

Abstract
The effects of desipramine (3 mg kg−1 i.p.) and clorgyline (1 mg kg−1 i.p.) on extracellular noradrenaline (NA) in the locus coeruleus (LC) and cingulate cortex were assessed in freely-moving rats by dual-probe microdialysis. Functional activities of α2-adrenoceptors regulating NA release in the LC and cingulate cortex were determined by systemic (0.3 mg kg−1 i.p.) or local (0.1–100 μM) clonidine administration.
Extracellular NA was increased in the LC and cingulate cortex following acute desipramine but not clorgyline treatment. Systemic clonidine decreased NA similarly in desipramine-, clorgyline-, and saline-treated animals, in both brain areas.
Long-term (twice daily, 14 days) but not short-term (twice daily, 7 days) desipramine, and long-term clorgyline (once daily, 21 days) treatments increased NA (3 fold) in cingulate cortex but not in the LC. Following long-term treatments, responses of NA to systemic clonidine were attenuated in the LC and cingulate cortex.
Clonidine perfusion by reverse dialysis into the cingulate cortex decreased local NA (−55±9%). The effect was attenuated by long-term desipramine (−31±9%) and clorgyline (−10±2%) treatments.
Clonidine perfusion by reverse dialysis into the LC decreased NA in the LC (−89±2%) and in cingulate cortex (−52±12%). This effect was attenuated in the LC following long-term desipramine (−72±4%) and clorgyline (−62±12%) treatments but it was not modified in the cingulate cortex (−57±10% and −68±6%, respectively).
These findings demonstrate that chronic desipramine or clorgyline treatments increase NA in noradrenergic terminal areas and desensitize α2-adrenoceptors modulating local NA release at somatodendritic and terminal levels. However, somatodendritic α2-adrenoceptors that control LC firing activity are not desensitized.
Keywords: α2-adrenoceptors, noradrenaline, microdialysis, desipramine, clorgyline, locus coeruleus, cingulate cortex
Introduction
One of the pathophysiological hypotheses of major depression relates to a decreased activity of central monoaminergic systems and more specifically of noradrenergic transmission (for reviews see Leonard, 1997; Charney, 1998; Ressler & Nemeroff, 1999).
The locus coeruleus (LC) is the main source of noradrenergic innervation to the brain and coeruleofugal noradrenergic fibres have been implicated in the pathophysiology of depressive disorders (Mongeau et al., 1997; Ressler & Nemeroff, 1999). The contribution of inhibitory α2-adrenoceptors located on noradrenergic terminals to the in vivo regulation of NA synthesis and release has been extensively evaluated (Pi & García-Sevilla, 1992; van veldhuizen et al., 1993; Dalley & Stanford, 1995; Meana et al., 1997). Somatodendritic α2-adrenoceptors also play an important role in the control of firing activity of LC neurones (Cedarbaum & Aghajanian, 1976) and, consequently, in NA release from noradrenergic fibres in terminal areas (van gaalen et al., 1997; Mateo et al., 1998). Furthermore, it has been demonstrated that α2-adrenoceptors regulate the somatodendritic release of NA in the LC area (Jorm & Stamford, 1993; Mateo et al., 1998; Callado & Stamford, 1999; 2000; Singewald et al., 1999).
Several studies have demonstrated that α2-adrenoceptor density is enhanced in post mortem brains of depressed suicide victims (Meana & García-Sevilla, 1987; Meana et al., 1992; González et al., 1994; Ordway et al., 1994; de paermentier et al., 1997; Callado et al., 1998; García-Sevilla et al., 1999). Down-regulation of α2-adrenoceptor density and desensitization of α2-adrenoceptor-mediated responses in the CNS are common responses to the chronic treatment with antidepressant drugs that increase synaptic NA concentration (Giralt & García-Sevilla, 1989; Menargues et al., 1990; Heal et al., 1991a, 1991b; Barturen & García-Sevilla, 1992; Mongeau et al., 1994; Esteban et al., 1999).
The sustained increase of extracellular NA concentrations following administration of antidepressant drugs has been proposed as the mechanism mediating down-regulation and desensitization of α2-adrenoceptors (Linnér et al., 1999). However, acute administration of the NA reuptake inhibitor desipramine enhances NA concentrations in the rat LC but decreases extracellular NA concentrations in terminal areas, reflecting an important in vivo tonic activation of autoinhibitory α2-adrenoceptors located on LC neurones (Mateo et al., 1998). Therefore, it seems that the in vivo antidepressant effects on extracellular NA concentrations in terminal areas exhibit important differences between acute and chronic treatments. Accordingly, it has been recently postulated that the time necessary to observe therapeutic efficacy of antidepressants reflects the time somatodendritic inhibitory α2-adrenoceptors need to become desensitized leading to a decrease in the inhibition of neurotransmitter release by nerve terminals (Linnér et al., 1999). In addition, desensitization of α2-adrenoceptors on noradrenergic terminals might contribute to increase NA availability in the area.
In the present study, the effects on noradrenergic transmission of two antidepressant drugs were investigated using in vivo microdialysis. The changes induced by acute and chronic administration of the NA reuptake inhibitor desipramine or the MAO inhibitor clorgyline on extracellular NA concentrations were monitored simultaneously in the LC and in the cingulate cortex, a projection area arising from the LC (van gaalen et al., 1997; Mateo et al., 1998). Dialysate NA concentrations reflect a balance between LC discharge rates affecting neurotransmitter release (Berridge & Abercrombie, 1999), inhibition of its reuptake and activation of inhibitory autoreceptors (Mateo et al., 1998). Therefore, the main objective of the study was to elucidate whether the putative α2-adrenoceptor desensitization induced by long-term antidepressant treatment contributes to modulate extracellular NA concentrations in the LC and in the cingulate cortex. For this purpose, the functional responses to systemic and local administration of the α2-adrenoceptor agonist drug clonidine were evaluated following saline, desipramine and clorgyline treatments. This approach allows us to examine separately the contribution of somatodendritic and terminal α2-adrenoceptors to extracellular NA concentrations under basal conditions.
Methods
Animals and treatment protocols
Experiments were performed on male Sprague-Dawley rats (University of the Basque Country, UPV/EHU, Spain). Animals were housed 4/5 per cage in a 12 h light-dark cycle at room temperature (22°C) with food and water available ad libitum. Animal care and all experimental protocols were performed in agreement with European Union regulations (O.J. of E.C. L 358/1 18/12/1986).
Animals were chronically treated with desipramine (3 mg kg−1 i.p., every 12 h for 7 or 14 days) or clorgyline (1 mg kg−1 i.p., every 24 h for 21 days). Control groups received 0.9% saline vehicle under similar conditions. Animals weighed 150–175 g at the start of treatment and 280–315 g at the time microdialysis experiments were carried out. Microdialysis probes were implanted under chloral hydrate anaesthesia (400 mg kg−1 i.p.) on day 8 or 15 for desipramine-treated groups and on day 22 for clorgyline-treated groups. Sample collection started 20–24 h later, to allow 36 and 48 h washout periods for desipramine and clorgyline treatments, respectively. Acute effects of desipramine (3 mg kg−1 i.p.) and clorgyline (1 mg kg−1 i.p.) were also tested by administration of a single injection of the drug. In these cases, drugs were given 2 h before the beginning of the collection period (Sato et al., 1994). For assessment of the functional status of α2-adrenoceptors following chronic or acute treatments, systemic (0.3 mg kg−1 i.p.) or local (0.1–100 μM) clonidine was administered during the collection period of dialysate samples.
Microdialysis procedures
Concentric dialysis probes were constructed as previously described (Adell & Artigas, 1991) using Cuprophan hollow fibers (Enka AG, Wuppertal, Germany). Rats were anaesthetized and placed in a David Kopf stereotaxic frame. Two microdialysis probes were implanted as previously described (Mateo et al., 1998). One probe (exposed tip 2.0 mm×0.25 mm) was implanted in the vicinity of the right LC (AP −3.7, L+1.3, V −8.2, taken from the λ suture point and the incisor bar lowered to a 15° angle) and the other one (exposed tip 4.0 mm×0.25 mm) in the ipsilateral cingulate cortex (AP +2.8, L+1.0, V −5.0, taken from bregma). The coordinates were chosen using the atlas of Paxinos & Watson (1986). Rats were allowed to recover from surgery overnight, one per cage, with food and water ad libitum.
Around 20 h after probe implantation, rats were placed individually in a CMA/120 system (CMA/Microdialysis AB, Sweden) for freely moving animals. The dialysis probes were connected to the system and then perfused with a modified CSF solution (mM): 148 NaCl, 2.7 KCl, 1.2 CaCl2 and 0.85 MgCl2, pH 7.4 at 1.0 μl min−1 flow rate (CMA/102 pump, CMA/Microdialysis AB). Dialysates were collected every 35 min into vials containing 5 μl perchloric acid 0.1 M. Following 1 h for stabilization, two or three consecutive samples were collected before clonidine administration and were considered as basal values. When administered locally, clonidine was dissolved in the artificial CSF and perfused by reverse dialysis through the probe in increasing concentrations 0.1 μM, 10 μM and 100 μM). Each concentration was perfused for two sampling periods of 35 min each. This period has been shown as a suitable time to produce a steady-state effect of a given dose of clonidine on extracellular NA concentrations (Mateo & Meana, 1999).
At the conclusion of the experiment, animals were killed with an overdose of chloral hydrate and brains were dissected out for histological examination to verify that microdialysis probes were correctly implanted. Only animals with correct microdialysis probe placement were considered. In vitro recovery for NA was in 10–16% range.
Analytical procedures
Immediately after collection, dialysate samples were assayed for NA by h.p.l.c. with electrochemical detection (injection volume 37 μl). Chromatographic analysis was made as previously reported (Mateo et al., 1998). The mobile phase composition was 12 mM citric acid, 1 mM EDTA, 1.2 mM octyl sodium sulphate (pH 5.0) and 12% (v v)−1 methanol, and was delivered at a flow rate of 0.8 ml min−1 by a Hewlett-Packard 1050 pump. NA was separated at room temperature (25°C) on a Chrospher 100RP-18, 5 μm (particle size), 125×4 mm column. Electrochemical detection of NA was achieved with an amperometric detector (Hewlett-Packard 1049A) at an oxidizing potential of+0.650 V. The limit of detection was 20–25 fmol per sample.
Statistical analysis
The mean values of the two or three dialysate samples obtained before clonidine or vehicle administration were considered as the 100% basal value. Extracellular NA concentrations of dialysate samples collected during an experiment were normalized as percentage of basal values. Values were not corrected for in vitro recovery.
The functional status of the α2-adrenoceptors evaluated as the time-course response of NA concentrations to clonidine administration was analysed. The modulation induced by desipramine and clorgyline treatments on extracellular NA concentrations and on effects induced by clonidine were assessed. One-way or two-way analysis of variance (ANOVA) for repeated measures, with treatment as between-subject factor and time as within-subject factor, were performed on normalized values. 2-tailed Student's t-test was used to evaluate differences between mean values. Results are expressed as mean±s.e.mean values. All statistical procedures were performed using the SYSTAT 8.0 software for Windows. Values of P less than 0.05 were considered statistically significant.
Drugs and reagents
Clonidine HCl, clorgyline HCl, desipramine HCl and NA bitartrate salt, were purchased from Sigma Chemical Co. (St. Louis, MO, U.S.A.). Chloral hydrate was provided by Fluka Chemie AG (Buchs, Switzerland). Reagents for h.p.l.c. analysis were of the highest purity available and were obtained from Merck (Darmstadt, Germany), Sigma Chemical Co. and SDS (Peypin, France). When applied, doses of the drugs are referenced as the salt forms.
Results
Effect of systemic clonidine administration on extracellular concentrations of NA in rats treated with a single dose of saline, desipramine or clorgyline
A preliminary set of dose-response experiments was undertaken to determine a dose of the α2-adrenoceptor agonist clonidine that would elicit a submaximal response on the regulation of extracellular NA concentrations. Thus, administration of different doses of clonidine (0.1, 0.3, 0.6 and 1.2 mg kg−1 i.p.) caused a dose-dependent decrease of extracellular NA in the cingulate cortex of control rats, with maximal observed decreases of −16%, −45%, −84% and −80%, respectively. The dose of 0.3 mg kg−1 i.p. clonidine was chosen for further experiments. Basal extracellular NA concentrations measured in the LC and in the cingulate cortex of control groups were in the range 3–6 nM (Table 1). In a saline-pretreated control group (1 ml kg−1 i.p., saline 2 h before the beginning of the sample collection) extracellular NA significantly decreased (F7,40=2.36, P<0.05) in the LC following systemic administration of clonidine (0.3 mg kg−1 i.p.), with a maximal observed effect of −50±10% (Figure 1a), and also in the cingulate cortex (F7,42=6.09, P<0.0001) with a maximal effect of −61±8% (Figure 1b). No significant effect of clonidine vehicle (saline) was observed in the LC (F7,16=0.82, P=0.59) and the cingulate cortex (F7,16=0.81, P=0.59) (Figure 1a,b).
Table 1.
Basal extracellular noradrenaline (NA) concentrations in locus coeruleus and cingulate cortex of rats treated with saline (control), desipramine or clorgyline
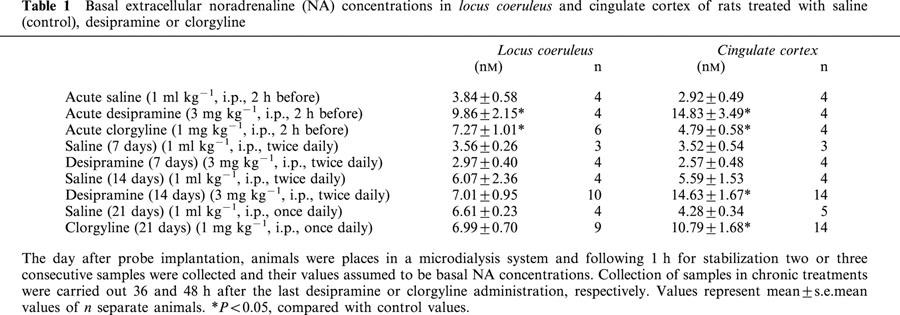
Figure 1.
Effects of systemic clonidine administration (0.3 mg kg−1, i.p.) on extracellular NA concentrations simultaneously evaluated in the LC (a) and cingulate cortex (b). Rats were treated with a single dose of desipramine (3 mg kg−1, i.p.), clorgyline (1 mg kg−1, i.p.) or saline (1 ml kg−1, i.p.) 2 h before the beginning of the sample collection. Arrows represent administration of clonidine or vehicle. Data are mean±s.e.mean values from four separate animals for each group and are expressed as percentages of the corresponding basal values.
In order to evaluate the acute effect of a single dose of desipramine (3 mg kg−1 i.p.) on the clonidine-induced decrease of NA, the drug was administered 2 h before the beginning of the collection period. There was a significant increase of basal extracellular NA concentrations in the LC (t=2.70, P<0.05) and in the cingulate cortex (t=3.38, P<0.05) when compared with the saline group (Table 1). Administration of clonidine (0.3 mg kg−1 i.p.) in this acutely desipramine-treated group decreased NA concentrations in the LC (F6,27=3.707, P<0.05) with a maximal effect of −63±13% (n=4) (Figure 1a). In the cingulate cortex, clonidine also decreased NA concentrations (F6,25=7.10, P<0.001) in the desipramine-treated group with an observed maximal response of −67±11% (n=4) (Figure 1b). No differences in the effects of clonidine were found between saline-, and desipramine-treated groups either in the LC (F1,8=0.09, P=0.77) or in the cingulate cortex (F1,8=0.31, P=0.59).
The acute effect of a single dose of clorgyline (1 mg kg−1 i.p., 2 h before the beginning of the collection period) on the clonidine-induced decrease of extracellular NA was also evaluated. In this clorgyline-treated group, basal NA concentrations in the LC (t=2.55, P<0.05) and the cingulate cortex (t=2.45, P<0.05) were significantly higher than values in the saline-treated group (Table 1). Following administration of clonidine (0.3 mg kg−1 i.p.) a decrease of NA concentration in the LC (F7,30=3.25, P<0.05) (Figure 1a) was observed, with a maximal effect of −40±17% (n=4). In the cingulate cortex, clonidine also decreased NA concentrations (F6,19=3.19, P<0.05) (Figure 1b) with a maximal response of −64±11% (n=4). There were no differences in the effects of clonidine between saline-, and clorgyline-treated groups both in the LC (F1,7=1.02, P=0.35) and the cingulate cortex (F1,7=1.66, P=0.24).
Effect of systemic clonidine administration on extracellular concentrations of NA in rats chronically treated with saline, desipramine or clorgyline
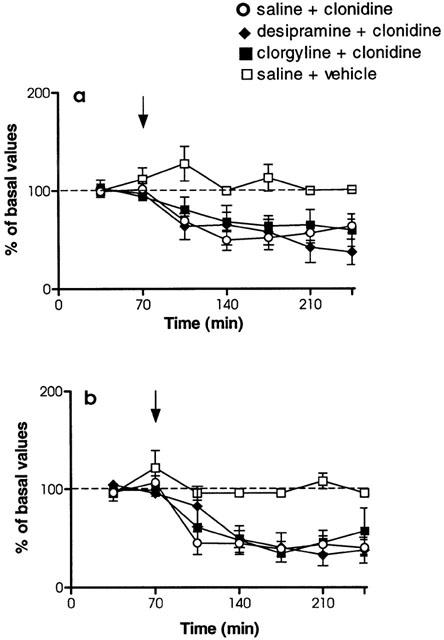
Basal extracellular concentrations of NA in the LC following chronic treatment with desipramine (3 mg kg−1 i.p., every 12 h, for 7 days) did not significantly differ from values observed in the respective saline group (t=1.13, P=0.30 in the LC; t=1.30, P=0.24 in cingulate cortex) (Table 1). Following administration of clonidine (0.3 mg kg−1 i.p.), approximately 36 h after the last desipramine injection, a decrease of NA was observed both in the LC (F6,53=4.04, P<0.001) with a maximal effect of −53±3% (n=4) (Figure 2a) and in the cingulate cortex (F6,54=4.92, P<0.0001) with a maximal effect of −50±6% (n=4) (Figure 2b). No significant differences in the effects of clonidine were found between saline-, and desipramine-treated groups in the LC (F1,8=0.53, P=0.49) and in the cingulate cortex (F1,8=2.04, P=0.19).
Figure 2.
Effects of systemic clonidine administration (0.3 mg kg−1, i.p.) on extracellular NA concentrations simultaneously evaluated in the LC (a) and cingulate cortex (b). Rats received short-term desipramine (3 mg kg−1, i.p., twice a day for 7 days), long-term desipramine (3 mg kg−1, i.p., twice a day for 14 days), long-term clorgyline (1 mg kg−1, i.p., once a day for 21 days) or saline (1 ml kg−1, i.p., 14-, and 21-days treatment represented as a single pool) treatment. Treatment was finished 36 h (desipramine) or 48 h (clorgyline) before the beginning of the sample collection. Arrows represent administration of clonidine. Data are mean±s.e.mean values from 4–7 separate animals for each treatment group and are expressed as percentages of the corresponding basal values. Data from clonidine vehicle are not included for clarification (see Figure 1 for details).
When treatment with desipramine was prolonged for 14 days, basal NA concentrations in the LC were not significantly different respect to values in the respective saline group (t=0.45, P=0.65). In contrast, NA concentrations in the cingulate cortex were significantly higher than values in the saline group (t=2.75, P<0.05) (Table 1). Administration of a single dose of clonidine induced a decrease of extracellular NA in the LC (F6,66=2.25 P<0.05) with a maximal effect of −27±11% (n=6) (Figure 2a). In the cingulate cortex, clonidine also induced a decrease in NA concentrations (F6,52=5.70, P<0.0001), with a maximal response of −27±16% (n=4) (Figure 2b). Significant differences in the effects of clonidine were observed between saline-, and desipramine-treated groups both in the LC (F1,10=10.29, P<0.01) and the cingulate cortex (F1,10=7.44, P<0.05).
A new group of animals was treated with clorgyline (1 mg kg−1 i.p., every 24 h, for 21 days). Following 48 h after the last clorgyline injection, basal concentrations of NA did not differ from the corresponding saline group values in the LC (t=0.34, P=0.73) but were significantly higher (t=2.26, P<0.05) in the cingulate cortex (Table 1). Extracellular NA was significantly decreased by systemic clonidine administration in the LC (F6,67=2.68, P<0.05) with a maximal effect of −40±17% (n=7) (Figure 2a) and in the cingulate cortex (F6,61=2,82, P<0.05) with a maximal effect of −38±11% (n=5) (Figure 2b). Significant differences in the effects of clonidine were also found between saline-, and clorgyline-treated groups in the LC (F1,11=11.05, P<0.01) and the cingulate cortex (F1,10=12.12, P<0.01).
Effect of local clonidine administration into the cingulate cortex on extracellular concentrations of NA in rats chronically treated with saline, desipramine or clorgyline
In order to discriminate whether observed effects of clonidine on NA in cingulate cortex were due to modulation of terminal autoreceptors, somatodendritic receptors controlling the LC firing activity, or both, further experiments with local clonidine administration were performed.
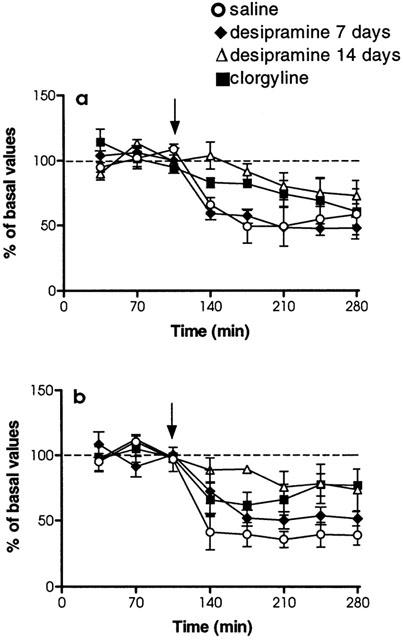
The effect of local administration of clonidine (0.1–100 μM), perfused through the dialysis probe located in the cingulate cortex, on extracellular concentrations of NA in the same brain area was evaluated. Clonidine locally applied caused a significant decrease in extracellular NA (F9,36=3.32, P<0.01) with a maximal observed effect of −55±9% (n=4) (Figure 3). When animals were treated with desipramine (3 mg kg−1 i.p., every 12 h, for 14 days), the local administration (approximately 36 h after the last desipramine dose) of clonidine through the dialysis probe did not produced a significant decrease of extracellular NA concentrations (F9,68=1.23, P=0.29) with a maximal effect of −31±9% (n=5) (Figure 3). A significant difference in the effects of clonidine was found between saline-, and desipramine-treated groups (F1,7=7.33, P<0.05). Following chronic treatment with clorgyline (1 mg kg−1 i.p., every 24 h, for 21 days), clonidine locally applied (approximately 48 h after the last clorgyline dose) in the cingulate cortex did not produced a significant decrease of extracellular NA concentrations (F9,65=0.60, P=0.78), the maximal effect being −10±2% (n=5) (Figure 3). Significant differences in the effects of clonidine were also found between saline-, and chronic clorgyline-treated groups (F1,7=11.66, P<0.05).
Figure 3.
Effects of local clonidine administration (0.1–100 μM) by reverse dialysis on extracellular NA concentrations in cingulate cortex. Rats received long-term desipramine (3 mg kg−1, i.p., twice a day for 14 days), long-term clorgyline (1 mg kg−1, i.p., once a day for 21 days) or saline (1 ml kg−1, i.p., 14-, and 21-days treatment represented as a single pool) treatment. Treatment was finished 36 h (desipramine) or 48 h (clorgyline) before the beginning of the sample collection. Arrows represent administration of clonidine. Data are mean±s.e.mean values from 4–5 separate animals for each treatment group and are expressed as percentages of the corresponding basal values. Data from clonidine vehicle are not included for clarification (see Figure 4b for details).
Effect of local clonidine administration into the LC on extracellular concentrations of NA evaluated simultaneously in the locus coeruleus and in the cingulate cortex of rats chronically treated with saline, desipramine or clorgyline
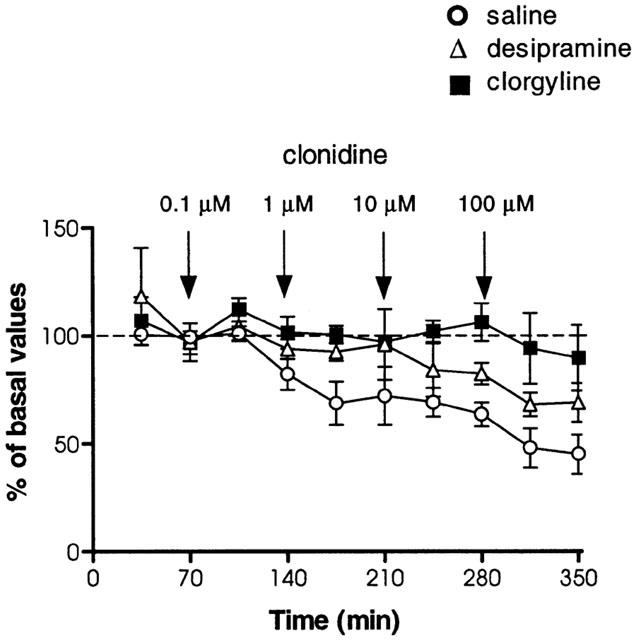
The effects of local administration of clonidine (0.1–100 μM), perfused through the dialysis probe located in the vicinity of the LC, on extracellular concentrations of NA in the LC and in the cingulate cortex were assessed simultaneously. In animals chronically treated with saline, administration of clonidine into the LC, significantly decreased extracellular NA in the LC (F9,39=2.48, P<0.05) with a maximal effect of −89±2% (n=4) (Figure 4a). Simultaneously, extracellular concentrations of NA evaluated in the cingulate cortex decreased significantly (F4,14=4.17, P<0.05) following local administration of clonidine into the LC, with a maximal decrease of −52±12% (n=4) (Figure 4b). Continuous perfusion of clonidine vehicle (artificial CSF) did not modify NA concentrations in the LC (F9,25=0.49, P=0.86) (Figure 4a) or the cingulate cortex (F9,23=0.88, P=0.55) (Figure 4b). In animals chronically treated with desipramine (3 mg kg−1 i.p., every 12 h, for 14 days), local administration of clonidine into the LC (approximately 36 h after the last desipramine injection) significantly decreased extracellular NA concentrations in the LC (F9,57=16.46, P<0.0001) with a maximal effect of −72±4% (n=4) (Figure 4b). In the same animals, extracellular NA concentrations in the cingulate cortex were significantly decreased in response to clonidine perfusion into the LC (F9,42=5.89, P<0.0001), with a maximal effect of −57±10% (n=4) (Figure 4b). The results in the LC displayed a significant difference in this effect of clonidine administered into the LC between the saline-, and the desipramine-treated groups (F1,6=7.63, P<0.05). A significant difference in the effect of clonidine between the same groups could not be observed in the cingulate cortex (F1,6=0.15, P=0.72). When animals were treated with clorgyline (1 mg kg−1 i.p., every 24 h, for 21 days), local administration of clonidine into the LC (approximately 48 h after the last clorgyline injection) significantly decreased extracellular NA concentrations in the LC (F9,50=3.13, P<0.001) with a maximal effect of −62±12% (n=4) (Figure 4a). When extracellular concentrations of NA in the same rats were analysed in the cingulate cortex, there was a significant decrease (F9,49=23.54, P<0.0001) induced by clonidine with a maximal effect of −68±6% (n=4) (Figure 4b). Similarly to results with desipramine treatment, the response of NA concentrations in the LC to clonidine was significantly different between saline-, and clorgyline-treated groups (F1,6=11.91, P<0.05). However, such a response evaluated in the cingulate cortex did not exhibit differences between saline-, and clorgyline-treated groups (F1,6=3.33, P=0.12).
Figure 4.
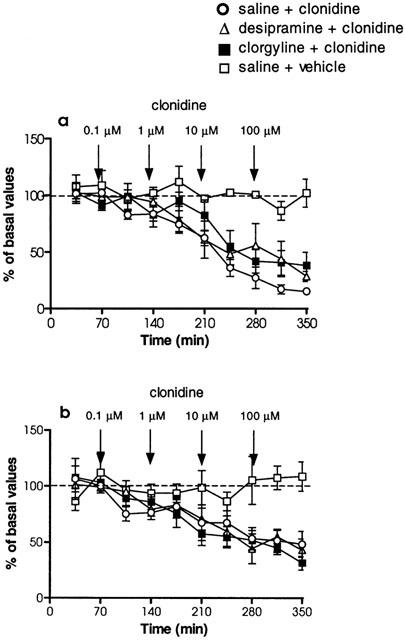
Effects of local clonidine administration (0.1–100 μM) by reverse dialysis into the LC area on extracellular NA concentrations simultaneously evaluated in the LC (a) and cingulate cortex (b). Rats received long-term desipramine (3 mg kg−1, i.p., twice a day for 14 days), long-term clorgyline (1 mg kg−1, i.p., once a day for 21 days) or saline (1 ml kg−1, i.p., 14-, and 21-days treatment represented as a single pool) treatment. Treatment was finished 36 h (desipramine) or 48 h (clorgyline) before the beginning of the sample collection. Arrows represent administration of clonidine or vehicle into the LC area. Data are mean±s.e.mean values from four separate animals for each treatment group and are expressed as percentages of the corresponding basal values.
Discussion
The present study demonstrates that in vivo modulation of central noradrenergic neurotransmission following chronic administration of desipramine or clorgyline, two drugs with different mechanisms of action, involves desensitization of inhibitory α2-adrenoceptors located on terminal areas. Under the same conditions, evidence was obtained that somatodendritic α2-adrenoceptors that modulate NA concentration in the cingulate cortex probably through the regulation of the firing activity of LC neurones are not desensitized. The dual-probe microdialysis technique makes possible the simultaneous investigation of extracellular NA concentration in the somatodendritic area of the LC and in projection areas arising from the LC, like the cingulate cortex (van gaalen et al., 1997, Mateo et al., 1998). Moreover, this approach allows us the evaluation of α2-adrenoceptor function differentiating between effects of local perfusion by reverse dialysis of the α2-adrenoceptor agonist clonidine into the LC and effects of similar perfusion into the cingulate cortex. Applying this methodology, the separate contribution of somatodendritic and terminal α2-adrenoceptors to the attenuation of clonidine effects induced by desipramine and clorgyline chronic administrations was clearly elucidated.
In agreement with earlier studies, acute desipramine administration (3 mg kg−1 i.p.) increased extracellular NA in the frontal cortex (L'heureux et al., 1986; Dennis et al., 1987; Tanda et al., 1996; Mateo et al., 1998; Seo et al., 1999) and in the LC (Mateo et al., 1998) (Table 1). Similarly, other NA re-uptake inhibitors augment extracellular NA concentrations in different brain regions following acute administration (Rowley et al., 1998; Linnér et al., 1999; Sacchetti et al., 1999; Wortley et al., 1999). Acute administration of desipramine or clorgyline does not affect α2-adrenoceptor sensitivity. Thus, the decrease of extracellular NA induced by clonidine exhibited curves in saline-, desipramine-, and clorgyline-treated animals that did not differ from each other and reached similar maximal effects (Figure 1). From these acute findings and given the enhancement of NA induced by acute desipramine treatment, it can be concluded that the presence of elevated extracellular NA concentrations in the LC and in the cingulate cortex does not disturb the evaluation of α2-adrenoceptor-mediated responses to systemic clonidine. That is, the attenuation of NA responsiveness to clonidine following long-term administration of desipramine or clorgyline does not represent a competition phenomenon between the synthetic agonist and the endogenous neurotransmitter for bind α2-adrenoceptors.
Overall, the results suggest that chronic desipramine (14 days) or clorgyline (21 days) treatments lead to an increased availability of NA at cortical level in the synaptic cleft which in turn induces desensitization of presynaptic α2-adrenoceptors regulating NA release by nerve terminals. This finding extends and, in general, agrees well with previously reported data about antidepressant and electroconvulsive shock effects on α2-adrenoceptor functional activity at presynaptic (Heal et al., 1991b; Thomas et al., 1992; Mongeau et al., 1994; Yoshioka et al., 1995; Esteban et al., 1999; Invernizzi et al., 2001; Sacchetti et al., 2001) and postsynaptic (Bill et al., 1989; Menargues et al., 1990; Heal et al., 1991a, 1991b) levels in the CNS. On the other hand, long-term treatment with NA reuptake or MAO inhibitors has been shown to reduce the density of brain α2-adrenoceptors (Smith et al., 1981; Cohen et al., 1982; Giralt & García-Sevilla, 1989; Barturen & García-Sevilla, 1992; Ribas et al., 1993). Therefore, the observed desensitization of clonidine-induced inhibition of NA release in cortex following these treatments appears to be the functional expression of a decrease in the density of α2-adrenoceptors at this specific brain level. The lack of significant modulations of cortical NA concentrations and α2-adrenoceptor sensitivity following short-term (7 days) treatment with desipramine (Table 1; Figure 2b) should be interpreted in the context of a longer time requirement, together with a maintained presence of antidepressant levels, as conditions needed to observe changes in noradrenergic transmission (Daniel & Melzacka, 1992); otherwise adaptive modulations are not observed (Sacchetti et al., 1999). Recent results with reboxetine have strengthened the idea that prolonged availability of high NA concentrations is necessary to induce α2-adrenoceptor desensitization (Invernizzi et al., 2001).
Long-term treatment with desipramine or clorgyline had little effect on NA concentrations in the LC whereas α2-adrenoceptors-mediated modulation of NA release at this level was markedly attenuated (Table 1; Figures 2a and 4a). Although there is a good evidence that NA concentrations in the LC are controlled by a population of α2-adrenoceptors located at this level (Jorm & Stamford, 1993; Mateo et al., 1998; Callado & Stamford, 1999; 2000), this functional response has not previously been employed to evaluate adaptive changes of α2-adrenoceptors induced by antidepressant treatments. The source of extracellular NA in the LC area is controversial and includes release from somatodendritic origin, release from nerve terminals of recurrent collaterals, as well as release from terminals of noradrenergic inputs arising from other cell groups (for a review, see Singewald & Philippu, 1998). Extracellular NA concentrations in the LC are sensitive to local reuptake or MAO inhibition (Palij & Stamford, 1994; Thomas et al., 1994; Mateo et al., 1998) but also to other multiple stimuli (Singewald & Philippu, 1998; Kaehler et al., 1999; Kawahara et al., 1999; Singewald et al., 1999) including antidepressant modulation of local 5-HT concentrations (Mateo et al., 2000). The present results show that the antidepressant-induced desensitization of α2-adrenoceptors located on noradrenergic terminals is mirrored by changes in the α2-adrenoceptor population that controls NA release in the LC area (Figures 2, 3 and 4a). Therefore, α2-adrenoceptors modulating NA release in the LC area could represent a local receptor population expressed on axon terminals and/or collaterals of noradrenergic inputs to the LC (Singewald & Philippu, 1998). Consistent with the location, these α2-adrenoceptors are desensitized by chronic antidepressant treatments. In contrast, α2-adrenoceptors also located in the LC but involved in the control of noradrenergic cell firing activity seem to represent a separate receptor population. Thus, long-term desipramine and clorgyline treatments did not attenuate the decrease of cortical NA concentrations induced by local administration of clonidine into the LC (Figure 4b). Since NA release in the cingulate cortex represents the firing activity of LC neurones (Florin-Lechner et al., 1996; Berridge & Abercrombie, 1999), the finding indicates that the susceptibilities for desensitization of somatodendritic and terminal α2-adrenoceptors are different. This apparent lack of desensitization is consistent with previous electrophysiological evidence showing that chronic desipramine or clorgyline treatments do not affect the sensitivity of α2-adrenoceptors that inhibit the firing activity of LC neurones (Blier & De Montigny, 1985; Lacroix et al., 1991). However, subsensitivity of this functional response has also been described, especially under short washout periods following chronic antidepressant treatment (Svensson & Usdin, 1978; Valentino et al., 1990). The finding is also in agreement with very recent autoradiography data showing that chronic desipramine treatment did not modify [3H]-RX821002 binding to α2-adrenoceptors in the rat locus coeruleus (Sacchetti et al., 2001). Whether the differential modulation of α2-adrenoceptors located in the LC following chronic antidepressant treatment represents actually the presence of two separate α2-adrenoceptor populations (terminal and somatodendritic autoreceptors) is a question that should be raised in subsequent studies.
A pharmacokinetic interaction between clonidine and desipramine or clorgyline is unlikely to explain the subsensitivity of NA responses to clonidine administration. First, clonidine does not show an important metabolism and it is cleared primarily by a renal mechanism, at least in humans (Lowenthal et al., 1988). Second, the local administration by reverse dialysis excludes clonidine from suffering systemic metabolism, and, therefore, from pharmacokinetic interactions at this level.
In conclusion, long-term but not short-term antidepressant treatment induces an in vivo desensitization of α2-adrenoceptors regulating the local release of NA both in the LC and in the cingulate cortex, whereas α2-adrenoceptors that modulate the firing activity of LC noradrenergic neurones remains unaltered. The present results are consistent with the delayed onset of action of antidepressant drugs and provide neurochemical support for the hypothesis that some of their clinical actions may be exerted by down-regulation of α2-adrenoceptors, in agreement with some previous evidence in brain of depressed subjects (de paermentier et al., 1997; García-Sevilla et al., 1999). Furthermore, the therapeutic activity of these antidepressants seems to be more related to the down-regulation of abnormally up-regulated α2-adrenoceptors in noradrenergic terminal areas of human brain (Meana et al., 1992; González et al., 1994; de paermentier et al., 1997; Callado et al., 1998; García-Sevilla et al., 1999) than to similar process on up-regulated α2-adrenoceptors in the LC area (Ordway et al., 1994).
Acknowledgments
Work supported by grants from FIS (98/0059) and the University of the Basque Country (G13/98). Y. Mateo and B. Fernández-Pastor are former and current recipients of Basque Government predoctoral fellowships, respectively.
Abbreviations
- 5-HT
5-hydroxytryptamine
- LC
locus coeruleus
- NA
noradrenaline
References
- ADELL A., ARTIGAS F. Differential effects of clomipramine given locally or systemically on extracellular 5-hydroxytryptamine in raphe nuclei and frontal cortex. Naunyn Schmiedeberg's Arch. Pharmacol. 1991;343:237–244. doi: 10.1007/BF00251121. [DOI] [PubMed] [Google Scholar]
- BARTUREN F., GARCÍA-SEVILLA J.A. Long-term treatment with desipramine increases the turnover of α2-adrenoceptors in the rat brain. Mol. Pharmacol. 1992;42:846–855. [PubMed] [Google Scholar]
- BERRIDGE C.W., ABERCROMBIE E.D. Relationship between locus coeruleus discharge rates and rates of norepinephrine release within neocortex as assessed by in vivo microdialysis. Neuroscience. 1999;93:1263–1270. doi: 10.1016/s0306-4522(99)00276-6. [DOI] [PubMed] [Google Scholar]
- BILL D.J., HUGHES I.E., STEPHENS R.J. The effects of acute and chronic desipramine on the thermogenic and hypoactivity responses to α2-adrenoceptor agonists in reserpinized and normal mice. Br. J. Pharmacol. 1989;96:144–152. doi: 10.1111/j.1476-5381.1989.tb11794.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BLIER P., DE MONTIGNY C. Serotonergic but not noradrenergic neurons in rat central nervous system adapt to long-term treatment with monoamine oxidase inhibitors. Neuroscience. 1985;16:949–955. doi: 10.1016/0306-4522(85)90107-1. [DOI] [PubMed] [Google Scholar]
- CALLADO L.F., MEANA J.J., GRIJALBA B., PAZOS A., SASTRE M., GARCÍA-SEVILLA J.A. Selective increase of α2A-adrenoceptor agonist binding sites in brains of depressed suicide victims. J. Neurochem. 1998;70:1114–1123. doi: 10.1046/j.1471-4159.1998.70031114.x. [DOI] [PubMed] [Google Scholar]
- CALLADO L.F., STAMFORD J.A. α2A- But not α2B/C-adrenoceptors modulate noradrenaline release in rat locus coeruleus: voltammetric data. Eur. J. Pharmacol. 1999;366:35–39. doi: 10.1016/s0014-2999(98)00889-9. [DOI] [PubMed] [Google Scholar]
- CALLADO L.F., STAMFORD J.A. Spatiotemporal interaction of α2-autoreceptors and noradrenaline transporters in the rat locus coeruleus: implications for volume transmission. J. Neurochem. 2000;74:2350–2358. doi: 10.1046/j.1471-4159.2000.0742350.x. [DOI] [PubMed] [Google Scholar]
- CEDARBAUM J.M., AGHAJANIAN G.K. Noradrenergic neurons of the locus coeruleus: inhibition by epinephrine and activation by the α-antagonist piperoxane. Brain Res. 1976;112:413–419. doi: 10.1016/0006-8993(76)90297-3. [DOI] [PubMed] [Google Scholar]
- CHARNEY D.S. Monoamine dysfunction and the pathophysiology and treatment of depression. J. Clin. Psychiatry. 1998;59:11–14. [PubMed] [Google Scholar]
- COHEN R.M., CAMPBELL I.C., DAUPHIN J.F., TALLMAN J.F., MURPHY D.L. Changes in α and β receptor densities in rat brain as a result of treatment with monoamine oxidase inhibiting antidepressants. Neuropharmacology. 1982;21:293–298. doi: 10.1016/0028-3908(82)90091-0. [DOI] [PubMed] [Google Scholar]
- DALLEY J., STANFORD S.C. Contrasting effects of the imidazol(in)e α2-adrenoceptor agonist, medetomidine, clonidine and UK14304 on extraneuronal levels of noradrenaline in the rat frontal cortex: evaluation using in vivo microdialysis and synaptosomal uptake studies. Br. J. Pharmacol. 1995;114:1717–1723. doi: 10.1111/j.1476-5381.1995.tb14962.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DANIEL W., MELZACKA M. A comparative study on desipramine pharmacokinetics in the rat brain after administration of desipramine or imipramine. J. Pharm. Pharmacol. 1992;44:429–432. doi: 10.1111/j.2042-7158.1992.tb03638.x. [DOI] [PubMed] [Google Scholar]
- DENNIS T., L'HEUREUX R., CARTER C., SCATTON B. Presynaptic alpha-2 adrenoceptors play a major role in the effects of idazoxan on cortical noradrenaline release (as measured by in vivo dialysis) in the rat. J. Pharmacol. Exp. Ther. 1987;241:642–649. [PubMed] [Google Scholar]
- DE PAERMENTIER F., MAUGER J.M., LOWTHER S., CROMPTON M.R., KATONA C.L.E., HORTON R.W. Brain α-adrenoceptors in depressed suicides. Brain Res. 1997;757:60–68. doi: 10.1016/s0006-8993(97)00138-8. [DOI] [PubMed] [Google Scholar]
- ESTEBAN S., LLADO J., SASTRE-COLL A., GARCÍA-SEVILLA J.A. Activation and desensitization by cyclic antidepressant drugs of α2-autoreceptors, α2-heteroreceptors and 5-HT1A-autoreceptors regulating monoamine synthesis in the rat brain in vivo. Naunyn-Schmiedeberg's Arch. Pharmacol. 1999;360:135–143. doi: 10.1007/s002109900045. [DOI] [PubMed] [Google Scholar]
- FLORIN-LECHNER S.M., DRUHAN J.P., ASTON-JONES G., VALENTINO R.J. Enhanced norepinephrine release in prefrontal cortex with burst stimulation of the locus coeruleus. Brain Res. 1996;742:89–97. doi: 10.1016/s0006-8993(96)00967-5. [DOI] [PubMed] [Google Scholar]
- GARCÍA-SEVILLA J.A., ESCRIBA P.V., OZAITA A., LA HARPE R., WALZER C., EYTAN A., GUIMON J. Up-regulation of immunolabeled α2A-adrenoceptors, Gi coupling proteins, and regulatory receptor kinases in the prefrontal cortex of depressed suicides. J. Neurochem. 1999;72:282–291. doi: 10.1046/j.1471-4159.1999.0720282.x. [DOI] [PubMed] [Google Scholar]
- GIRALT M.T., GARCÍA-SEVILLA J.A. Acute and long-term regulation of brain α2-adrenoceptors after manipulation of noradrenergic transmission in the rat. Eur. J. Pharmacol. 1989;164:455–466. doi: 10.1016/0014-2999(89)90253-7. [DOI] [PubMed] [Google Scholar]
- GONZÁLEZ A.M., PASCUAL J., MEANA J.J., BARTUREN F., DEL ARCO C., PAZOS A., GARCÍA-SEVILLA J.A. Autoradiographic demonstration of increased α2-adrenoceptor agonist binding sites in the hippocampus and frontal cortex of depressed suicide victims. J. Neurochem. 1994;63:256–265. doi: 10.1046/j.1471-4159.1994.63010256.x. [DOI] [PubMed] [Google Scholar]
- HEAL D.J., PROW M.R., BUCKETT W.R. Determination of the role of noradrenergic and 5-hydroxytryptaminergic neurones in postsynaptic α2-adrenoceptor desensitization by desipramine and ECS. Br. J. Pharmacol. 1991a;103:1865–1870. doi: 10.1111/j.1476-5381.1991.tb12343.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HEAL D.J., PROW M.R., BUCKETT W.R. Effects of antidepressant drugs and electroconvulsive shock on pre- and postsynaptic α2-adrenoceptor function in the brain: rapid down-regulation by sibutramine hydrochloride. Psychopharmacology. 1991b;103:251–257. doi: 10.1007/BF02244212. [DOI] [PubMed] [Google Scholar]
- INVERNIZZI R.W., PARINI S., SACCHETTI G., FRACASSO C., CACCIA S., ANNONI K., SAMANIN R. Chronic treatment with reboxetine by osmotic pumps facilitates its effect on extracellular noradrenaline and may desensitize α2-adrenoceptors in the prefrontal cortex. Br. J. Pharmacol. 2001;132:183–188. doi: 10.1038/sj.bjp.0703821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JORM C.M., STAMFORD J.A. Actions of the hypnotic anaesthetic, dexmedetomidine, on noradrenaline release and cell firing in rat locus coeruleus slices. Br. J. Anaesthesia. 1993;71:447–449. doi: 10.1093/bja/71.3.447. [DOI] [PubMed] [Google Scholar]
- KAEHLER S.T., SINGEWALD N., PHILIPPU A. The release of catecholamines in hypothalamus and locus coeruleus is modulated by peripheral chemoreceptors. Naunyn-Schmiedeberg's Arch. Pharmacol. 1999;360:428–434. doi: 10.1007/s002109900094. [DOI] [PubMed] [Google Scholar]
- KAWAHARA Y., KAWAHARA H., WESTERINK B.H.C. Comparison of effects of hypotension and handling stress on the release of noradrenaline and dopamine in the locus coeruleus and medial prefrontal cortex of the rat. Naunyn-Schmiedeberg's Arch. Pharmacol. 1999;360:42–49. doi: 10.1007/s002109900042. [DOI] [PubMed] [Google Scholar]
- LACROIX D., BLIER P., CURET O., DE MONTIGNY C. Effects of long-term desipramine administration on noradrenergic neurotransmission: electrophysiological studies in the rat brain. J. Pharmacol. Exp. Ther. 1991;257:1081–1090. [PubMed] [Google Scholar]
- LEONARD B. The role of noradrenaline in depression: A review. J. Psychopharmacol. 1997;11:S39–S47. [PubMed] [Google Scholar]
- L'HEUREUX R., DENNIS T., CURET O., SCATTON B. Measurement of endogenous noradrenaline release in the rat cerebral cortex in vivo by transcortical dialysis: effects of drugs affecting noradrenergic transmission. J. Neurochem. 1986;46:1794–1801. doi: 10.1111/j.1471-4159.1986.tb08498.x. [DOI] [PubMed] [Google Scholar]
- LINNÉR L., ARBORELIUS L., NOMIKOS G.G., BERTILSSON L., SVENSSON T.H. Locus coeruleus neuronal activity and noradrenaline availability in the frontal cortex of rats chronically treated with imipramine: effect of α2-adrenoceptor blockade. Biol. Psychiatry. 1999;46:766–774. doi: 10.1016/s0006-3223(99)00126-2. [DOI] [PubMed] [Google Scholar]
- LOWENTHAL D.T., MATZEK K.M., MACGREGOR T.R. Clinical pharmacokinetics of clonidine. Clin. Pharmacokinet. 1988;14:287–310. doi: 10.2165/00003088-198814050-00002. [DOI] [PubMed] [Google Scholar]
- MATEO Y., MEANA J.J. Determination of the somatodendritic α2-adrenoceptor subtype located in rat locus coeruleus that modulates cortical noradrenaline release in vivo. Eur. J. Pharmacol. 1999;379:53–57. doi: 10.1016/s0014-2999(99)00488-4. [DOI] [PubMed] [Google Scholar]
- MATEO Y., PINEDA J., MEANA J.J. Somatodendritic α2-adrenoceptors in the locus coeruleus are involved in the in vivo modulation of cortical noradrenaline release by the antidepressant desipramine. J. Neurochem. 1998;71:790–798. doi: 10.1046/j.1471-4159.1998.71020790.x. [DOI] [PubMed] [Google Scholar]
- MATEO Y., RUIZ-ORTEGA J.A., PINEDA J., UGEDO L., MEANA J.J. Inhibition of 5-hydroxytryptamine reuptake by the antidepressant citalopram in the locus coeruleus modulates the rat brain noradrenergic transmission in vivo. Neuropharmacology. 2000;39:2036–2043. doi: 10.1016/s0028-3908(00)00041-1. [DOI] [PubMed] [Google Scholar]
- MEANA J.J., BARTUREN F., GARCÍA-SEVILLA J.A. α2-adrenoceptors in the brain of suicide victims: increased receptor density associated with major depression. Biol. Psychiatry. 1992;31:471–490. doi: 10.1016/0006-3223(92)90259-3. [DOI] [PubMed] [Google Scholar]
- MEANA J.J., GARCÍA-SEVILLA J.A. Increased α2-adrenoceptor density in the frontal cortex of depressed suicide victims. J. Neural Transm. 1987;70:377–381. doi: 10.1007/BF01253612. [DOI] [PubMed] [Google Scholar]
- MEANA J.J., HERRERA-MARSCHITZ M., GOINY M., SILVEIRA R. Modulation of catecholamine release by α2-adrenoceptors and I1-imidazoline receptors in rat brain. Brain Res. 1997;744:216–226. doi: 10.1016/s0006-8993(96)01080-3. [DOI] [PubMed] [Google Scholar]
- MENARGUES A., OBACH R., GARCÍA-SEVILLA J.A. Modulation by antidepressant drugs of CNS postsynaptic α2-adrenoceptors mediating mydriasis in the rat. Naunyn-Schmiedeberg's Arch. Pharmacol. 1990;341:101–107. doi: 10.1007/BF00195065. [DOI] [PubMed] [Google Scholar]
- MONGEAU R., BLIER P., DE MONTIGNY C. The serotonergic and noradrenergic systems of the hippocampus: their interactions and the effects of antidepressant treatments. Brain Res. Rev. 1997;23:145–195. doi: 10.1016/s0165-0173(96)00017-3. [DOI] [PubMed] [Google Scholar]
- MONGEAU R., DE MONTIGNY C., BLIER P. Electrophysiologic evidence for desensitization of α2-adrenoceptors on serotonin terminals following long-term treatment with drugs increasing norepinephrine synaptic concentration. Neuropsychopharmacology. 1994;10:41–51. doi: 10.1038/npp.1994.6. [DOI] [PubMed] [Google Scholar]
- ORDWAY G.A., WIDDOWSON P.S., SMITH K.S., HALARIS A. Agonist binding to α2-adrenoceptors is elevated in the locus coeruleus from victims of suicide. J. Neurochem. 1994;63:617–624. doi: 10.1046/j.1471-4159.1994.63020617.x. [DOI] [PubMed] [Google Scholar]
- PALIJ P., STAMFORD J.A. Rauwolscine potentiates the effect of desipramine on limbic noradrenaline efflux. Neuroreport. 1994;7:1121–1124. doi: 10.1097/00001756-199604260-00003. [DOI] [PubMed] [Google Scholar]
- PAXINOS G., WATSON C. The Rat Brain in Stereotaxic Coordinates 1986Academic Press, Orlando; 2nd edition [Google Scholar]
- PI F., GARCÍA-SEVILLA J.A. α2-Autoreceptor-mediated modulation of tyrosine hydroxylase activity in noradrenergic regions of the rat brain in vivo. Naunyn-Schmiedeberg's Arch. Pharmacol. 1992;345:653–660. doi: 10.1007/BF00164579. [DOI] [PubMed] [Google Scholar]
- RESSLER K.J., NEMEROFF C.B. Role of norepinephrine in the pathophysiology and treatment of mood disorders. Biol. Psychiatry. 1999;46:1219–1233. doi: 10.1016/s0006-3223(99)00127-4. [DOI] [PubMed] [Google Scholar]
- RIBAS C., MIRALLES A., GARCÍA-SEVILLA J.A. Acceleration by chronic treatment with clorgyline of the turnover of brain α2-adrenoceptors in normotensive but not in spontaneous hypertensive rats. Br. J. Pharmacol. 1993;110:99–106. doi: 10.1111/j.1476-5381.1993.tb13777.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROWLEY H.L., KILPATRICK I.C., NEEDHAM P.L., HEAL D.J. Elevation of extracellular cortical noradrenaline may contribute to the antidepressant activity of zotepine: an in vivo microdialysis study in freely moving rats. Neuropharmacology. 1998;37:937–944. doi: 10.1016/s0028-3908(98)00094-x. [DOI] [PubMed] [Google Scholar]
- SACCHETTI G., BERNINI M., BIANCHETTI A., PARINI S., INVERNIZZI R.W., SAMANIN R. Studies on the acute and chronic effects of reboxetine on extracellular noradrenaline and other monoamines in the rat brain. Br. J. Pharmacol. 1999;128:1332–1338. doi: 10.1038/sj.bjp.0702926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SACCHETTI G., BERNINI M., GOBBI M., PARINI S., PIRONA L., MENNINI T., SAMANIN R. Chronic treatment with desipramine facilitates its effect on extracellular noradrenaline in the rat hippocampus: studies on the role of presynaptic α2-adrenoceptors. Naunyn-Schmiedeberg's Arch. Pharmacol. 2001;363:66–72. doi: 10.1007/s002100000334. [DOI] [PubMed] [Google Scholar]
- SATO Y., SHIBANOKI S., SUGAHARA M., ISHIKAWA K. Measurement and pharmacokinetic analysis of imipramine and its metabolite by brain microdialysis. Br. J. Pharmacol. 1994;112:625–629. doi: 10.1111/j.1476-5381.1994.tb13120.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SEO D.O., SHIN C.Y., LEE C.J., DAILEY J.W., REITH M.E.A., JOBE P.C., KO K.H. Effect of alterations in extracellular norepinephrine on adrenoceptors: a microdialysis study in freely moving rats. Eur. J. Pharmacol. 1999;365:39–46. doi: 10.1016/s0014-2999(98)00856-5. [DOI] [PubMed] [Google Scholar]
- SINGEWALD N., KAEHLER S.T., PHILIPPU A. Noradrenaline release in the locus coeruleus of conscious rats is triggered by drugs, stress and blood pressure changes. NeuroReport. 1999;10:1583–1587. doi: 10.1097/00001756-199905140-00035. [DOI] [PubMed] [Google Scholar]
- SINGEWALD N., PHILIPPU A. Release of neurotransmitters in the locus coeruleus. Prog. Neurobiol. 1998;56:237–267. doi: 10.1016/s0301-0082(98)00039-2. [DOI] [PubMed] [Google Scholar]
- SMITH C.B., GARCÍA-SEVILLA J.A., HOLLINGSWORTH P.J. α2-adrenoceptors in rat brain are decreased after long-term tricyclic antidepressant drug treatment. Brain Res. 1981;210:413–418. doi: 10.1016/0006-8993(81)90919-7. [DOI] [PubMed] [Google Scholar]
- SVENSSON T.H., USDIN T. Feedback inhibition of brain noradrenaline neurons by tricyclic antidepressants: α-receptor mediation. Science. 1978;202:1089–1091. doi: 10.1126/science.213833. [DOI] [PubMed] [Google Scholar]
- TANDA G., FRAU R., DI CHIARA G. Chronic desipramine and fluoxetine differentially affect extracellular dopamine in the rat prefrontal cortex. Psychopharmacology. 1996;127:83–87. doi: 10.1007/BF02805978. [DOI] [PubMed] [Google Scholar]
- THOMAS D.N., NUTT D.J., HOLMAN R.B. Effects of acute and chronic electroconvulsive shock on noradrenaline release in the rat hippocampus and frontal cortex. Br. J. Pharmacol. 1992;106:430–434. doi: 10.1111/j.1476-5381.1992.tb14351.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THOMAS D.N., POST R.M., PERT A. Focal and systemic cocaine differentially affect extracellular norepinephrine in the locus coeruleus, frontal cortex and hippocampus of the anaesthetized rat. Brain Res. 1994;645:135–142. doi: 10.1016/0006-8993(94)91646-2. [DOI] [PubMed] [Google Scholar]
- VALENTINO R.J., CURTIS A.L., PARRIS D.G., WEHBY R.G. Antidepressant actions on brain noradrenergic neurons. J. Pharmacol. Exp. Ther. 1990;253:833–840. [PubMed] [Google Scholar]
- VAN GAALEN M., KAWAHARA H., KAWAHARA Y., WESTERINK B.H.C. The locus coeruleus noradrenergic system in the rat brain studied by dual-probe microdialysis. Brain Res. 1997;763:56–62. doi: 10.1016/s0006-8993(97)00416-2. [DOI] [PubMed] [Google Scholar]
- VAN VELDHUIZEN M.J.A., FEENSTRA M.G.P., HEINSBROEK R.P.W., BOER G.J. In vivo microdialysis of noradrenaline overflow: effects of α-adrenoceptor agonists and antagonists measured by cumulative concentration-response curves. Br. J. Pharmacol. 1993;109:655–660. doi: 10.1111/j.1476-5381.1993.tb13623.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WORTLEY K.E., HEAL D.J., STANFORD S.C. Modulation of sibutramine-induced increases in extracellular noradrenaline concentration in rat frontal cortex and hypothalamus by α2-adrenoceptors. Br. J. Pharmacol. 1999;128:659–666. doi: 10.1038/sj.bjp.0702859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YOSHIOKA M., MATSUMOTO M., NUMAZAWA R., TOGASHI H., SMITH C.B., SAITO H. Changes in the regulation of 5-hydroxytryptamine release by α2-adrenoceptors in the rat hippocampus after long-term desipramine treatment. Eur. J. Pharmacol. 1995;294:565–570. doi: 10.1016/0014-2999(95)00582-x. [DOI] [PubMed] [Google Scholar]