

Abstract
This study was designed to address the controversy related to the involvement of phospholipase C in the signalling pathway linked to CCKA receptor stimulation by the cholecystokinin analogue JMV-180, a full agonist for amylase release, in rat pancreatic acini.
JMV-180 was shown to stimulate phospholipase C by measuring the incorporation of [32P]-orthophosphoric acid ([32P]-Pi) into phosphatidic acid (PtdOH) and phosphatidylinositol (PtdIns). Both responses elicited by JMV-180 were time and concentration dependent. Maximal effects elicited by JMV-180 were 39.08±0.72 and 8.02±0.40% for the labelling of [32P]-PtdIns and [32P]-PtdOH, respectively, as compared to the maximal effects of CCK-8, a full agonist of the CCKA receptor.
[32P]-Pi incorporation into PtdOH and PtdIns was sensitive to lithium, demonstrating that both responses are a consequence of phospholipase C activation. However, since lithium blocks the phosphoinositide cycle by an uncompetitive mechanism, its effect was only apparent at high concentrations of CCK-8 (>10 pM), which elicited stimuli above 20 and 60% of the maximal [32P]-PtdOH and [32P]-PtdIns labelling, respectively.
JMV-180 inhibited the incorporation of [32P]-Pi into PtdOH and PtdIns as stimulated by CCK-8, down to its own maximal effect. The estimated IC50 values for the inhibition curves were not significantly different from those calculated assuming the same single binding site for both agonists. These results indicated that the well established role of JMV-180 as a partial agonist for CCKA receptor-linked signalling responses, also applies for the stimulation of phospholipase C.
The comparison of CCK-8 and JMV-180 dose-response curves of amylase release to those of PtdIns and PtdOH labelling with [32P]-Pi showed the existence of an amplification mechanism between phospholipase C and amylase release for both agonists.
In conclusion, we show that JMV-180, as well as CCK-8, stimulate phospholipase C upon interaction with the same binding site at the CCKA receptor in rat pancreatic acini.
Keywords: Cholecystokinin receptor CCKA, JMV-180, CCK-8, lithium, phospholipase C, phosphoinositide cycle, amylase release, pancreatic acini, rat
Introduction
Cholecystokinin (CCK) acts as hormone and neuropeptide to regulate secretion and motility in the gastrointestinal tract. Two cloned receptors, CCKA and CCKB, mediate the actions of this peptide (Wank, 1998; Alexander & Peters, 1999).
In rat pancreatic acini, CCK regulates amylase secretion. Studies using the CCK analogue CCK-8, have established that this effect takes place through CCKA receptor (Jensen, 1994), showing an intriguing bell-shaped concentration-effect curve. Binding experiments have established two different affinity states (high and low) of the CCKA receptor for CCK-8, and it has been proposed that the high and low affinity states are responsible for the stimulation and inhibition of amylase release, respectively (Jensen, 1994).
CCKA receptor is coupled to a Gq/ll protein (Williams, 1997; Piiper et al., 1997). Consistent with this, phosphoinositide phospholipase C (PLC) stimulation by CCK-8 has been widely characterized (Yule & Williams, 1994; Wank, 1998; Bianchi et al., 1994). The activation of PLC is considered to be responsible for the stimulation of amylase secretion by low doses of CCK-8 in rat pancreatic acini (Jensen, 1994; Wank, 1998), and also for its inhibition with high doses of the agonist (Matozaki et al., 1990; Bianchi et al., 1994), which may take place after PKC activation and a subsequent receptor desensitization (Ozcelebi et al., 1996).
JMV-180, another CCK analogue, stimulates fully amylase secretion in a saturable fashion through the CCKA receptor in rat pancreatic acini (Stark et al., 1989; Matozaki et al., 1990; Tsunoda et al., 1996). However, JMV-180 effects on PLC have been a matter of controversy. Some authors have reported an enhancement of 3H-inositol phosphates (3H-InsPs) production by JMV-180 (Bianchi et al., 1994; Gaisano et al., 1994), but this observation was not confirmed by others using a similar approach (Matozaki et al., 1990; Saluja et al., 1992; Tsunoda et al., 1996) or measuring inositol 1,4,5-trisphosphate (Ins1,4,5P3) mass (Talkad et al., 1994). Moreover, since JMV-180 clearly stimulates PLC in mouse pancreatic acini (Bianchi et al., 1994) or in CHO cells (Yule et al., 1993), it has been suggested that signal transduction events triggered by JMV-180 may be a matter of species or tissue. On the other hand, some inhibitory effects of JMV-180 on CCK-8 elicited responses have also been described: JMV-180 inhibited Ins(1,4,5)P3 production in rat pancreatic acini (Matozaki et al., 1990), intracellular Ca2+ increase in guinea-pig pancreatic acinar cells (Sjödin et al., 1997), and inhibition of amylase secretion by high CCK-8 concentrations in rat pancreatic acini (Padfield & Panesar, 1998). Provided that both cholecystokinin analogues CCK-8 and JMV-180 bind to the same sites at CCKA receptor (Stark et al., 1989), one of the current interpretations for the different biochemical and physiological profiles of CCK-8 and JMV-180 assumes that the latter behaves as an agonist for the high affinity state, and as an antagonist for the low affinity state (Tsunoda et al., 1996). It has been suggested, however, that the lack of ability of JMV-180 to inhibit amylase secretion may be due to its low efficacy in stimulating PLC (Bianchi et al., 1994).
As mentioned, PLC stimulation can be monitored using different strategies. In addition to quantifying the rapid and transient increase of Ins(1,4,5)P3 (Berridge, 1983), a common approach measures the 3H-InsPs that accumulate in the presence of lithium, which inhibits inositol monophosphatases allowing the amplification of the signal (Berridge et al., 1982; Jope & Williams, 1994). However, the use of lithium can be particularly misleading when PLC stimulation is very weak. Lithium inhibits inositol monophosphatases through an uncompetitive mechanism, which results in a reduction of both apparent kinetic parameters, Kmapp and Vmaxapp, of the enzymes (Nahorski et al., 1991). Therefore, the inhibitory strength of lithium depends on substrate concentration, which in turn is directly related to the robustness of PLC stimuli. Another approach to evaluate PLC activation relies on the concomitant acceleration of the phosphoinositide cycle (Michell, 1975; Abdel-Latif, 1986; Claro et al., 1993), where an increase of labelling in the phosphate moiety of phosphatidylinositol (Ptdlns), in the absence of lithium, reflects the recycling of the PLC-catalyzed reaction products. Diacylglycerol (DAG) is first phosphorylated to phosphatidic acid (PtdOH), and then PtdOH is activated by CTP to form CDP-DAG, which in turn condenses with inositol, derived from the Ins(1,4,5)P3 dephosphorylation to form PtdIns.
In this work we have measured the increase of [32P]-orthophosphoric acid ([32P]-Pi) labelling of PtdIns and PtdOH in rat pancreatic acini, as stimulated by CCK-8 and JMV-180, showing that both CCK analogues stimulate PLC through CCKA receptor activation. The two responses are sensitive to lithium, although this sensitivity is only apparent at values of the response above a certain threshold due to the uncompetitive inhibition mechanism of lithium. CCK-8 and JMV-180 interact with the same single binding site to fully and partially stimulate PLC, respectively. For both agonists it is shown an amplification mechanism between phospholipase C activation and amylase release. We conclude that, as previously described for CCK-8, phospholipase C may be involved in JMV-180-stimulated amylase release in rat pancreatic acini.
Methods
Materials
We purchased CCK-8 from Peninsula Laboratories, Inc. (Belmont, CA, U.S.A.), JMV-180 from Research Plus, Inc. (Bayonne, NJ, U.S.A.), [32P]-orthophosphoric acid (carrier free) from DuPont New England Nuclear (ITISA, Madrid, Spain), and purified collagenase (type CLSPA) from Worthington Biochemical Corporation (Lakewood, NJ, U.S.A.). Soybean trypsin inhibitor (type II-S), BME Amino Acids solution, BME Vitamins solution, carbamoylcholine (Cch), quinacrine and phospholipid standards were from Sigma (St. Louis, MO, U.S.A.), silica gel-60 TLC plates from Merck (Barcelona, Spain); and Phadebas® Amylase Test from Pharmacia AB (Uppsala, Sweden). All other chemicals used were of analytical grade.
Tissue preparation
Wistar rats (200–250 g) were killed by cervical dislocation and dispersed pancreatic acini were isolated by collagenase digestion according to the modifications (Jensen et al., 1982) of the procedure published previously (Peikin et al., 1978).
Unless otherwise indicated, the standard incubation solution contained (in mM): HEPES, 25 (pH adjusted to 7.45 with concentrated NaOH; Na Cl 98; KCl 6; sodium pyruvate 5; sodium fumarate 6; sodium L-glutamate 5; glucose 11.5; KH2PO4 2; MgCl2 1.2; CaCl2 0.5; L-glutamine 2; Soybean trypsin inhibitor 1% (w v−1); amino acid mixture 1% (w v−1); vitamins mixture 1% (v v−1); and bovine serum albumin 1% (w v−1). The incubation solution was equilibrated with 100% O2, which consisted of the gas phase in all the incubations performed.
[32P]-Pi incorporation into PtdIns and PtdOH
Prior to stimulation, pancreatic acini from one rat were suspended in 4 ml of the HEPES-buffered incubation solution detailed above without added phosphate, in a flat-bottomed flask. 100 μCi ml−1 of [32P]-Pi were added to the acini suspension and it was incubated for 2 h at 37°C. This allowed acini to reestablish energy charge and [32P]-Pi-label to reach a pre-equilibrium state. Then, acini were washed by centrifugation and resuspended with the same fresh incubation solution in a final volume of 6 ml. Aliquots of the acini suspension (100 μl) were pipetted into tubes containing agonists and other additions, when indicated, to a final volume of 250 μl and incubated for 1 h at 37°C, unless otherwise stated. At the end of incubation 1.2 ml of chloroform/methanol (1 : 2, v v−1) were added to stop the reaction. After 30 min on ice, 0.5 ml each of chloroform and water were added, and the tubes were shaken vigorously. After a 5-min centrifugation at 2000 × g, the upper (aqueous) phases were removed, and the lower (organic) phases containing 32P-lipids were washed with 1.55 ml methanol/water (1 : 1 v v−1). Aliquots (0.6 ml) of the washed organic phases were centrifuged under vacuum to evaporate the solvent. To separate 32P-lipids, these were resuspended in 15 μl chloroform/methanol (4 : 1, v v−1) and spotted onto silica gel-60 TLC plates that were developed with chloroform/methanol/acetone/acetic acid/water (50 : 10 : 20 : 10 : 5, by vol.). Areas corresponding to PtdIns and PtdOH were identified with authentic standards after staining with I2 vapor, and 32P radioactivity was quantified using a Packard Instant Imager system.
Amylase release
Prior to stimulation, pancreatic acini from one rat were resuspended in approximately 25 ml of standard incubation solution and incubated for 2 h at 37°C. Then acini were washed by centrifugation and resuspended with fresh standard incubation solution in a total volume of 300 ml. Amylase release was measured using the procedure described previously (Jensen et al., 1982; Peikin et al., 1978). Amylase activity released into the extracellular medium was routinely determined after 30 min incubation using the Phadebas® reagent as substrate and was expressed as per cent of total cellular amylase activity.
Data analysis
Concentration-response and inhibition curves were analysed by non-linear regression with the program GRAPHPAD PRISM (GraphPad Software Inc). Each concentration-response curve was fitted to a logistic function of the form: E=Emax[A]n/([A]n+EC50n), where [A] is the agonist concentration, E the agonist effect, Emax the maximal response, n the Hill coefficient, and EC50 the agonist concentration giving half-maximal response. Similarly, individual inhibition curves were fitted to a function of the form: Per cent inhibition=100[B]/([B]+IC50) where [B] is the antagonist concentration and IC50 that causing 50% of the maximal inhibition effect. Both EC50 and IC50 values were actually estimated as logarithms. The computed parameter estimates for each group of n-replicated experiments were expressed as mean±s.e.mean and, for display purposes, they were used to generate a logistic curve that was drawn through the experimental data.
The expected IC50 value for the antagonistic action of a partial agonist when it is challenged in front of a full agonist was calculated by using the equation: IC50=EC50(pa) (1+([L]/EC50(fa))), where [L] is the full agonist concentration, EC50(pa) is the estimated EC50 for the partial agonist, and EC50(fa) is that for the full agonist (Cheng & Prusoff, 1973).
Results
The first set of experiments, shown in Figure 1, determined the time-course of [32P]-Pi incorporation into PtdIns and PtdOH, after stimulation of rat pancreatic acini with the cholecystokinin analogues, CCK-8 and JMV-180, which are full agonists for amylase release. [32P]-PtdIns increase (a) elicited by 1 nM CCK-8 was approximately linear for the first 30 min, and declined slowly thereafter. However, this same response as stimulated by 1 μM JMV-180 was linear during the first 60 min, being at this point 30–40% of that elicited by CCK-8. On the other hand, the stimulation of [32P]-Pi incorporation into PtdOH (b) was rapid for CCK-8 and JMV-180, and reached a plateau after 10 min that remained constant for the following 50 min. In contrast to the PtdIns effect, this PtdOH effect as stimulated by JMV-180 was only about 10% of that stimulated by CCK-8. [32P]-Pi labelling of PtdIns and PtdOH as stimulated by both agonists was not modified by the presence of 100 μM quinacrine, a non-specific inhibitor of phospholipase A2 activity, which is able to block intracellular arachidonic acid release in pancreas (How et al., 1996; 1997) (not shown). Also, inclusion of 200 mM ethanol, which might switch part of [32P]-PtdOH formation to [32P]-phosphatidylethanol accumulation, due to the transphosphatidylation reaction catalyzed by phospholipase D (Exton, 1997) had no effect (results not shown). These latter results suggest that neither phospholipase A2 nor phospholipase D are involved in the responses.
Figure 1.
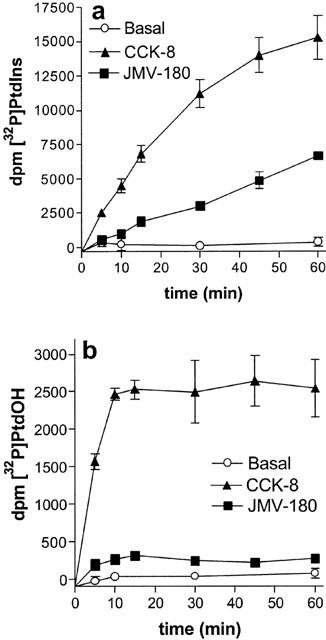
Time course of [32P]-PtdIns (a) and [32P]-PtdOH (b) increase in rat pancreatic acini. [32P]-Pi-prelabelled rat pancreatic acini were incubated for the times indicated with 1 nM CCK-8, 1 μM JMV-180, or under basal conditions. Results are mean±s.e.mean of three experiments in triplicate.
Shown in Figure 2 are the dose-response curves in the absence of lithium (filled symbols) for [32P]-Pi labelling of PtdIns (Figure 2a) and of PtdOH (Figure 2b) after 60-min stimulation with CCK-8 and JMV-180. By fitting the concentration effect curves for the [32P]-Pi incorporation into PtdIns for both agonists to a sigmoid curve, as stated in methods, we estimated the EC50 values (17.5±3.3 pM and 29.4±8.3 nM for CCK-8 and JMV-180, respectively), the Emax values, which for JMV-180 was 39.08±0.72% of that elicited by CCK-8, and the Hill slope values, which were not significantly different (P>0.05) from the unit (0.92±0.10 and 0.90±0.08 for CCK-8 and JMV-180, respectively). Regarding [32P]-Pi incorporation into PtdOH, the estimated EC50 values were 216±53 pM and 23.8±7.4 nM for CCK-8 and JMV-180, respectively, the Emax obtained with JMV-180 was only 8.2±0.4% of that obtained with CCK-8, and again, the estimated Hill slope values were not significantly different (P>0.05) from the unit (0.85±0.11 and 1.09±0.32 for CCK-8 and JMV-180, respectively). At this point, it should be stressed that JMV-180 did not elicit a full response of [32P]-Pi incorporation into PtdIns or into PtdOH, and that the concentration-effect curves for the two responses elicited by JMV-180 and CCK-8 were best fitted to a one binding site model.
Figure 2.
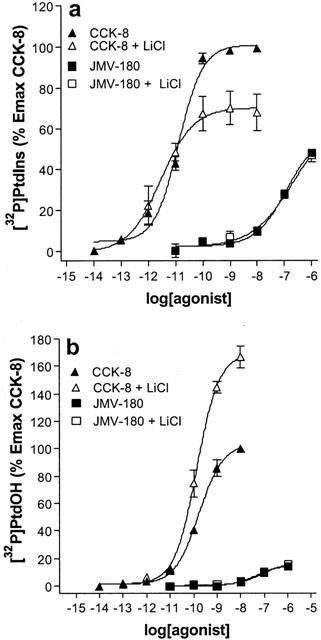
Dose-response curves for [32P]-Pi incorporation into PtdIns (a) and PtdOH (b) in the absence and in the presence of lithium. [32P]-Pi-prelabelled rat pancreatic acini were incubated for 60 min with the indicated concentrations of CCK-8 or JMV-180 in the absence or presence of 10 mM LiCl. Values are mean±s.e.mean of at least three experiments performed in triplicate and are represented as percentages of maximal effect of CCK-8 in the absence of LiCl.
Lithium inhibits the phosphoinositide cycle at the inositol monophosphatases step, leading to a depletion of free inositol and therefore to the blockade of PtdIns resynthesis (Berridge et al., 1982; Jope & Williams, 1994), which most likely would be mirrored by the accumulation of PtdOH. We therefore investigated the effect of lithium on both the increase of [32P]-PtdIns and [32P]-PtdOH. These results are also depicted in Figure 2 (open symbols). Shown in Figure 2a, the dose-response curve elicited by CCK-8 in the presence of 10 mM lithium indicated that [32P]-Pi labelling of PtdIns was sensitive to the ion, although the expected effect of lithium, i.e. a decrease in [32P]-PtdIns, was only apparent at high concentrations (>10 pM) of CCK-8, when the response reached a value that exceeded 60% of the maximal effect. In accordance with the results obtained with CCK-8, the effect of 10 mM lithium was not apparent on the [32P]-PtdIns labelling evoked by JMV-180, since the response to this agonist did not reach 60% of the maximal effect of CCK-8. In Figure 2b it is shown that [32P]-Pi labelling of PtdOH is also sensitive to lithium. In this case, the stimulus threshold for the appearance of the expected lithium effect, i.e. an increase in [32P]-PtdOH, was aproximately above 20% of the maximal effect of CCK-8, which again was not reached by JMV-180, and therefore the response elicited by JMV-180 was apparently non-sensitive to 10 mM lithium. It is worth mentioning here that amylase release, which was fully stimulated by either CCK-8 or JMV-180, was not affected by the presence of lithium (results not shown).
The lack of effect of lithium on the PtdIns and PtdOH responses to JMV-180 might not depend on a threshold of stimulation, inherent to its uncompetitive inhibitory mechanism. Rather, it might simply reflect that the PtdIns and PtdOH responses to JMV-180 are not due to the stimulation of phosphoinositide-PLC. This possibility seems unlikely, but in order to rule it out we evaluated the effect of lithium on [32P]-Pi incorporation into PtdIns (Figure 3a) and PtdOH (Figure 3b) after stimulation of the muscarinic acetylcholine receptor by the agonist carbamoylcholine (Cch), which is also coupled to phosphoinositide hydrolysis in rat pancreatic acini (Jensen, 1994). We chose the muscarinic acetylcholine receptor, as it was previously shown that JMV-180 does not interact with Cch-induced responses, such as Ins1,4,5P3 or intracellular calcium release, in rat pancreatic acini (Matozaki et al., 1990; Sjödin et al., 1997). Moreover, in our hands JMV-180 did not inhibit the effects on [32P]-PtdIns and [32P]-PtdOH labelling triggered by Cch, and consistently, the responses of JMV-180 and submaximal concentrations of Cch were additive (not shown). The responses obtained with Cch, expressed as percentage of the maximal effect of CCK-8 indicated that, again, the effect of lithium was apparent only when the stimulatory effect of Cch was above the thresholds defined before, 60 and 20% for [32P]-PtdIns (a) and [32P]-PtdOH labelling (b), respectively.
Figure 3.
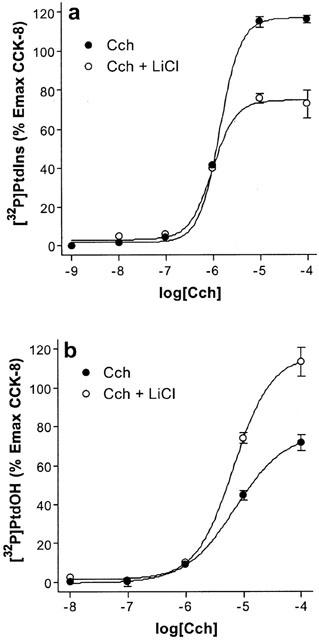
Effect of lithium on the dose-response curves of [32P]-PtdIns (a) and [32P]-PtdOH (b) increase stimulated by Cch. [32P]-Pi-prelabelled rat pancreatic acini were incubated for 60 min with the indicated concentrations of Cch in the absence or presence of 10 mM LiCl. Values are mean±s.e.mean of three experiments performed in triplicate, and are represented as a percentage of the maximal effect of CCK-8 in the absence of LiCl (Figure 2).
The results obtained with lithium up to this point clearly suggest that the measured effects on PtdIns and PtdOH [32P]-Pi-labelling were actually mirroring phosphoinositide-PLC stimulation. Further, our results shed some light into the controversy around JMV-180 and PLC measurement, as the absence of lithium effects on JMV-180 responses may well arise from its weak stimulation of PLC, which prevents lithium either to deplete free inositol and amplify the accumulation of inositol phosphates.
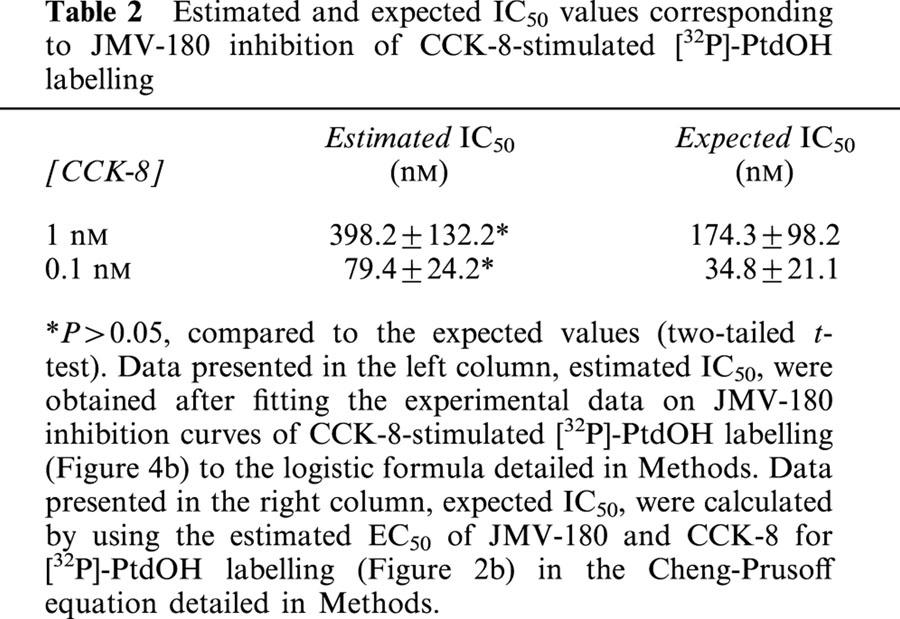
We performed further experiments to investigate the JMV-180 binding site linked to its responses, and for that purpose we evaluated the interaction of both CCKA receptor agonists. Figure 4 shows inhibition curves obtained after stimulation of pancreatic acini with CCK-8 in the presence of increasing concentrations of JMV-180. Regarding [32P]-PtdIns labelling (Figure 4a), we show that the effect of all concentrations of CCK-8 tested was inhibited by JMV-180 down to a level that was approximately coincident with the Emax elicited by JMV-180 alone (35.07±6.34, 44.99±6.46, 39.23±2.87, and 34.50±4.51% for 1, 0.3, 0.1, and 0.03 nM CCK-8, respectively). Table 1 shows, for each inhibition curve, the values of IC50 estimated after fitting the experimental data to the equation indicated in Methods, and also the expected IC50 values, calculated from estimated EC50 values for CCK-8 and JMV-180, by using the equation detailed in Methods, which assumes a single binding site for both agonists. There were no significant differences between the pairs of values shown in both columns of the table. The interaction of both agonists was also evaluated for the PtdOH response (Figure 4b), and again JMV-180 inhibited [32P]-Pi incorporation into PtdOH as stimulated by the two concentrations of CCK-8 tested to a level similar to the estimated Emax value for JMV-180 alone (11.46±5.46 and 8.34±1.76%, for 1 and 0.1 nM CCK-8, respectively). In analogy with Table 1, Table 2 shows the comparison of estimated and expected IC50 values for the inhibition of the PtdOH response by JMV-180. Just as with the PtdIns response, estimated and expected IC50 values, corresponding to each CCK-8 concentration used, were not significantly different (P>0.05).
Figure 4.
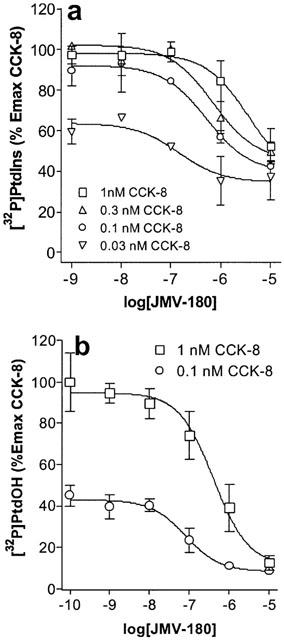
Competition curves for [32P]-Pi incorporation into PtdIns (a) and PtdOH (b) [32P]-Pi-prelabelled rat pancreatic acini were incubated for 60 min with increasing concentrations of JMV-180 in the presence of an indicated concentration of CCK-8. Values are mean±s.e.mean of at least three experiments performed in triplicate and are represented as percentages of maximal effect of CCK-8.
Table 1.
Estimated and expected IC50 values corresponding to JMV-180 inhibition of CCK-8-stimulated [32P]-PtdIns labelling
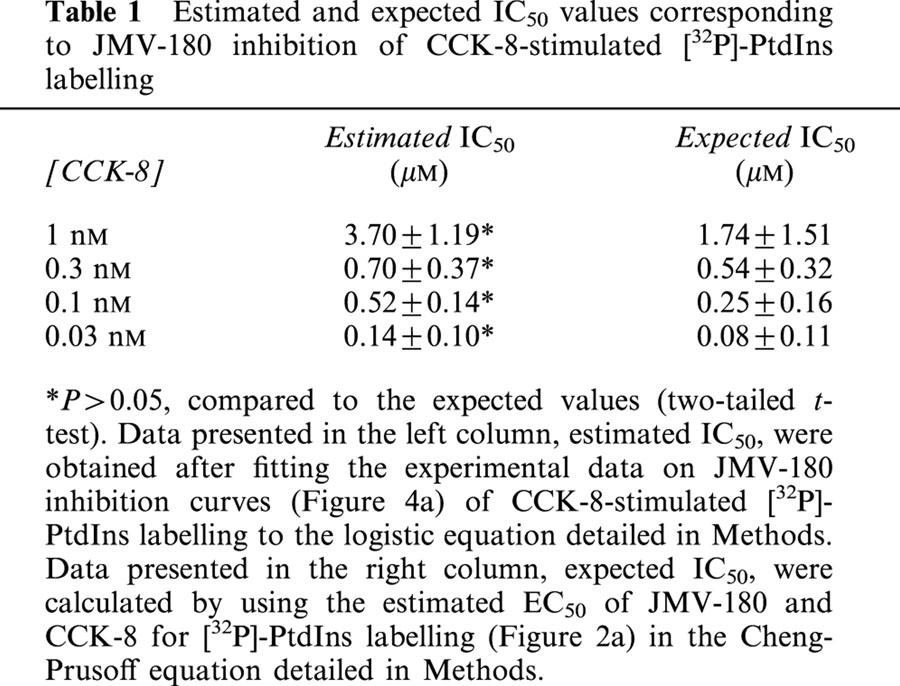
Table 2.
Estimated and expected IC50 values corresponding to JMV-180 inhibition of CCK-8-stimulated [32P]-PtdOH labelling
We represent in Figure 5, for each agonist, the concentration-effect curves for the two responses described above together with that for the stimulation of amylase release (CCK-8 in (a) and JMV-180 in (b)). As it is apparent from the figure, in the case of CCK-8 the EC50 values for the three responses follow the rank order: [32P]-PtdOH>[32P]-PtdIns >amylase release. In contrast, EC50 values for the three responses triggered by JMV-180 were all similar. Moreover, the Emax values of the three responses to JMV-180, expressed as percentage of the maximal CCK-8 effect of each response, were clearly different, in the rank order: amylase release >[32P]-PtdIns>[32P]-PtdOH.
Figure 5.
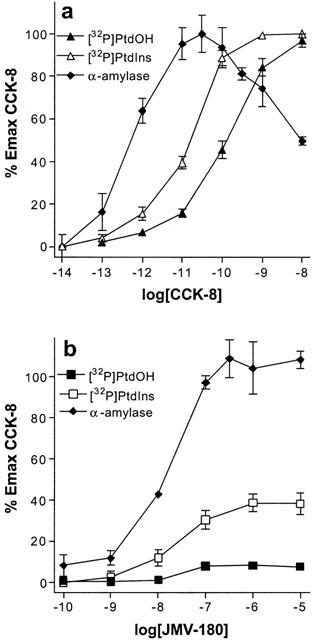
Dose-response curves for [32P]-Pi incorporation into PtdOH and PtdIns, and amylase release stimulated by CCK-8 (a) and JMV-180 (b) [32P]-Pi-prelabelled, except for the amylase release experiments, rat pancreatic acini were incubated for 60 min with the indicated concentrations of CCK-8 or JMV-180. Values are mean±s.e.mean of at least three experiments performed in triplicate, and are represented as a percentage of the maximal effect of CCK-8 for each response.
Discussion
The results presented in this work show that JMV-180 acts through CCKA receptor to stimulate PLC, which may take part in the signalling mechanism involved in its full stimulation of amylase secretion in rat pancreatic acini.
At first, we clearly observed an increase of [32P]-PtdIns and [32P]-PtdOH labelling, which was time (Figure 1) and concentration (Figure 2) dependent, after stimulation with either CCK-8 and JMV-180. This effect was documented for the first time in pancreas by Hokin & Hokin (1953), and constituted the initial clue to unravel the role of phosphoinositide PLC as a general signal transducing system. In our preparation, we did not observe any effect on [32P]-Pi incorporation into PtdIns or PtdOH stimulated by CCK-8 or JMV-180, after a treatment with quinacrine, a non-specific inhibitor of phospholipase A2. Quinacrine was previously shown to inhibit arachidonic acid release in pancreas (How et al., 1996; 1997), a response that may well be elicited by the cytosolic isoform of phospholipase A2, since it is specific for the cleavage of arachidonic acid, rather than other ester-linked fatty acids in the position 2 of glycerol in glycerophospholipids (Leslie, 1997). On the other hand, we have shown previously that PLD in rat pancreatic acini is not stimulated by CCK-8 (Sarri et al., 2000). Consistent with this, inclusion of 200 mM ethanol, which would reduce some [32P]-PtdOH formed by the action of phospholipase D, did not affect PtdIns and PtdOH responses. These results, although not absolutely conclusive, pointed toward the only involvement of PLC in the responses we measured.
The expected results from the experiments with lithium were an increase of [32P]-PtdOH and a decrease in [32P]-PtdIns, due to the depletion of free inositol after the inhibition of inositol monophosphatases (Berridge et al., 1982; Jope & Williams, 1994), and the subsequent blockade at the PtdIns synthase step of the phosphoinositide cycle. However, the effect of lithium, as shown in Figures 2 and 3, was only patent at a certain level of the [32P]-PtdIns and [32P]-PtdOH after stimulation of either CCKA or muscarinic acetylcholine receptors, by CCK-8 and Cch, respectively, indicating that the effect of lithium was related to the robustness of PLC response, rather than to the type of receptor. That threshold of stimulation was not reached when JMV-180 was the agonist, which interacts with CCKA, but not with muscarinic acetylcholine receptors. These results suggest that the absence of [3H]-InsPs accumulation after stimulation of rat pancreatic acini by JMV-180, described in several studies (Matozaki et al., 1990; Saluja et al., 1992; Tsunoda et al., 1996), might be due to the uncompetitive mechanism of lithium: the low PLC stimulation triggered by JMV-180 is likely not enough for the InsPs formed to reach the necessary level to allow lithium to block their recycling to inositol. Moreover, the fact that the lithium-sensitive level of PLC activity, either measured as [32P]-Pi labelling of PtdIns or PtdOH, is higher than that necessary to fully stimulate amylase release (Figure 5) seems a likely explanation for the absence of lithium effect on amylase release promoted by any of the two agonists, as this response is already saturated when the metabolism of PLC-derived InsPs became sensitive to inhibition by lithium.
The experimental approach used in this work, i.e. [32P]-Pi-labelling of PtdOH and PtdIns, therefore, reflects unambiguously PLC activity in dispersed rat pancreatic acini, and it turned out to be a method with higher sensitivity than the measurement of [3H]-InsPs accumulation or Ins1,4,5P3 mass increase for weak PLC stimulations.
JMV-180 displayed a clear partial agonist profile for PLC stimulation through CCKA receptor, either measured as PtdIns or PtdOH labelling with [32P]-Pi (Figures 2 and 4): first, its maximal effects estimated from the dose-response curves were lower than those of CCK-8, and second, it acted as an antagonist when incubated together with CCK-8, inhibiting the CCK-8 responses to a level coincident with its own maximal effect level. Moreover, the estimated IC50 values of these inhibition-curves were substantially coincident with those calculated by using the estimated EC50 values of each agonist and assuming that JMV-180 is a partial agonist acting at the CCK-8 binding site (Tables 1 and 2) (Cheng & Prusoff, 1973). At this point, it is important to note that the dose-response curves of both [32P]-Pi incorporation into PtdIns and PtdOH for CCK-8 and JMV-180 fitted to a one site model. Taken together, the data show that JMV-180 and CCK-8 interact with the same binding site of the CCKA receptor to stimulate PLC. This conclusion is strengthened after examining the results shown in Figure 5: the potency of CCK-8 to trigger three different responses increased clearly as the measured response was more distant to the initial event of receptor activation of PLC, but in contrast, the potency of JMV-180 was not changed and its maximal effect increased likewise. These results illustrate that only a very small fraction of the maximally stimulatable PLC activity is needed to stimulate amylase release fully, in agreement with other authors who have previously postulated an amplification mechanism between phosphoinositide breakdown and amylase release after cholecystokinin receptor stimulation (Lin et al., 1986).
In conclusion, we demonstrate that JMV-180 stimulates PLC through its interaction with the CCKA receptor at the same binding site for CCK-8, which is likely the one coupled to amylase release. Furthermore, as high levels of PLC stimulation have been related to amylase release inhibition (Matozaki et al., 1990; Bianchi et al., 1994), the same binding site may be the responsible for both responses to CCK-8. However, JMV-180 may not elicit the inhibitory effect on amylase release, in contrast to CCK-8, just due to its partial agonist effect in stimulating PLC.
Acknowledgments
This work was supported by DGESIC grant PB97-0370. Belén Ramos is recipient of a doctoral fellowship from the Spanish Ministry of Science and Technology.
Abbreviations
- Cch
carbamoylcholine
- CCK-8
cholecystokinin octapeptide (26–33), Asp-Tyr(SO3H)-Met-Gly-Trp-Met-Asp-Phe-NH2
- DAG
diacylglycerol
- InsPs
inositol phosphates
- Ins(1,4,5)P3
inositol 1,4,5 trisphosphate
- JMV-180
Boc-Tyr(SO3H)-Nle-Gly-Trp-Nle-Asp-2-phenylethylester
- [32P]-Pi
[32P]-orthophosphoric acid
- PLC
phospholipase C
- PtdIns
phosphatidylinositol
- PtdOH
phosphatidate
References
- ABDEL-LATIF A.A. Calcium-mobilizing receptors, polyphosphoinositides, and the generation of second messengers. Pharmacol. Rev. 1986;38:227–272. [PubMed] [Google Scholar]
- ALEXANDER S.P.H., PETERS J.A.Cholecystokinin and gastrin receptors TiPS Receptor and Ion Nomenclature. 1999SupplElsevier Trends Journals, Cambridge, U.K; 28–29.10th ed. pp [Google Scholar]
- BERRIDGE M.J. Rapid accumulation of inositol trisphosphate reveals that agonists hydrolyse polyphosphoinositides instead of phosphatidylinositol. Biochem. J. 1983;212:849–858. doi: 10.1042/bj2120849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BERRIDGE M.J., DOWNES C.P., HANLEY M.R. Lithium amplifies agonist-dependent phosphatidylinositol responses in brain and salivary glands. Biochem. J. 1982;206:587–595. doi: 10.1042/bj2060587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BIANCHI B.R., MILLER T.R., WITTE D.G., LIN C.W. Novel CCK analogues and bombesin: a detailed analysis between phosphoinositide breakdown and high-dose inhibition of pancreatic enzyme secretion in three rodent species. J. Pharmacol. Exp. Ther. 1994;268:996–1002. [PubMed] [Google Scholar]
- CHENG Y.C., PRUSOFF W.H. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 percent inhibition (IC50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- CLARO E., FAIN J.N., PICATOSTE F. Noradrenaline stimulation unbalances the phosphoinositide cycle in rat cerebral cortical slices. J. Neurochem. 1993;60:2078–2086. doi: 10.1111/j.1471-4159.1993.tb03492.x. [DOI] [PubMed] [Google Scholar]
- EXTON J. Phospholipase D: enzymology, mechanisms of regulation and function. Physiol. Rev. 1997;77:303–320. doi: 10.1152/physrev.1997.77.2.303. [DOI] [PubMed] [Google Scholar]
- GAISANO H.Y., WONG D., SHEU L., FOSKETT J.K. Calcium release by cholecystokinin analogue OPE is IP3 dependent in single rat pancreatic acinar cells. Am. J. Physiol. 1994;267:C220–C228. doi: 10.1152/ajpcell.1994.267.1.C220. [DOI] [PubMed] [Google Scholar]
- HOKIN L.E., HOKIN M.R. Enzyme secretion and the incorporation of P32 into phospholipids of pancreas slices. J. Biol. Chem. 1953;203:967–977. [PubMed] [Google Scholar]
- HOW W., ARITA Y., MORISSET J. Caerulein-stimulated arachidonic acid release in rat pancreatic acini: a diacylglycerol lipase affair. Am. J. Physiol. 1996;271:C1735–C1742. doi: 10.1152/ajpcell.1996.271.5.C1735. [DOI] [PubMed] [Google Scholar]
- HOW W., ARITA Y., MORISSET J. Endogenous arachidonic acid release and pancreatic amylase secretion. Pancreas. 1997;14:301–308. doi: 10.1097/00006676-199704000-00014. [DOI] [PubMed] [Google Scholar]
- JENSEN R.T., LEMP G.F., GARDNER J.D. Interaction of COOH-terminal fragments of cholecystokinin with receptors on dispersed acini from guinea pig pancreas. J. Biol. Chem. 1982;257:5554–5559. [PubMed] [Google Scholar]
- JENSEN R.T.Receptors on pancreatic acinar cells Physiology of the Gastrointestinal Tract. 1994Raven Press, New York; 1377–1446.3rd ed. (Johnson LR, ed.) pp [Google Scholar]
- JOPE R.S., WILLIAMS M.B. Lithium and brain signal transduction systems. Biochem. Pharmacol. 1994;47:429–441. doi: 10.1016/0006-2952(94)90172-4. [DOI] [PubMed] [Google Scholar]
- LESLIE C.C. Properties and regulation of cytosolic phospholipase A2. J. Biol. Chem. 1997;272:16709–16712. doi: 10.1074/jbc.272.27.16709. [DOI] [PubMed] [Google Scholar]
- LIN C.W., BIANCHI B.R., GRANT D., MILLER T., DANAHER E.A., TUFANO M.D., KOPECKA H., NADZAN A.M. Cholecystokinin receptors: relationships among phosphoinositide breakdown, amylase release and receptor affinity in pancreas. J. Pharmacol. Exp. Ther. 1986;236:729–734. [PubMed] [Google Scholar]
- MATOZAKI T., GÖKE B., TSUNODA Y., RODRIGUEZ M., MARTINEZ J., WILLIAMS J.A. Two functionally distinct cholecystokinin receptors show different modes of action on Ca2+ mobilization and phospholipid hydrolysis in isolated rat pancreatic acini. Studies using a new cholecystokinin analog, JMV-180. J. Biol. Chem. 1990;265:6247–6254. [PubMed] [Google Scholar]
- MICHELL R.H. Inositol phospholipids and cell surface receptor function. Biochim. Biophys. Acta. 1975;415:81–147. doi: 10.1016/0304-4157(75)90017-9. [DOI] [PubMed] [Google Scholar]
- NAHORSKI S.R., RAGAN C.I., CHALLISS R.A. Lithium and the phosphoinositide cycle: an example of uncompetitive inhibition and its pharmacological consequences. Trends. Pharmacol. Sci. 1991;12:297–303. doi: 10.1016/0165-6147(91)90581-c. [DOI] [PubMed] [Google Scholar]
- OZCELEBI F., RAO R.V., HOLICKY E., MADDEN B.J., MCCORMICK D.J., MILLER L.J. Phosphorylation of cholecystokinin receptors expressed on Chinese hamster ovary cells. J. Biol. Chem. 1996;271:3750–3755. doi: 10.1074/jbc.271.7.3750. [DOI] [PubMed] [Google Scholar]
- PADFIELD P.J., PANESAR N. Cholecystokinin octapeptide inhibits Ca2+-dependent amylase secretion from permeabilized pancreatic acini by blocking the MgATP-dependent priming of exocytosis. Biochem. J. 1998;330:329–334. doi: 10.1042/bj3300329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PEIKIN S.R., ROTTMAN A.J., BATZRI S., GARDNER J.D. Kinetics of amylase release by dispersed acini prepared from guinea pig pancreas. Am. J. Physiol. 1978;235:E743–E749. doi: 10.1152/ajpendo.1978.235.6.E743. [DOI] [PubMed] [Google Scholar]
- PIIPER A., STRYJEK-KAMINSKA D., KLENGEL R., ZEUZEM S. CCK, carbachol, and bombesin activate distinct PLC-β isoenzymes via Gq/ll in rat pancreatic acinar membranes. Am. J. Physiol. 1997;272:G135–G140. doi: 10.1152/ajpgi.1997.272.1.G135. [DOI] [PubMed] [Google Scholar]
- SALUJA A.K., DAWRA R.K., LERCH M.M., STEER M.L. CCK-JMV-180, an analog of cholecystokinin, releases intracellular calcium from an inositol trisphosphate-independent pool in rat pancreatic acini. J. Biol. Chem. 1992;267:11202–11207. [PubMed] [Google Scholar]
- SARRI E., RAMOS B., SALIDO G., CLARO E. Cholecystokinin octapeptide CCK-8 and carbachol reduce [32P]orthophosphate labelling of phosphatidylcholine without modifying phospholipase D activity in rat pancreatic acini. FEBS Lett. 2000;486:63–67. doi: 10.1016/s0014-5793(00)02233-x. [DOI] [PubMed] [Google Scholar]
- SJÖDIN L., VIITANEN E., GYLFE E. Cholecystokinin-JMV-180 and pilocarpine are potent inhibitors of cholecystokinin and carbachol actions on guinea pig pancreatic acinar cells. Naunyn Schmiedebergs Arch. Pharmacol. 1997;355:631–637. doi: 10.1007/pl00004994. [DOI] [PubMed] [Google Scholar]
- STARK H.A., SHARP C.M., SUTLIFF V.E., MARTINEZ J., JENSEN R.T., GARDNER J.D. CCK-JMV-180: a peptide that distinguishes high-affinity cholecystokinin receptors from low-affinity cholecystokinin receptors. Biochim. Biophys. Acta. 1989;1010:145–150. doi: 10.1016/0167-4889(89)90154-7. [DOI] [PubMed] [Google Scholar]
- TALKAD V.D., PATTO R.J., METZ D.C., TURNER R.J., FORTUNE K.P., BHAT S.J., GARDNER J.D. Characterization of the three different states of the cholecystokinin (CCK) receptor in pancreatic acini. Biochim. Biophys. Acta. 1994;1224:103–116. doi: 10.1016/0167-4889(94)90118-x. [DOI] [PubMed] [Google Scholar]
- TSUNODA Y., YOSHIDA H., OWYANG C. Structural requirements of CCK analogues to differentiate second messengers and pancreatic secretion. Am. J. Physiol. 1996;271:G8–G19. doi: 10.1152/ajpgi.1996.271.1.G8. [DOI] [PubMed] [Google Scholar]
- WANK S.A. G-Protein-coupled receptors in gastrointestinal physiology I. CCK receptors: an exemplary family. Am. J. Physiol. 1998;274:G607–G613. doi: 10.1152/ajpgi.1998.274.4.g607. [DOI] [PubMed] [Google Scholar]
- WILLIAMS J.A. Pancreatic acinar cell intracellular signalling mechanisms. Curr. Opin. Gastroenterol. 1997;13:369–374. [Google Scholar]
- YULE D.I., TSENG M.J., WILLIAMS J.A., LOGSDON C.D. A cloned CCK-A receptor transduces multiple signals in response to full and partial agonists. Am. J. Physiol. 1993;265:G999–G1004. doi: 10.1152/ajpgi.1993.265.5.G999. [DOI] [PubMed] [Google Scholar]
- YULE D.I., WILLIAMS J.A.Stimulus-secretion coupling in the pancreatic acinus Physiology of the Gastrointestinal Tract. 1994Raven Press, New York; 1447–1471.3rd ed. (Johnson LR, ed.) pp [Google Scholar]