther. 1996;276:1136–1142. [PubMed] [Google Scholar]
- OCAÑA M., DEL POZO E., BARRIOS M., BAEYENS J.M. Subgroups among μ-opioid receptor agonists distinguished by ATP-sensitive K+ channel-acting drugs. Br. J. Pharmacol. 1995;114:1296–1302. doi: 10.1111/j.1476-5381.1995.tb13346.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OCAÑA M., DEL POZO E., BARRIOS M., ROBLES L.I., BAEYENS J.M. An ATP-dependent potassium channel blocker antagonises morphine analgesia. Eur. J. Pharmacol. 1990;186:377–378. doi: 10.1016/0014-2999(90)90466-j. [DOI] [PubMed] [Google Scholar]
- RAFFA R.B., CODD E.E. Lack of glibenclamide or TEA affinity for opioid receptors: further evidence for in vivo modulation of antinociception at K+ channels. Brain Res. 1994;650:146–148. doi: 10.1016/0006-8993(94)90217-8. [DOI] [PubMed] [Google Scholar]
- RANDALL L.D., SELITTO J.J. A method for measurement of analgesic activity on inflamed tissues. Arch. Int. Pharmacodyn. 1957;113:233–249. [PubMed] [Google Scholar]
- REEVE H.L. , VAUGHAN P.F.T., PEERS C. Glibenclamide inhibits a voltage-gated K+ current in the human neuroblastoma cell line SH-SY5Y. Neurosci. Lett. 1992;135:37–40. doi: 10.1016/0304-3940(92)90130-y. [DOI] [PubMed] [Google Scholar]
- ROANE D.S., BOYD N.E. Reduction of food intake and morphine analgesia by central glibenclamide. Pharmacol. Biochem. Behav. 1993;46:205–207. doi: 10.1016/0091-3057(93)90341-p. [DOI] [PubMed] [Google Scholar]
- ROMEY G., HUGHES M., SCHMID-ANTOMARCHI H., LAZDUNSKI M. Apamin: a specific toxin to study a class of Ca2+-dependent K+ channels. J. Physiol., Paris. 1984;79:259–264. [PubMed] [Google Scholar]
- ROSATI B., ROCHETTI M., ZAZA A., WANKE E. Sulfonylureas blockade of neural and cardiac HERG channels. FEBS Lett. 1998;440:125–130. doi: 10.1016/s0014-5793(98)01444-6. [DOI] [PubMed] [Google Scholar]
- SATOH M., MINAMI M. Molecular pharmacology of the opioid receptors. Pharmacol. Ther. 1995;68:343–364. doi: 10.1016/0163-7258(95)02011-x. [DOI] [PubMed] [Google Scholar]
- SMITH T.W., BUCHAN P., PARSONS D.N., WILKINSON S. Peripheral antinociceptive effects of N-methyl morphine. Life Sci. 1982;31:1205–1208. doi: 10.1016/0024-3205(82)90343-5. [DOI] [PubMed] [Google Scholar]
- STEIN C. Peripheral mechanisms of opioid analgesia. Anesth. Analg. 1993;76:182–191. doi: 10.1213/00000539-199301000-00031. [DOI] [PubMed] [Google Scholar]
- STEIN C., HASSAN A.H.S., PRZEWLOCKI R., GRAMSCH C., PETER K., HERZ A. Opioids from immunocytes interact with receptors on sensory nerves to inhibit nociception in inflammation. Proc. Natl. Acad. Sci. U.S.A. 1990;87:5935–5939. doi: 10.1073/pnas.87.15.5935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STRETTON D., MIURA M., BELVISI M.G., BARNES P.J. Calcium-activated potassium channels mediate prejunctional inhibition of peripheral sensory nerves. Proc. Natl. Acad. Sci. U.S.A. 1992;89:1325–1329. doi: 10.1073/pnas.89.4.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VERGONI A.V., SCARANO A., BERTOLINI A. Pinacidil potentiates morphine analgesia. Life Sci. 1992;50:135–138. doi: 10.1016/0024-3205(92)90467-4. [DOI] [PubMed] [Google Scholar]
- WILD K.D., VANDERAH T., MOSBERG H.I., PORRECA F. Opioid δ receptor subtypes are associated with different potassium channels. Eur. J. Pharmacol. 1991;193:135–136. doi: 10.1016/0014-2999(91)90215-c. [DOI] [PubMed] [Google Scholar]
- YAKSH T.L., RUDY T.A. Analgesia mediated by a direct spinal action of narcotics. Science. 1976;192:1357–1358. doi: 10.1126/science.1273597. [DOI] [PubMed] [Google Scholar]
- YONEHARA N., TAKIUCHI S. Involvement of calcium-activated potassium channels in the inhibitory prejunctional effect of morphine on peripheral sensory nerves. Regulat. Peptides. 1997;68:147–153. doi: 10.1016/s0167-0115(96)02102-7. [DOI] [PubMed] [Google Scholar]
- ZIMMERMAN D.M., LEANDER J.D., REEL J.K., HYNES M.D. Use of β-funaltrexamine to determine mu opioid receptor involvement in the analgesic activity of various opioid ligands. J. Pharmacol. Exp. Ther. 1987;241:374–378. [PubMed] [Google Scholar]