

Abstract
Recent studies demonstrated that inhibition or genetic inactivation of the enzyme poly (ADP-ribose) polymerase (PARP) is beneficial in myocardial reperfusion injury. PARP activation in the reperfused myocardium has been assumed, but not directly demonstrated. Furthermore, the issue whether pharmacological PARP inhibition affords long-term functional benefit in the reperfused myocardium has not been explored. These questions were addressed in the present study.
In a rat model of myocardial ischemia (1 h) and reperfusion (up to 24 h), there was a marked and significant activation of PARP in the ischemic borderzone, as determined by poly(ADP-ribose) (PAR) immunohistochemistry. PAR localized to the nuclei of myocytes and infiltrating mononuclear cells. In the core of the infarction, necrotic tissues and diffuse PAR staining were observed. PARP activation remained markedly detectable 24 h after reperfusion. The PARP inhibitor 3-aminobenzamide (20 mg kg−1 intraperitoneally 10 min before reperfusion, and every 2 h thereafter for 6 h) markedly reduced the activation of the enzyme in myocytes.
3-aminobenzamide significantly protected against myocardial morphological and functional alterations at 24 h post-reperfusion. Notably, infarct size was reduced, circulating creatine kinase activity was attenuated, and myocardial contractility (dP dt−1) was restored by 3-aminobenzamide.
Our results demonstrate a significant and prolonged activation of PARP in the reperfused myocardium, localizing to the necrotic area and the ischaemic borderzone. Furthermore, the studies demonstrate that PARP inhibition affords long-term beneficial morphological and functional effects in the reperfused myocardium. These data strengthen the notion that pharmacological PARP inhibition is a viable novel experimental approach for protection against myocardial reperfusion injury.
Keywords: Poly(ADP-ribose) polymerase (PARP), ischaemia, reperfusion, heart, 3-aminobenzamide
Introduction
Poly (ADP-ribose) polymerase (PARP), also known as poly (ADP-ribose) synthetase (PARS) is an abundant nuclear enzyme of eukaryotic cells. DNA single-strand breaks have been shown to induce the activation of PARP. Activated PARP catalyses an energy consuming cycle by transferring ADP ribose units to nuclear proteins. The results of this process are a rapid depletion of the intracellular NAD+ and ATP pools which slows the rate of glycolysis and mitochondrial respiration leading to cell necrosis (see: Szabó et al., 1996; 1998; Le Rhun et al., 1998; Ha & Snyder, 1999; Szabó, 2000).
PARP has recently been proposed to play a role in the pathogenesis of myocardial reoxygenation injury. Myocardial reperfusion injury in vitro or in vivo have been shown to be ameliorated by pharmacological inhibitors of PARP (Gilad et al., 1997; Thiemermann et al., 1997; Zingarelli et al., 1997; Bowes et al., 1998a, 1998b; Docherty et al., 1999; Grupp et al., 1999), and in genetically engineered mice, which lack functional PARP enzyme (PARP−/−) (Zingarelli et al., 1998; Pieper et al., 2000; Yang et al., 2000).
Although the activation of PARP in the reperfused myocardium has been assumed in previous studies, the phenomenon has not yet been directly demonstrated, nor has the localization of PARP activation been defined or its time-course delineated. The issue whether or not pharmacological PARP inhibition affords long-term functional beneficial effects in the reperfused myocardium in vivo has not yet been addressed either. The aim of the present study was to address these questions. Addressing these issues has obvious implications for designing novel, innovative approaches for the experimental therapy of heart attacks.
Methods
The investigation conformed to the Guide for the Care and Use of Laboratory Animals published by U.S. National Institutes of Health (NIH Publication No. 85-23, revised 1985) and was performed with the approval of the local Institutional Animal Care and Use Committee.
Surgical preparation
Male Wistar rats (Charles River) (111 animals in total) weighing 300–330 g were anaesthetized with intraperitoneal (i.p.) thiopentone sodium (60 mg kg−1). The animals were either tracheotomized (2 h reperfusion, protocol 1, see below) or intubated (23 h reperfusion, protocol 2, see below) under direct vision with polyethylene tubing (PE 205) and placed on mechanical ventilation (75 strokes min−1, tidal volume (Vt)=8 ml kg−1, inspired fraction of oxygen (FiO2)=0.5) using a Harvard model 683 rodent respirator (Harvard Apparatus, Holliston, MA, U.S.A.). A positive end-expiratory pressure of 5 cmH2O was applied to minimize atelectasis. The animals were placed on controlled heating pads, and core temperature measured via a rectal probe was maintained at 37°C. In the 23 h reperfusion protocol (see below), the animals were given an analgesic (buprenorphine 0.25 mg kg−1 subcutaneously) just prior to surgery, and a second identical dose 6 h later.
Myocardial infarction model
Coronary artery ligation was performed as previously described (Maclean et al., 1976; Horstick et al., 1999). Following a left thoracotomy at the fourth intercostal space, the pericardium was opened and the heart briefly exteriorized. The left anterior descending (LAD) coronary artery was then ligated by an intramural 6.0 silk suture. Immediate palor of the left ventricular free wall was indicative of successful occlusion. It is worth mentioning that the procedure of heart exteriorization may by itself induce myocardial ischaemia, that could result in a phenomenon of preconditioning. As previously reported (Horstick et al., 1999) the time required to complete the procedure is critical in determining the occurrence of such alterations, and it is advised that the duration of the surgical procedure does not exceed 120 s. In our laboratory, the procedure is realized in 90 s, and the time elapsed from the pericardiotomy to the ligation of the LAD and repositioning of the heart is less than 20 s.
One hour of myocardial ischaemia was followed by 2 or 23 h of reperfusion, depending on the experimental protocol as detailed below. In the animals used in the 23 h reperfusion protocol, the chest wall was closed in layers using 4.0 silk suture, and the animals were rapidly weaned from the ventilator and placed in cages with free access to food and water.
Measurements
Determination of left ventricular function
Analysis of left ventricular performance was done in rats exposed to 24 h of myocardial ischaemia-reperfusion. The animals were re-anaesthetized with thiopentone sodium (60 mg kg−1, i.p), tracheotomized and mechanically ventilated (Vt=8 ml kg−1, 75 strokes min−1, FiO2=0.21). A microtip catheter transducer (Millar Instruments, Houston, TX, U.S.A.) was inserted into the right carotid artery and advanced into the left ventricle under pressure control. The pressure signal was continuously recorded using a MacLab A/D converter (AD Instruments, Mountain View, CA, U.S.A.), and stored and displayed on an Apple Macintosh personal computer. The left ventricular systolic and end-diastolic pressures were measured and the maximal slope of systolic pressure increment (dP dt−1 max), an index of contractility, was calculated. After these measurements, the catheter was pulled back into the aorta for the measurement of arterial blood pressure.
Determination of myocardial infarct size
Area at risk (AAR) and infarct size were determined using the triphenyl tetrazolium chloride (TTC)-Evans blue technique (Zingarelli et al., 1997). At the conclusion of the experiment, the animals were killed by exsanguination. The chest was opened and the ascending aorta cannulated in situ with polyethylene tubing (PE 90), and the left ventricle was retrogradely lavaged with 5 ml of 10 mM phosphate buffered saline (PBS) at 37°C. Then, the ligature around the LAD was retightened and the heart perfused through the aortic cannula with 0.3 ml of 2% Evans blue. The area at risk was therefore determined by negative staining. The hearts were then immediately washed with water, trimmed off right ventricle and atria, and frozen at −20°C for 20 min followed by sectioning into 6–7 transverse slices. The blue (i.e. perfused) and non-blue (i.e. AAR) portions of left ventricle were then separated. The sections of ischaemic left ventricle were then incubated in a 2% solution of TTC stain in 10 mM PBS (pH 7.4) at 37°C for 20 min. The non-infarcted zone was coloured brick red due to the formation of a precipitate resulting from the reaction of TTC with dehydrogenase enzymes. Loss of these enzymes from infarcted myocardium prevents formation of the precipitate; thus the infarcted area within the risk region remained pale yellow (i.e., necrotic area). The myocardial slices were dissected using microsurgical methods and weighed by an investigator (E.S) blinded to the treatment administered. The three colours (white, red and blue) represent the infarction area, non-infarcted ischaemic area, and non-ischaemic area, respectively. The non-infarcted ischaemic area and the necrotic area together yield the area of risk. The normal, area at risk and infarcted areas were then expressed as percentage values according to conventional methods (area at risk/left ventricle and infarcted area/area at risk).
Determination of serum creatine phosphokinase activity (MB fraction)
Creatine phosphokinase (myocardial specific MB fraction, CKMB) activity was measured in the serum using a commercial kit obtained from Sigma (St Louis, MO, U.S.A.) according to the manufacturer's instructions.
Immunohistochemical analysis of PARP activation in the reperfused myocardium
In a subset of animals (see below, experimental protocols), hearts were harvested at the conclusion of the experiments and paraffin-embedded for PAR immunohistochemistry, which was performed as previously described (Liaudet et al., 2000). Paraffin sections (3 μm) were deparaffinized in xylene and rehydrated in decreasing concentrations (100, 95 and 70%) of ethanol followed by a 10 min incubation in PBS (pH7.4). Sections were treated with 0.3% hydrogen peroxide for 15 min to block endogenous peroxidase activity and then rinsed briefly in 10 mM PBS. Non-specific binding was blocked by incubating the slides for 1 h in PBS containing 2% horse serum. Mouse monoclonal anti-poly(ADP-ribose) antibody (Alexis, San Diego, CA, U.S.A.) and isotype-matched control antibody were applied in a dilution of 1 : 100 for 2 h at room temperature. Following extensive washing (5×5 min) with PBS, immunoreactivity was detected with a biotinylated horse anti-mouse secondary antibody and the avidin–biotin–peroxidase complex (ABC) both supplied in the Vector Elite kit (Vector Laboratories, Burlingame, CA, U.S.A.). Colour was developed using Ni-DAB substrate (95 mg diaminobenzidine, 1.6 g NaCl, 2 g nickel sulphate in 200 ml 0.1 M acetate buffer). Sections were then counterstained with nuclear fast red, dehydrated and mounted in Permount. Photomicrographs were taken with a Zeiss Axiolab microscope equipped with a Fuji HC-300C digital camera.
Experimental protocols
Protocol 1 (1 h ischemia/2 h reperfusion)
Forty rats were used in this protocol. The animals were assigned to receive an i.p. injection of 3-aminobenzamide (20 mg kg−1; 3-AB group) or vehicle only (isotonic saline, 1 ml; control group), given 10 min before reperfusion and repeated 2 h later. The perioperative mortality was 30% (12 animals) and was not different between groups (see results). Among the surviving animals, 10 rats in each group were used for the determination of infarct size, and four animals in each group were used for immunohistochemical studies.
Protocol 2 (1 h ischemia/23 h reperfusion)
Seventy-one rats were used in this protocol. The animals received an i.p. injection of either 3-AB (20 mg kg−1; 3-AB group) or vehicle only (isotonic saline, 1 ml; control group), given 10 min before reperfusion and repeated 2, 4 and 6 h later. A third group of rats had the same surgical preparation as the other animals, except that the suture around the LAD was not ligated (sham group). The total mortality was 44% (31 animals), and was comparable between 3-AB and control groups, while no animal died in the sham group (see Results). All the surviving animals (n=15 animals in the 3-AB and control groups and n=10 in the sham group) were used for the determination of haemodynamic parameters. A subset of animals (n=11 rats in the 3-AB and control groups) was also used for the determination of infarct size, and another subset (four animals in each group) was also used for immunohistochemical studies.
Materials
3-aminobenzamide and all other chemicals were purchased from Sigma chemicals (St-Louis, MO, U.S.A.), unless otherwise stated.
Data analysis
All values in the figures and text are expressed as mean±standard errors of the mean (s.e.mean) of n observations, where n represents the number of animals studied. The area at risk and infarct size were compared between 3-AB and control groups of rats exposed to LAD occlusion by unpaired t-test. The data of CKMB, as well as all the haemodynamic data were compared by analysis of variance between the sham, control and 3-AB groups of rats. When the relevant F-value was significant at the 5% level, further pair-wise comparisons were made using the Bonferroni multiple-comparison procedure. A value of P<0.05 was considered significant.
Results
Mortality
Twelve animals died in protocol 1, from which seven died during ischaemia before any treatment and five died after reperfusion, including three control rats, and two rats treated with 3-AB. In protocol 2, no death was observed in the 10 sham rats. Thirty-one deaths occurred in the 61 animals exposed to LAD occlusion. Twelve animals died before reperfusion and any treatment. Nineteen rats died during the 23 h of reperfusion, including 11 control animals and eight animals treated with 3-AB (P=NS, chi-square test).
Patterns of myocardial PARP activation in reperfusion injury: effect of 3-aminobenzamide
Whole hearts sections from representative animals in both 3-AB and control groups are shown in Figure 1a. A massive staining indicative of PARP activation was evident in the left ventricular free wall of control animals (left panel), both after 2 h (top) and 23 h (bottom) reperfusion. The intensity of staining was markedly reduced in the hearts from rats treated with 3-AB, independently from the duration of reperfusion (right panel).
Figure 1.
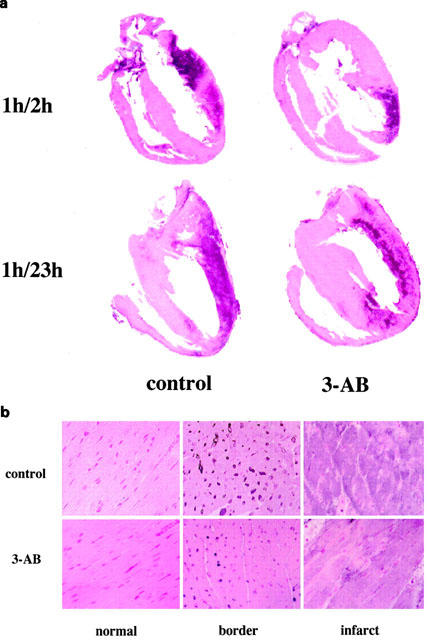
Immunohistochemical localization of PARP activation. Poly(ADP-ribose) formation, an indicator of PARP activation, as determined in whole heart sections (a) from rats exposed to 1 h ischaemia and either 2 or 23 h reperfusion. In both conditions, a massive staining is evident in the left ventricular free wall of control animals. The staining was clearly reduced in rats treated with 3-aminobenzamide. Higher magnification (×40) shows that PARP activation was mainly located in the nuclei of myocytes in the peri-infarction (border) zone. In the infarcted myocardium, severe architectural alterations are coexisting with a more diffuse pattern of PAR staining. Treatment with 3-AB attenuated PARP activation in the border zone, while limiting PAR formation in the infarcted myocardium. Immunohistochemical pictures represent n⩾4 sections per group.
Figure 1b shows three distinctive histological patterns noted in hearts from rats exposed to 2 h reperfusion (control group, top sections; 3-AB group, bottom sections). Sections from the ventricular septum (left panel), illustrate the normal myocardial architecture, with absence of PAR staining. The peri-infarction (border) zone (middle panel) shows an intense PAR staining, mostly located in the nuclei of cardiac myocytes. A more diffuse PAR staining is apparent in the necrotic area (right panel), where the myocardial architecture is severely altered. Treatment with 3-AB suppressed the nuclear staining in the peri-infarction zone and also attenuated the staining in the infarcted area. Similar results were obtained in hearts harvested from rats exposed to 23 h myocardial reperfusion (not shown).
Effect of 3-aminobenzamide on infarct size and serum creatine–phosphokinase activity
Area at risk and infarct size measured in the short-term experiments (protocol 1) are illustrated in Figure 2a. AAR was not statistically significantly different between controls (42.6±3.9%) and 3-AB-treated rats (45.1±3.1%). In contrast, infarct size was significantly reduced in rats treated with 3-AB (in per cent of AAR: 52.1±4.9% vs 67.2±3.5%, P<0.05). Similar observations were made in rats exposed to the long-term experiments (protocol 2), as shown in Figure 2b. AAR was comparable between control and 3-AB groups, while the infarct size was reduced in 3-AB animals (in per cent of AAR: 47.1±5.6% vs 62.0±3.8%, P<0.05). In agreement with these findings, the increase in serum CKMB activity observed in the control group was significantly reduced by 3-AB treatment (Sham: 246±27 UI l−1; control: 597±60 UI L−1, P<0.05 vs sham; 3-AB: 381±47 UI l−1, P<0.05 vs control, P=NS vs sham), as depicted in Figure 3.
Figure 2.
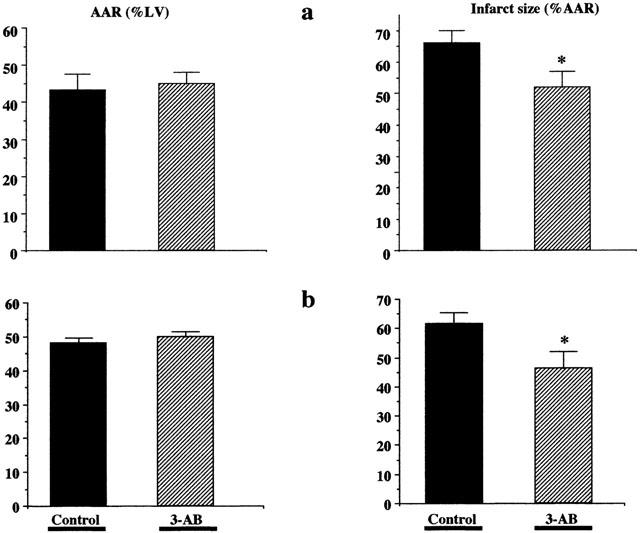
3-aminobenzamide reduces myocardial infarct size. Rats were exposed to 1 h LAD occlusion followed by 2 h (a) or 23 h (b) reperfusion. Area at risk (AAR) and infarct size were determined using the triphenyl-tetrazolium chloride (TTC)-Evans blue technique. Treatment of the animals with 3-AB (20 mg kg−1 i.p. 10 min before reperfusion, 2 h later, and in the 24 h group every 2 h thereafter for 6 h) significantly reduced infarct size (expressed as a percentage of the area at risk), both after 2 or 23 h reperfusion. *P<0.05 control vs 3-AB (unpaired t-test; n=10–11 animals in each group).
Figure 3.
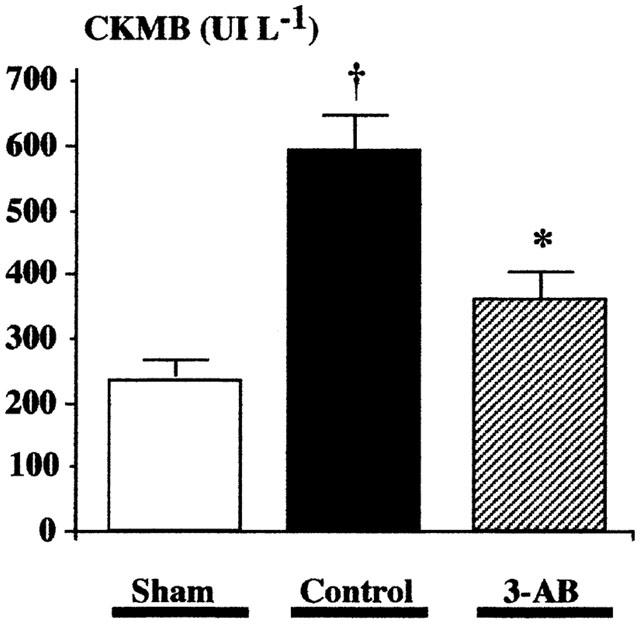
Effect of 3-aminobenzamide on serum CKMB after myocardial infarction. Serum creatine-phosphokinase (myocardial specific MB fraction, CKMB) was measured in sham rats (n=10) as well as in rats exposed to 1 h LAD occlusion and 23 h reperfusion. A significant increase in CKMB activity was observed after myocardial infarction in control rats (n=15), which was significantly reduced by treatment with 3-AB (20 mg kg−1 i.p. 10 min before reperfusion and every 2 h thereafter for 6 h, n=15). †P<0.05 vs sham; *P<0.05 control vs 3-AB (ANOVA followed by Bonferroni).
Effect of 3-aminobenzamide on haemodynamic parameters following myocardial ischaemia-reperfusion
In the control group of rats, myocardial contractility (assessed by dP dt−1 max) was significantly reduced when compared to sham animals (3670±278 mm Hg s−1 vs 5800±211 mm Hg s−1). Treatment with 3-AB suppressed this systolic dysfunction (5125±150 mm Hg s−1; P<0.05 vs controls; P=NS vs sham). Furthermore, the significant decreases in left ventricular maximal systolic pressure and mean arterial blood pressure complicating the course of myocardial infarction in control rats were also prevented in animals treated with 3-AB. Finally, a moderate increase in left ventricular end-diastolic pressure was noted after myocardial infarction in both treatment groups, which tended to be less pronounced in the 3-AB-treated animals (but this difference did not reach statistical significance) (Figure 4).
Figure 4.
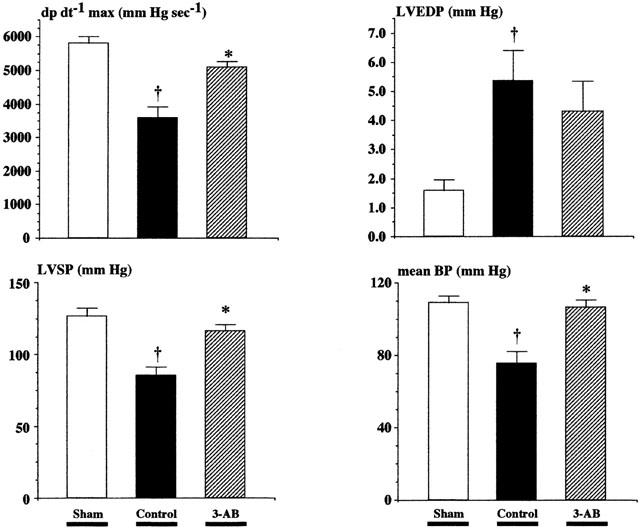
Effect of 3-AB on haemodynamic parameters after myocardial infarction. Control rats (n=15) exposed to 1 h LAD occlusion and 23 h reperfusion had a severe reduction in dP dt−1 max, left ventricular systolic pressure (LVSP) and mean blood pressure (mean BP), as well as a significant increase in left ventricular end-diastolic pressure (LVEDP) when compared to sham-operated animals (n=10). Treatment with 3-AB (20 mg kg−1 i.p. 10 min before reperfusion and every 2 h thereafter for 6 h, n=15) significantly reduced the left ventricular systolic dysfunction, restored normal blood pressure, and blunted the increase in LVEDP. †P<0.05 vs sham *P<0.05 control vs 3-AB (ANOVA followed by Bonferroni).
Discussion
Recent studies demonstrated that pharmacological inhibition or genetic inactivation of PARP attenuates myocardial necrosis in the acute and delayed stages of myocardial infarction (see Introduction), and that several mechanisms account for these cardioprotective actions. Experimental studies using isolated heart preparations (Thiemermann et al., 1997; Grupp et al., 1999; Pieper et al., 2000; Szabados et al., 2000) have shown that this protection was related to a reduced catabolism of myocardial NAD+ as well as preservation of the myocardial ATP stores. In addition, in vivo studies have indicated that these beneficial effects were associated with a suppression of neutrophil infiltration in the absence of functional PARP (Zingarelli et al., 1998; Yang et al., 2000), as well as with a down-regulation of inflammatory mediator production (Yang et al., 2000).
The current study is the first one to directly demonstrate that PARP is activated in the reperfused myocardium. The time-course of PARP activation is rather prolonged: it is present at 2 h after the start of reperfusion, and is still present as late as 24 h after reperfusion. This delayed pattern of PARP activation may be related to the continuing presence of free radical and oxidant production in the reperfused myocardium (which would, therefore, maintain a continuing trigger for DNA single-strand breakage). Alternatively, it is conceivable that a massive, early DNA single-strand breakage, which remains unrepaired for prolonged periods of time, is responsible for the prolonged pattern of PARP activation. The fact that PARP activation is a continuing process which is present even in delayed myocardial reperfusion would suggest that it is worthwhile to test, in future studies, whether or not delayed administration of PARP inhibitors (e.g. several hours after the start of reperfusion) would continue to exert cardioprotective effects.
The site of the most pronounced PARP activation is the area of necrosis and peri-infarct zone (latter zone is likely to coincide with the area at risk). Most of the staining was seen in cardiac myocytes, indicating that the heart tissue itself, rather than the infiltrating mononuclear cells, is the main site of PARP activation (and related pathophysiological processes). The reduction in PAR staining in the 3-aminobenzamide-treated animals directly demonstrates that the dosing regimen of the PARP inhibitor is sufficient to suppress PARP activation in the heart. A more diffuse staining pattern can be seen in the area of necrosis: this pattern is likely to reflect the fact that the cellular content (and thus the poly-ADP-ribosylated proteins, which were initially localized in the nucleus) are now more-or-less uniformly distributed in the necrotic area, due to myocardial necrosis and the associated breakdown of the cell membrane permeability. Because PARP activation triggers cellular necrosis due to cellular energetic collapse (Virág et al., 1998; Ha & Snyder, 1999), we believe that the primary mode of PARP inhibitors' protective effects is related to a direct inhibition of myocyte necrosis in myocardial reperfusion. The peri-infarct zone (AAR), which contains viable cells, in which PARP is markedly activated, is the likely site of the PARP inhibitors' beneficial effects in vivo.
In a prior study in thymocytes exposed to cytotoxic oxidants, the protection against cell necrosis by PARP inhibitors was also associated with a significant increase in the portion of the apoptotic cells (Virág et al., 1998). Further studies will be required to determine whether PARP inhibition results in a similar tendency in the present in vivo model. The frequently employed TUNEL assay (terminal deoxynucleotidyl transferase dUTP nick-end labelling) essentially only recognizes free DNA ends, and thus will detect both DNA single-strand breakage (i.e. necrotic or pre-necrotic cells, where PARP can become activated) and DNA double-strand breaks (i.e. apoptotic cells). Thus, TUNEL staining, alone, will not be helpful in distinguishing between the pre-necrotic and the apoptotic populations of cells.
The second novel conclusion of the current study is that the reduction of myocardial necrosis by PARP inhibition translates into functional improvement of the heart, especially when considering left ventricular contractility, in a chronic therapeutic regimen. Myocardial infarction was, indeed, associated with a significant systolic dysfunction (reduced dP dt−1), that was suppressed by 3-AB. In contrast, the moderate diastolic dysfunction complicating myocardial infarction (increased left ventricular end-diastolic pressure) was not significantly influenced by 3-AB. This finding is somewhat expected from prior short-term, functional in vitro studies, where contractile function was improved in hypoxic/reoxygenated Langendorff hearts in the presence of PARP inhibitors (e.g. Docherty et al., 1999; Grupp et al., 1999). Nevertheless, the current findings are reassuring, in the context of designing potential future therapeutic approaches with novel pharmacological inhibitors of PARP.
It is noteworthy that the beneficial effects of 3-AB were not associated with a significant improvement in survival after myocardial infarction. Since most of the deaths occurring in this setting are related to the development of ventricular fibrillation, this suggests that 3-AB, in spite of its positive influence on myocardial morphology and function, did not reduce electrical instability and the incidence of malignant ventricular tachyarrhythmias. However, it should be pointed out that the model used in the present study (1 h ischaemia) was particularly severe. We can not rule out that a shorter ischaemic period might have revealed stronger effects of the drug, notably regarding survival. Further studies will be necessary to address this issue as well as the potential effects of PARP inhibitors on post-infarction ventricular arrhythmias.
Taken together, the current work (1) directly demonstrates the prolonged and confined activation of PARP in the reperfused myocardium and (2) demonstrates that pharmacological inhibition of PARP provides both morphological and functional improvements in a rat model of myocardial reperfusion injury. These observations, coupled with the facts that (3) in the absence of functional PARP there is a down-regulation of pro-inflammatory mediators and adhesion molecules, and (4) there is suppression of neutrophil infiltration into the reperfused heart in the absence of functional PARP, support the view that potent and selective PARP inhibitors – which can be viewed as agents which interrupt several, interrelated pathways of myocardial reperfusion injury – should be further explored in the context of experimental therapy of heart attacks.
Acknowledgments
This work was supported by grants from the National Institutes of Health (R01HL59266 and R01GM60915) to Dr Szabó. Dr Liaudet is on leave from the Critical Care Division, Department of Internal Medicine, University Hospital, Lausanne, Switzerland and is supported by a grant from the ADUMED foundation, Switzerland.
Abbreviations
- 3-AB
3-aminobenzamide
- AAR
area at risk
- ADP
adenosine diphosphate
- ATP
adenosine triphosphate
- CK
creatine kinase
- DNA
desoxyribonucleic acid
- LAD
left anterior descending coronary artery
- NAD
nicotinamide adenine dinucleotide
- PAR
poly(ADP-ribose)
- PARP
poly(ADP-ribose) polymerase
- TTC
triphenyl-tetrazolium chloride
- TUNEL
terminal deoxyribonucleotidyl transferase-mediated dUTP nick-end labelling
References
- BOWES J., PIPER J., THIEMERMANN C. Inhibitors of the activity of poly (ADP-ribose) synthetase reduce the cell death caused by hydrogen peroxide in human cardiac myoblasts. Br. J. Pharmacol. 1998a;124:1760–1766. doi: 10.1038/sj.bjp.0702009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOWES J., RUETTEN H., MARTORANA P.A., STOCKHAUSEN H., THIEMERMANN C. Reduction of myocardial reperfusion injury by an inhibitor of poly (ADP-ribose) synthetase in the pig. Eur. J. Pharmacol. 1998b;359:143–150. doi: 10.1016/s0014-2999(98)00638-4. [DOI] [PubMed] [Google Scholar]
- DOCHERTY J.C., KUZIO B., SILVESTER J.A., BOWES J., THIEMERMANN C. An inhibitor of poly (ADP-ribose) synthetase activity reduces contractile dysfunction and preserves high energy phosphate levels during reperfusion of the ischaemic rat heart. Br. J. Pharmacol. 1999;127:1518–1524. doi: 10.1038/sj.bjp.0702705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GILAD E., ZINGARELLI B., SALZMAN A.L., SZABÓ C. Protection by inhibition of poly (ADP-ribose) synthetase against oxidant injury in cardiac myoblasts in vitro. J. Mol. Cell. Cardiol. 1997;29:2585–2597. doi: 10.1006/jmcc.1997.0496. [DOI] [PubMed] [Google Scholar]
- GRUPP I.L., JACKSON T.M., HAKE P., GRUPP G., SZABÓ C. Protection against hypoxia-reoxygenation in the absence of poly (ADP-ribose) synthetase in isolated working hearts. J. Mol. Cell. Cardiol. 1999;31:297–303. doi: 10.1006/jmcc.1998.0864. [DOI] [PubMed] [Google Scholar]
- HA H.C., SNYDER S.H. Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc. Natl. Acad. Sci. U.S.A. 1999;96:13978–13982. doi: 10.1073/pnas.96.24.13978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HORSTICK G., BERG O., HEIMANN A., DARIUS H., LEHR H.A., BHAKDI S., KEMPSKI O., MEYER J. Surgical procedure affects physiological parameters in rat myocardial ischemia: need for mechanical ventilation. Am. J. Physiol. 1999;276:H472–H479. doi: 10.1152/ajpheart.1999.276.2.H472. [DOI] [PubMed] [Google Scholar]
- LE RHUN Y.L., KIRKLAND J.B., SHAH G.M. Cellular responses to DNA damage in the absence of poly (ADP-ribose) polymerase. Biochem. Biophys. Res. Comm. 1998;245:1–10. doi: 10.1006/bbrc.1998.8257. [DOI] [PubMed] [Google Scholar]
- LIAUDET L., SORIANO F.G., SZABÓ E., VIRAG L., MABLEY J.G., SALZMAN A.L., SZABÓ C. Protection against hemorrhagic shock in mice genetically deficient in poly(ADP-ribose)polymerase. Proc. Natl. Acad. Sci. U.S.A. 2000;97:10203–10208. doi: 10.1073/pnas.170226797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MACLEAN D., FISHBEIN M.C., MAROKO P.R., BRAUNWALD E. Hyaluronidase-induced reductions in myocardial infarct size. Science. 1976;194:199–200. doi: 10.1126/science.959848. [DOI] [PubMed] [Google Scholar]
- PIEPER A.A., WALLES T., WEI G., CLEMENTS E.E., VERMA A., SNYDER S.H., ZWEIER J.L. Myocardial postischemic injury is reduced by polyADPribose polymerase-1 gene disruption. Mol. Med. 2000;6:271–282. [PMC free article] [PubMed] [Google Scholar]
- SZABADOS E., LITERATI-NAGY P., FARKAS B., SUMEGI B. BGP-15, a nicotinic amidoxime derivate protecting heart from ischemia reperfusion injury through modulation of poly(ADP-ribose) polymerase. Biochem. Pharmacol. 2000;59:937–945. doi: 10.1016/s0006-2952(99)00418-9. [DOI] [PubMed] [Google Scholar]
- SZABÓ C. Cell death: The role of PARP. 2000CRC Press: Boca Raton, Florida, USA; Szabó, C (ed) [Google Scholar]
- SZABÓ C., ZINGARELLI B., O'CONNOR M., SALZMAN A.L. DNA strand breakage, activation of poly-ADP ribosyl synthetase, and cellular energy depletion are involved in the cytotoxicity in macrophages and smooth muscle cells exposed to peroxynitrite. Proc. Natl. Acad. Sci. U.S.A. 1996;93:1753–1758. doi: 10.1073/pnas.93.5.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SZABÓ C., VIRÁG L., CUZZOCREA S., SCOTT G.J., HAKE P., O'CONNOR M.P., ZINGARELLI B., SALZMAN A.L., KUN E. Protection against peroxynitrite-induced fibroblast injury and arthritis development by inhibition of poly (ADP-ribose) synthetase. Proc. Natl. Acad. Sci. U.S.A. 1998;95:3867–3872. doi: 10.1073/pnas.95.7.3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THIEMERMANN C., BOWES J., MYINT F.P., VANE J.R. Inhibition of the activity of poly(ADP ribose) synthase reduces ischemia-reperfusion injury in the heart and skeletal muscle. Proc. Natl. Acad. Sci. U.S.A. 1997;94:679–683. doi: 10.1073/pnas.94.2.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VIRÁG L., SCOTT G.S., MARMER D., SALZMAN A.L., SZABÓ C. Peroxynitrite-induced thymocyte apoptosis: the role of caspases and poly-(ADP-ribose) synthetase (PARS) activation. Immunology. 1998;94:345–355. doi: 10.1046/j.1365-2567.1998.00534.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG Z., ZINGARELLI B., SZABÓ C. Effect of genetic disruption of poly (ADP-ribose) synthetase on delayed production of inflammatory mediators and delayed necrosis during myocardial ischemia-reperfusion injury. Shock. 2000;13:60–66. doi: 10.1097/00024382-200013010-00011. [DOI] [PubMed] [Google Scholar]
- ZINGARELLI B., CUZZOCREA S., ZSENGELLER Z., SALZMAN A.L., SZABÓ C. Inhibition of poly (ADP ribose) synthetase protects against myocardial ischemia and reperfusion injury. Cardiovasc. Res. 1997;36:205–212. doi: 10.1016/s0008-6363(97)00137-5. [DOI] [PubMed] [Google Scholar]
- ZINGARELLI B., SALZMAN A.L., SZABÓ C. Genetic disruption of poly (ADP ribose) synthetase inhibits the expression of P-selectin and intercellular adhesion molecule-1 in myocardial ischemia-reperfusion injury. Circ. Res. 1998;83:85–94. doi: 10.1161/01.res.83.1.85. [DOI] [PubMed] [Google Scholar]