

Abstract
The ability of a series of homologues and analogues of palmitoylethanolamide to inhibit the uptake and fatty acid amidohydrolase (FAAH)-catalysed hydrolysis of [3H]-anandamide ([3H]-AEA) has been investigated.
Palmitoylethanolamide and homologues with chain lengths from 12–18 carbon atoms inhibited rat brain [3H]-AEA metabolism with pI50 values of ∼5. Homologues with chain lengths ⩽eight carbon atoms gave <20% inhibition at 100 μM.
R-palmitoyl-(2-methyl)ethanolamide, palmitoylisopropylamide and oleoylethanolamide inhibited [3H]-AEA metabolism with pI50 values of 5.39 (competitive inhibition), 4.89 (mixed type inhibition) and 5.33 (mixed type inhibition), respectively.
With the exception of oleoylethanolamide, the compounds did not produce dramatic inhibition of [3H]-WIN 55,212-2 binding to human CB2 receptors expressed on CHO cells. Palmitoylethanolamide, palmitoylisopropylamide and R-palmitoyl-(2-methyl)ethanolamide had modest effects upon [3H]-CP 55,940 binding to human CB1 receptors expressed on CHO cells.
Most of the compounds had little effect upon the uptake of [3H]-AEA into C6 and/or RBL-2H3 cells. However, palmitoylcyclohexamide (100 μM) and palmitoylisopropylamide (30 and 100 μM) produced more inhibition of [3H]-AEA uptake than expected to result from inhibition of [3H]-AEA metabolism alone.
In intact C6 cells, palmitoylisopropylamide and oleoylethanolamide inhibited formation of [3H]-ethanolamine from [3H]-AEA to a similar extent as AM404, whereas palmitoylethanolamide, palmitoylcyclohexamide and R-palmitoyl-(2-methyl)ethanolamide were less effective.
These data provide useful information upon the ability of palmitoylethanolamide analogues to act as ‘entourage' compounds. Palmitoylisopropylamide may prove useful as a template for design of compounds that reduce the cellular accumulation and metabolism of AEA without affecting either CB1 or CB2 receptors.
Keywords: Anandamide, fatty acid amidohydrolase, palmitoylethanolamide
Introduction
It is now well established that the endogenous fatty acid amide anandamide (AEA) has, as a result of its actions upon cannabinoid and vanilloid receptors, a number of interesting pharmacological properties including effects on nociception (Calignano et al., 1998; Jaggar et al., 1998; Richardson et al., 1998; Raffa et al., 1999; Gauldie et al., 2001), memory processes (Terranova et al., 1995; Mallet & Beninger, 1998), lung function (Calignano et al., 2000), spasticity (Baker et al., 2001) and cell proliferation (de petrocellis et al., 1998; Sarker et al., 2000; MacCarrone et al., 2000) (for a recent review of the therapeutic potential of AEA and related endocannabinoids, see Piomelli et al., 2000).
AEA is metabolised by a fatty acid amide hydrolase (Deutsch & Chin, 1993), and inhibition of this enzyme by phenylmethylsulphonyl fluoride and palmitylsulphonyl fluoride, have been shown to potentiate the pharmacological actions of AEA both in vitro and in vivo (Childers et al., 1994; Pertwee et al., 1995; Compton & Martin, 1997; Gifford et al., 1999). This raises the possibility that compounds interfering with the metabolism of AEA may be therapeutically useful. A number of FAAH inhibitors have been described, often based on the structure of AEA itself (see e.g. Koutek et al., 1994; Bisogno et al., 1998; Boger et al., 2000b), although an inherent disadvantage in this approach is that some of the compounds are per se active at cannabinoid receptors. An alternative approach is to prevent the metabolism of AEA with compounds related to palmitoylethanolamide, since this fatty acid amide, although a substrate for FAAH (Natarajan et al., 1984) is inactive at cannabinoid receptors (Lambert et al., 1999). Indeed, the sulphonyl fluoride analogue of palmitoylethanolamide is a potent inhibitor of AEA metabolism by rat brain FAAH (IC50 7 nM) with rather weak effects on the binding of the CB agonist [3H]-CP 55,940 to rat forebrain CB1 receptors (Deutsch et al., 1997).
An alternative to transition state inhibitors such as sulphonyl fluoride analogues of fatty acid amides is to use homologues of palmitoylethanolamide to compete with [3H]-AEA for the FAAH active site. In a recent study, a number of homologues and analogues of palmitoylethanolamide were synthesized and demonstrated not to be active towards either CB1 or CB2 receptors at concentrations of 1 μM, (Lambert et al., 1999; see also Lang et al., 1999 for data with stearoylethanolamide and oleoylethanolamide). In the present study, we have investigated the abilities of these compounds to prevent the FAAH-catalysed metabolism of AEA and their interference with cellular AEA uptake, in the hope of identifying possible therapeutically useful ‘entourage' compounds. In addition, we have investigated a novel N-methyl analogue of palmitoylethanolamide, R-palmitoyl-(2-methyl)ethanolamide. The structures of palmitoylethanolamide, R-palmitoyl-(2-methyl)ethanolamide and the two compounds found to show the best activity, palmitoylisopropylamide (C16:0) and oleoylethanolamide (C18:1), are shown in Figure 1.
Figure 1.
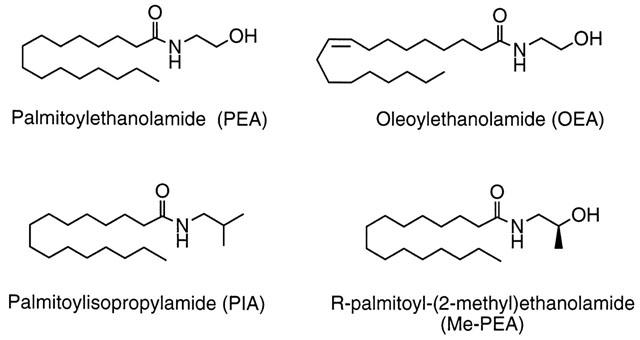
Structures of palmitoylethanolamide (C16:0), oleoylethanolamide (C18:1), palmitoylisopropylamide (C16:0) and R-palmitoyl-(2-methyl)ethanolamide (C16:0).
Methods
Compounds
Anandamide [ethanolamine-1-3H] ([3H]-AEA; specific activity 60 Ci mmol−1) was obtained from American Radiolabeled Chemicals, Inc. (St. Louis, MO, U.S.A.). [3H]-WIN 55,212-2 (specific activity 45.5 Ci mmol−1) and [3H]-CP55,940 (specific activity 101.1 Ci mmol−1) were obtained from New England Nuclear, Boston, MA, U.S.A. Decanoylethanolamide (C10:0), lauroylethanolamide (C12:0), myristoylethanolamide (C14:0), palmitoylethanolamide (C16:0), palmitoylbutylamide (C16:0), palmitoylisopropylamide (C16:0), palmitoylcyclohexamide (C16:0) and oleoylethanolamide (C18:1) were synthesized as earlier described by Lambert et al. (1999). Non-radioactive anandamide (arachidonoylethanolamide, AEA), (R)-(+)-arachidonyl-1′-hydroxy-2′-propylamide (R1-methanandamide), heptadecanoylethanolamide (C17:0), stearoylethanolamide (C18:0), palmitoyltrifluoromethyl ketone (PTMK) and arachidonyl-serotonin were purchased from Cayman Chemical Company (Ann Arbor, MI, U.S.A.). R-palmitoyl-(2-methyl)ethanolamide (C16:0) was custom synthesized by the same company on our request and given the catalogue number 90357. N-(4-hydroxyphenyl)arachidonylamide (AM404) was purchased from Tocris Cookson (Bristol, U.K.). Fatty acid free bovine serum albumin (BSA) was obtained from the Sigma Chemical Co. (St Louis, MO, U.S.A.). All cell culture media, sera and supplements were from Gibco/Life Technologies.
Synthesis of propanoylethanolamide (C3:0)
In a two-neck flask, 14.06 g of ethanolamine (0.23 mol) were poured into 20 ml of dry methylene chloride. The solution was cooled in an ice bath and magnetically stirred. Propionyl chloride (5.325 g, 57 mmol) was added dropwise. The reaction mixture was stirred for 12 h at room temperature, filtered, and the filtrate was evaporated under reduced pressure. The residue of filtrate was chromatographed on silica gel (eluting with ethyl acetate: methyl alcohol 8 : 2) to give 5.031 g (74%) of a colourless oil. TLC (ethyl acetate: methyl alcohol 8 : 2, v v−1) Rf=0.5; spectroscopic data: 1H NMR (d6-DMSO): δ(p.p.m.) 1.11 (t, J=24 Hz, 3H); 2.13-2.6 (m, 6H); 3.72–3.76 (m, NH); 2.64 (OH); 13C NMR (d6-DMSO): δ(p.p.m.) 10.64 (CH3); 29.37, 42.37, 60.90 (CH2); 174.28 (C=O) mass spectrometry; [M+.]=117; IR ν(cm−1): 1648 (C=O); 3304 (OH).
Synthesis of butanoylethanolamide (C4:0)
In a two-neck flask, 11.76 g of ethanolamine (0.192 mol) were poured into 20 ml of dry methylene chloride. The solution was cooled in an ice bath and magnetically stirred. Butyryl chloride (5.11 g, 48 mmol) was added dropwise. The reaction mixture was stirred for 12 h at room temperature, filtered, and the filtrate was evaporated under reduced pressure. The residue of filtrate was chromatographed on silica gel (eluting with ethyl acetate: methyl alcohol 8 : 2, v v−1) to give 4.57 g (73%) of a yellowish oil. TLC (ethyl acetate: methyl alcohol 8 : 2, v v−1) Rf=0.68; spectroscopic data: 1H NMR (d6-DMSO): δ(p.p.m.) 0.85 (t, J=12 Hz, 3H); 1.48–1.55 (m, 2H); 2.06 (t, J=15 Hz, 2H); 3.11–3.16 (m, 2H); 3.39–3.44 (m, 2H); 4.67–4.7 (m, NH); 3.50 (OH); 13C NMR (d6 - DMSO): δ(p.p.m.) 14.02 (CH3); 19.13, 37.83, 41.90, 60.54 (CH2); 172.79 (C=O); mass spectrometry [M+]=131; IR ν(cm−1): 1648 (C=O); 3302 (OH).
Synthesis of hexanoylethanolamide (C6:0)
In a two-neck flask, 1.294 g of 1,1′-carbonyldiimidazole (7.98 mmol) were poured into 20 ml of dry methylene chloride. The solution was cooled in an ice bath and magnetically stirred. Hexanoic acid (0.92 g, 7.98 mmol) was added dropwise. Ethanolamine (1.95 g, 31.9 mmol) was then added to the reaction mixture, which was stirred for 24 h at room temperature. After this incubation, the reaction mixture was washed with water, 10% citric acid solution and NaCl saturated solution and dried (MgSO4) and the solvent was evaporated under reduced pressure. The residue was chromatographed on silica gel (eluting with ethyl acetate: methyl alcohol 8 : 2, v v−1) to give 1.02 g (80%) of a white solid. Mp: 44–45°C (uncorrected); TLC (ethyl acetate: methyl alcohol 8 : 2, v v−1): Rf=0.68; 1H NMR (d6-DMSO): δ(p.p.m.) 0.8 (t, J=12 Hz, 3H); 1.18–1.23 (m, 4H); 1.40–1.44 (m, 2H); 1.99 (t, J=15 Hz, 2H); 3.03–3.07 (m, 2H); 3.37–3.40 (m, 2H); 4.58–4.60 (m, NH); 3.63 (OH); 13C NMR (d6-DMSO): δ(p.p.m.) 13.80 (CH3); 21.86, 24.94, 30.91, 35.34; 41.4; 60 (CH2); 172.31 (C=O); mass spectrometry, [M+.]=159; IR ν(cm−1): 1639 (C=O); 3297 (OH).
Synthesis of octanoylethanolamide (C8:0)
In a two-neck flask, 7.35 g of ethanolamine (0.12 mol) were poured into 15 ml of dry methylene chloride. The solution was cooled in an ice bath and magnetically stirred. Octanoyl chloride (1.95 g, 12 mmol) was added dropwise. The reaction mixture was stirred for 12 h at room temperature and then filtered. The solid was recrystallized in ethanol-water to give 2.06 g (92%). TLC (acetone: methylene chloride 9 : 1, v v−1) Rf=0.67; spectroscopic data: 1H NMR (d-CDCl3): δ(p.p.m.) 0.86(t, J=7 Hz, 3H); 1.11–1.32 (m, 8H); 1.59–1.61 (m, 2H); 2.17–2.23 (t, J=15 Hz, 2H); 3.36–3.42 (m, 2H); 3.67–3.7 (m, 2H); 4.02 (NH); 13C NMR (d-CDCl3): δ(p.p.m.) 14.06 (CH3); 22.62, 25.81, 29.05, 29.29, 31.72, 36.70, 42.42, 61.98 (CH2); 174.74 (C=O); mass spectrometry [M+.]=187; IR ν(cm−1): 1639 (C=O); 3307 (OH). m.p.=57–58°C.
Membrane preparation for FAAH assay
Frozen brains (minus cerebellum) stored at −70°C from adult rats were thawed and homogenized at 4°C in 20 mM HEPES buffer, pH 7.0, with 1 mM MgCl2 using a glass homogenizer. The homogenates were centrifuged twice at 36,000×g for 20 min at 4°C after which the tissue pellets were re-suspended in homogenization buffer and incubated at 37°C for 15 min. After centrifugation at 36,000×g for 20 min at 4°C, membranes were re-suspended in 50 mM Tris-HCl buffer, pH 7.4, containing 1 mM EDTA and 3 mM MgCl2. The protein content in the membrane preparations were determined according to the method of Harrington (1990), using bovine serum albumin as standard, and the preparations were stored at −70°C until used for assay.
FAAH assay
The method used was first described by Omeir et al. (1995) who used [14C]-labelled AEA as substrate. In the present study we used [3H]-AEA (Fowler et al., 1997). Briefly, membranes (10 μg protein assay−1 unless otherwise stated), test compounds or ethanol carrier (10 μl) and assay buffer (10 mM Tris-HCl, 1 mM EDTA, 1% (w v−1) BSA, pH 7.6) (final assay volume of 200 μl) were preincubated for 0–180 min, as indicated, at 37°C. [3H]-AEA (25 μl) (2 μM final concentration unless otherwise stated) was added and the samples were incubated at 37°C for 10 min, unless otherwise stated. Reactions were stopped by placing the tubes in ice and adding 400 μl of chloroform : methanol (1 : 1 v v−1). After vortex mixing of the tubes the phases were separated by centrifugation in a bench centrifuge. Aliquots (200 μl) of the methanol/buffer phase were removed and analysed for radioactivity by liquid scintillation spectroscopy with quench correction. Blanks contained distilled water instead of the homogenate preparations.
Radioligand binding experiments
Human CB1- (‘CHO-CB1') and CB2- (‘CHO-CB2') transfected CHO cells, kindly donated by Drs Nokin and Detheux (Euroscreen, Belgium) were maintained in culture using Ham's F12 medium containing 10% fetal bovine serum, 100 μg/ml streptomycin, 100 U ml−1 penicillin and 200 μg/ml G418. Membranes (40 μg) and test compounds were incubated at 30°C with 1 nM [3H]-CP55,940 (CHO-CB1) or 1 nM [3H]-WIN 55,212-2 (CHO-CB2) for 1 h in 50 mM Tris-HCl with MgCl2 and EDTA (pH 7.4) in the presence of 50 μM PMSF. Non-specific binding was determined with 10 μM HU-210 (CHO-CB1) or 10 μM WIN 55,212-2 (CHO-CB2). After incubation, the membrane suspension was rapidly filtered through 0.5% polyethyleneimine-pretreated GF/B glass fibre filters (Whatman), and the radioactivity trapped on the filters was measured by liquid scintillation spectroscopy. Assays were undertaken in quadruplicate.
Cell cultures
Rat basophilic leukaemia (RBL-2H3) cells (passage range 14–22) and rat C6 glioma cells (passage range 52–65) were obtained from the American Type Culture Collection, MD, U.S.A. The cells were grown in 75 cm2 culturing flasks at 37°C, 5% CO2 in air at normal atmospheric pressure. The cells were cultured in Eagle's minimum essential medium, 2 mM L-glutamine supplemented with 15% foetal bovine serum and 100 units ml−1 penicillin+100 μg ml−1 streptomycin (RBL-2H3) or Ham's F10 medium, supplemented with 10% foetal bovine serum and 100 units ml−1 penicillin+100 μg ml−1 streptomycin (C6). The culture media were changed three times a week.
[3H]-AEA uptake in RBL-2H3 and C6 cells
The method developed by Rakhshan et al. (2000) was used. RBL-2H3 or C6 cells were plated on 24 well plates at an initial density of 1.5–2×105 cells.well−1 and incubated at 37°C for 18 h under an atmosphere of 5% CO2 in air at normal atmospheric pressure. After the incubation period, cells were washed once with 0.5 ml warm assay buffer (mM: NaCl 120, KCl 4.7, CaCl2 2.2, HEPES 10, KH2PO4 1.2, MgSO4 1.2, pH 7.4; gassed with 95% O2, 5% CO2) and preincubated in 350 μl buffer with the test compounds or ethanol carrier at 37°C for 10 min. [3H]-AEA (50 μl, final concentration 10 μM) was added to the wells and uptake at 37°C was allowed for 15 min. After the incubation, the plates were placed on ice after which the cells were rinsed three times with ice-cold buffer containing 1% BSA to terminate the transport. The cells were aspirated of the buffer and incubated at 75°C in 0.2 M NaOH (500 μl well−1) for 15 min. Aliquots (300 μl) of the solubilized cells were transferred to scintillation vials and assayed for tritium content by liquid scintillation spectroscopy with quench correction. Test substances and AEA were diluted in ethanol and buffer and the ethanol concentration was constant between the different wells and never exceeded 5%. Previous experiments using RBL-2H3 cells have demonstrated that this concentration of ethanol does not significantly affect the uptake of [3H]-AEA (Jacobsson & Fowler, 2001).
Metabolism of [3H]-AEA in intact C6 glioma cells
The method used was a modification of the [3H]-AEA uptake assay but where [3H]-ethanolamine was collected. C6 cells were seeded in 24-well flat bottom plates (initial density of 0.12×106 cells well−1) in Ham's F10 medium, supplemented with 1% foetal bovine serum and 100 units ml−1 penicillin+100 μg ml−1 streptomycin. After incubation overnight, the wells were washed once with 400 μl warm assay buffer (mM: NaCl 120, KCl 4.7, CaCl2 2.2, HEPES 10, KH2PO4 1.2, MgSO4 1.2, pH 7.4; gassed with 95% O2, 5% CO2) and pre-incubated for 10 min at 37°C in 350 μl buffer plus 25 μl of either carrier (ethanol), test compound or phenylmethylsulphonyl fluoride (PMSF, 1.5 mM final concentration). [3H]-AEA (25 μl, final concentration 3 μM) was added to the wells. The plates were then incubated for 30 min at 37°C with the lids on, after which they were placed on ice and aliquots (400 μl) of methanol added to each well. The wells were scraped and the contents placed in test tubes. Chloroform (200 μl) was added, the test tubes were vortex mixed and centrifuged to separate the phases. Aliquots (200 μl) of the aqueous phase (containing the [3H]-ethanolamine) were taken and counted for radioactivity by scintillation spectroscopy with quench correction. Blanks were defined as the radioactivity recovered for samples on a separate plate preincubated with PMSF.
Determination of pI50, Ki(slope) and Ki(intercept) values
pI50 values [−log10(IC50 value)] were analysed using the built-in equation ‘sigmoid dose-response (variable slope)' of the GraphPad Prism computer programme (GraphPad Software Inc., San Diego, CA, U.S.A.). Two different approaches were used. In order to determine the maximal inhibition produced by the compounds, the data (expressed as per cent of control) were analysed using ‘top' (i.e. uninhibited) values fixed at 100. For compounds where the ‘bottom' values (i.e. minimum activity remaining) were significantly greater than 0 (i.e. when the 95% confidence intervals of the value were both greater than 0), the pI50 values (i.e. reflecting the potency with respect to the inhibitable fraction of the [3H]-AEA metabolism) were taken from these analyses. For compounds where the 95% confidence intervals of the minimum activity remaining straddled zero, the pI50 values were taken from analyses where the bottom value was set to 0. This approach was taken to avoid artefactually low pI50 values resulting from the use of unrealistic ‘bottom values'. An example of this would be decanoylethanolamide (C10:0), where the calculated ‘bottom' value was −332±1288 (95% confidence limit −3011 to +2347). Apparent Km and Vmax values for inhibition of [3H]-AEA metabolism by palmitoylisopropylamide (C16:0) and oleoylethanolamide (C18:1) were calculated from the mean data using the Direct Linear Plot analysis (Eisenthal & Cornish-Bowden, 1974) using the Enzyme Kinetics v 1.4 software package, Trinity Software, Campton, NH, U.S.A. These values were then used in secondary replots to determine Ki(slope) and Ki(intercept) values.
Statistics
ANOVA tests for repeated measures and Bonnferroni-Dunn post hoc tests were conducted using the Statview™ computer programme (SAS Institute Inc., Cary, NC, U.S.A.).
Results
Validation of blank value for assay of FAAH
In the FAAH assay used, blanks are defined as the tritium recovered in the aqueous phase following incubation of [3H]-anandamide ([3H]-AEA) in the absence of homogenate (see Methods section). In view of the finding that some of the palmitoylethanolamide analogues and homologues do not produce complete inhibition of [3H]-anandamide metabolism (see below), it was considered to be of importance to confirm that the blanks determined in this way are valid. To this end, three compounds known to inhibit FAAH, namely arachidonoyl-serotonin, AM404 and arvanil (Bisogno et al., 1998; Melck et al., 1999; Jarrahian et al., 2000) and one presumed inhibitor (palmitoyl trifluoromethyl ketone, PTMK; ‘presumed' since the arachidonoyl- and oleoyl- trifluoromethyl ketones are highly potent transition-state FAAH inhibitors, Koutek et al., 1994; Patterson et al., 1996). All four compound produced a complete inhibition of [3H]-AEA hydrolysis with calculated minimum activity remaining (‘bottom') values that were not different from zero (Figure 2), thereby confirming the validity of the blank. In the case of PTMK, the pI50 value was 7.10±0.02.
Figure 2.
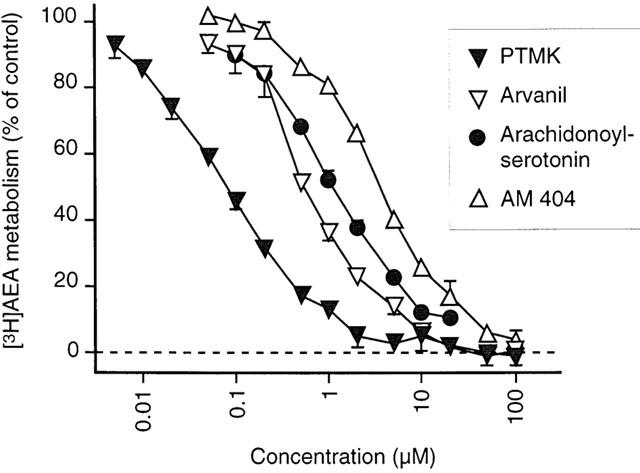
Inhibition of the hydrolysis by rat brain membranes of 2 μM [3H]-AEA by arachidonoyl-serotonin, AM404, arvanil and PTMK. Data are means±s.e. mean (when not enclosed by the symbol), n=3–6, with no preincubation with the compounds and membranes prior to addition of [3H]-AEA. Analysis of the data using a constant ‘top' value of 100 and variable slope (see Methods) gave ‘bottom' values (i.e. the plateau value of the remaining activity not inhibited by the compounds) of: arachidonoyl-serotonin, 3.7±2.2% (−0.8 to +8%); AM404, 1.3±2.1% (−3.1 to +5.7%); arvanil, 0.9±1.9% (−2.9 to +4.7%); PTMK, 0.3±1.4% (−2.5 to +3.0%) (means±s.e.mean, with 95% confidence intervals in parentheses). With the ‘bottom' values set to zero, the pI50 values (means±s.e.mean) calculated from these data were 5.93±0.02, 5.44±0.02, 6.20±0.03 and 7.10±0.02 for arachidonoyl-serotonin, AM404, arvanil and PTMK, respectively.
Inhibition of FAAH catalysed [3H]-AEA hydrolysis by homologues and analogues of palmitoylethanolamide
A series of homologues of palmitoylethanolamide, where the side-chain was reduced in length from C16:0 (palmitoylethanolamide) to C3:0 (propanoylethanolamide) and increased to C18:0 (stearoylethanolamide) were tested for their ability to prevent [3H]-AEA hydrolysis (Figure 3a,b). The observed inhibition will reflect both the potency and the solubility of the compound, the latter particularly in the case of the long-chain compounds. Indeed, our experience is that the solubility of these saturated fatty acid amides can vary from preparation to preparation, due to temperature-dependent polymorphism of the pure crystals (J. Wouters, S. Vandevoorde & D. Lambert, unpublished results). In order to calculate potencies, the maximum inhibition was calculated and the pI50 values determined for this inhibitable component. Using this approach, it was seen that complete inhibition of [3H]-AEA could be attained for the compounds with side-chain lengths of 10, 12 and 14 carbon atoms, whereas only partial inhibition (78, 55 and 33% for 16, 17 and 18 carbon atoms, respectively) was seen at longer chain lengths (Table 1). This partial inhibition presumably reflects limited solubility of the compounds rather than the presence of an FAAH enzyme subtype with which the compounds do not interact. In the absence of a preincubation phase, the potencies of the compounds towards the inhibitable component of the activity were similar for the compounds with side chains ranging from 18 to 12 carbon atoms (pI50 values 4.77–4.95) with the possible exception of palmitoylethanolamide itself (pI50 value of 5.30). This value for palmitoylethanolamide corresponds to an IC50 value of 5.1 μM, which is in good agreement with the KM value of this compound as a substrate for rat brain FAAH using the same assay conditions (range 1.3–8.5 μM, Tiger et al., 2000). For the 10 carbon atom, a lower potency was seen (pI50 value of 4.26), and for the four compounds with side chain lengths of 8, 6, 4 and 3 carbon atoms, <20% inhibition of [3H]-AEA metabolism was seen at the highest concentration tested (100 μM). Oleoylethanolamide, the mono-unsaturated analogue of stearoylethanolamide, had sufficient solubility and was able completely to inhibit [3H]-AEA metabolism with a pI50 value of 5.33.
Figure 3.
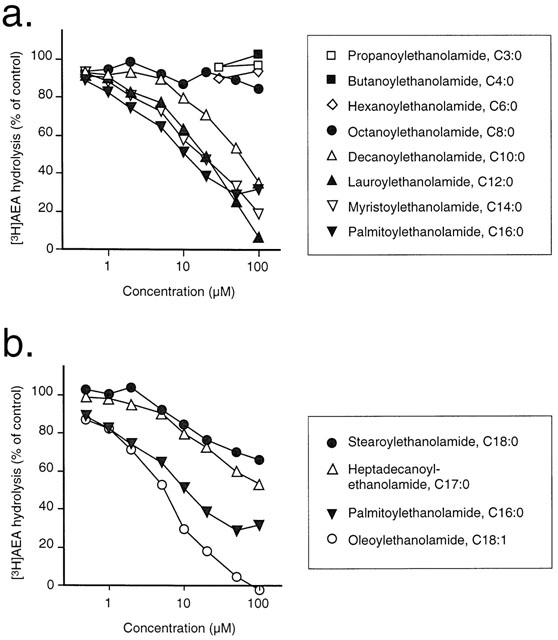
Inhibition of the hydrolysis by rat brain membranes of 2 μM [3H]-AEA by a series of palmitoylethanolamide homologues (a) C16:0–C3:0 (b) C16:0–C18:0 and C18:1. Shown are means of 3–6 experiments, with no preincubation with the compounds and membranes prior to addition of [3H]-AEA. The data for palmitoylethanolamide are the same in both graphs.
Table 1.
Effect of compounds related to palmitoylethanolamide upon the hydrolysis of [3H]-AEA by rat brain FAAH

Four analogues of palmitoylethanolamide where the ethanolamide group was replaced with butylamide, isopropylamide, cyclohexamide and (2-methyl)ethanolamide groups were also investigated for their abilities to prevent [3H]-AEA metabolism (Figure 4). The isopropylamide, butylamide and (2-methyl)ethanolamide analogues were active with pI50 values similar to that of palmitoylethanolamide, although complete inhibition of [3H]-AEA metabolism was not seen with any of them, presumably for reasons of solubility (Table 1). The cyclohexamide analogue produced 11% inhibition at the highest concentration tested (100 μM).
Figure 4.
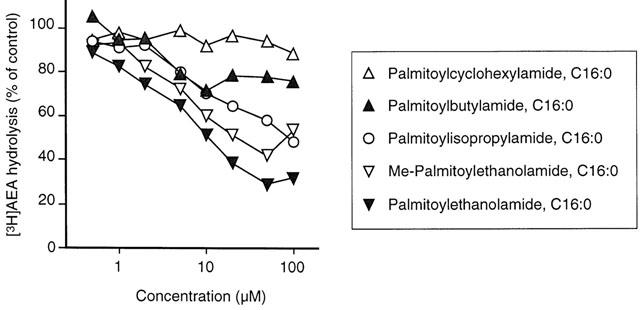
Inhibition of the hydrolysis by rat brain membranes of 2 μM [3H]-AEA by a series of palmitoylethanolamide analogues. Shown are means of 3–4 experiments, with no preincubation with the compounds and membranes prior to addition of [3H]-AEA. The data for palmitoylethanolamide are the same as that shown in Figure 3.
The above experiments were undertaken in the absence of preincubation. Two series of experiments were undertaken to determine whether a preincubation of homogenate with test compound affected the observed inhibition of [3H]-AEA uptake. In the first series, dose-response experiments (analogous to those shown in Figures 3 and 4) for all the compounds except stearoyl- and heptadecanolethanolamide were undertaken using a 60 min preincubation phase. With two exceptions, this treatment did not affect the observed maximum inhibition or the potency of the compounds (data not shown). The two exceptions were for palmitoylethanolamide and palmitoylisopropylamide, where the 95% confidence intervals of the minimum activity remaining straddled zero. In the case of palmitoylethanolamide, the apparent residual activity was 15±8% [95% confidence intervals −2 to 33] (as opposed to 22±7% (95% confidence intervals +7 to +37) in the absence of preincubation). The pI50 value for palmitoylethanolamide was calculated to be 5.12±0.13 assuming a residual inhibition and 4.90±0.06 assuming no residual inhibition. This difference underlines the importance of regarding the pI50 values as approximate rather than exact estimates of potency. For palmitoylisopropylamide, the apparent pI50 value (assuming maximal inhibition) was 4.71±0.05 following a 60 min preincubation period.
In a second experiment, the 10 : 0–16 : 0 ethanolamide compounds, oleoylethanolamide, palmitoylisopropylamide, palmitoylbutylamide and R-palmitoyl-(2-methyl)ethanolamide were pre-incubated for 0, 60, 120 and 180 min at 37°C with a stronger homogenate concentration (50 μg protein assay−1) before being incubated with [3H]-AEA for a shorter time (3 min). Single concentrations of the compounds, chosen to be on the steep part of the concentration-response curve were tested (data not shown). Only lauroylethanolamide (20 μM) exhibited a small, but significant reduction in its ability to prevent AEA hydrolysis as the preincubation time was increased. Thus, the per cent activity remaining following 0, 60, 120 and 180 min of incubation with laurylethanolamide were 51±3, 51±5, 57±1 and 62±4%, respectively (means±s.e.mean, n=3, one-way ANOVA for repeated measures (preincubation time) gave F3,6=8.4 (P<0.05)). For comparison, 15 μM palmitoylethanolamide gave values of 50±3, 53±1, 58±3 and 53±5%, respectively (means±s.e.mean, n=3, P>0.05). The same pattern was seen with lauryethanolamide when a 10 min incubation time and a more dilute homogenate was used (data not shown).
Mode of inhibition of [3H]-AEA metabolism by palmitoylisopropylamide, oleoylethanolamide and R-palmitoyl-(2-methyl)ethanolamide
The modes of inhibition of [3H]-AEA metabolism by palmitoylisopropylamide and oleoylethanolamide are shown in Figure 5. Both compounds acted as mixed-type inhibitors with Ki(slope) and Ki(intercept) values, respectively of 15 μM and 87 μM (palmitoylisopropylamide) and 3.9 μM and 11 μM for oleoylethanolamide. R-palmitoyl-(2-methyl)ethanolamide was also investigated and found to be a competitive inhibitor with a Ki value of 6.6 μM (data not shown).
Figure 5.
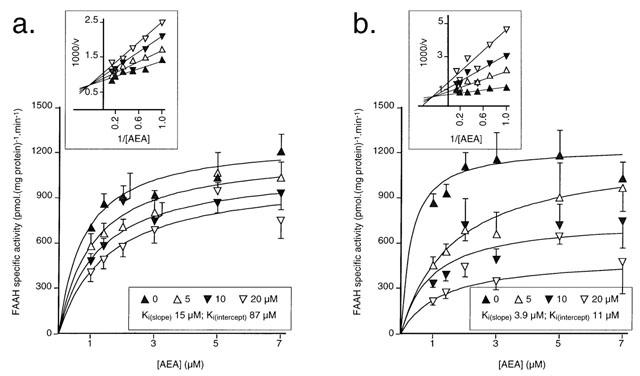
Mode of inhibition of rat brain [3H]-AEA metabolism by (a) palmitoylisopropylamide and (b) oleoylethanolamide. Shown are means±s.e. mean of three experiments. Secondary replots of the mean data to illustrate the mixed-type nature of the inhibition are shown as inserts. The Ki(slope) and Ki(intercept) values were calculated from the mean data as described in Methods.
Interaction of the analogues and homologues of palmitoylethanolamide with cannabinoid CB receptors expressed on CHO cells
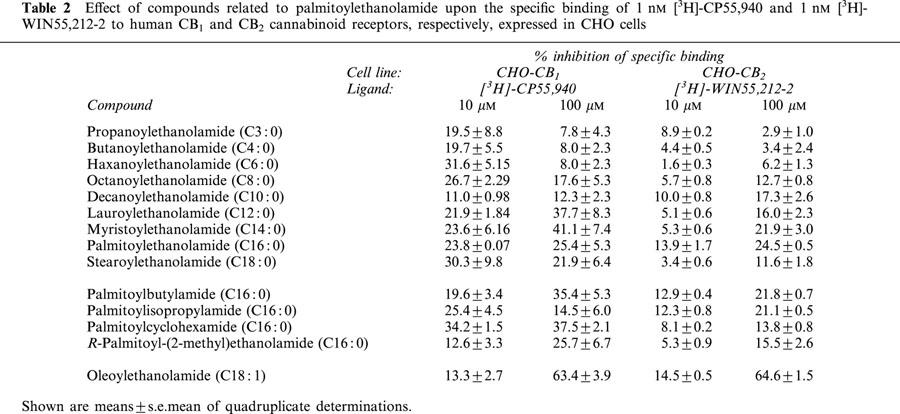
As reported in the introduction, most of the compounds selected for this study have been shown not to interact with CB receptors (Lambert et al., 1999). However, the highest concentration used in that study was 1 μM, raising the possibility that interactions can occur at higher concentrations of the compounds. In consequence, the effects of the compounds at 10 and 100 μM upon the specific binding of the synthetic cannabinoid receptor agonist radioligand [3H]-WIN 55,212-2 to recombinant human CB2 receptors expressed on CHO cells was investigated. With the exception of oleoylethanolamide (65% inhibition at 100 μM), all of the compounds produced <15% inhibition of binding at 10 μM and <25% inhibition at 100 μM (Table 2). For the binding of the agonist [3H]-CP55,940 to recombinant human CB1 receptors expressed on CHO cells, the data was less clear, although <26% inhibition of binding was found at both concentrations for palmitoylethanolamide, R-palmitoyl-(2-methyl)ethanolamide and palmitoylisopropylamide and the C4.0 and C10.0-homologues (Table 2). As with the CB2 receptors, oleoylethanolamide produced a large (63%) inhibition of binding at 100 μM (Table 2).
Table 2.
Effect of compounds related to palmitoylethanolamide upon the specific binding of 1 nM [3H]-CP55,940 and 1 nM [3H]-WIN55,212-2 to human CB1 and CB2 cannabinoid receptors, respectively, expressed in CHO cells
Inhibition of [3H]-AEA uptake in RBL-2H3 and C6 cells
The ability of the compounds to inhibit the uptake of [3H]-AEA into RBL-2H3 and C6 cells is shown in Figure 6. RBL-2H3 cells were chosen since [3H]-AEA transport mechanisms in these cells have been extensively characterized (Bisogno et al., 1997; de petrocellis et al., 2000; Rakhshan et al., 2000; Jacobsson & Fowler, 2001). Less is known about the uptake of AEA into C6 cells, although there are some apparent differences in the properties of the transporters in the two cell lines (such as sensitivity to nitric oxide donors, Bisogno et al., 2001; Jacobsson & Fowler, 2001). The initial data, shown in Figure 6c, investigated the effects of the palmitoylethanolamide homologues and analogues at concentrations of 1, 10 and 100 μM, to inhibit the uptake of 10 μM [3H]-AEA into RBL-2H3. [3H]-AEA binds avidly to cell culture wells, and it was proposed to compensate for this by defining non-specific effects as those inhibitable by 100 μM AM 404. However, this assumes that AM 404 at this concentration does not affect the binding of [3H]-AEA to the wells, which turned out to be the case (Figure 6a). The other monounsaturated compounds, oleoylethanolamide and R-methanandamide also affected the binding of [3H]-AEA to the wells, whereas the saturated compounds did not (see Figure 6a–c).
Figure 6.
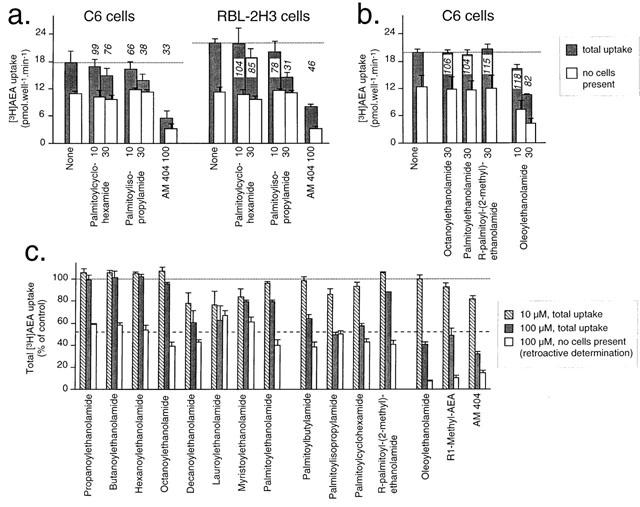
Effect of palmitoylethanolamide and related compounds upon the uptake of 10 μM [3H]-AEA into rat C6 glioma and RBL-2H3 basophilic leukaemia cells (a,b) shown are means±s.e.mean (n=3) of the total uptake in the absence and presence of cells. The numbers given above (or enclosed within) the columns are the specific uptake remaining in the presence of the test compounds, calculated as 100×(mean total uptake - mean uptake, no cells present)/(mean total uptake in the absence of compound - mean uptake, no cells present in the absence of compound). The dotted line shows the mean total uptake in the absence of test compound (i.e. ‘none'). (c) effects on the total uptake of [3H]-AEA into RBL-2H3 cells. Shown are means±s.e.mean, n=3. The dotted line shows the mean total uptake in the absence of test compound (i.e. 100%). The dashed line is a retroactive estimation of the mean percentage of the total [3H]-AEA uptake due to binding to the cell culture wells, determined from the data shown as ‘none' for the RBL-2H3 cells in panel a. The unfilled columns in the experiments shown in panel c were calculated from means of three separate experiments where the effects of the compounds upon the binding to [3H]-AEA to culture wells was determined. The observed figure was then normalized to 52% (i.e. the dashed line). Thus, for example, a compound producing a 34% inhibition of the binding to the well would have an unfilled column of 52×(0.66)=34% of total [3H]-AEA uptake.
In these subsequent experiments, the percentage of the total apparent uptake in RBL-2H3 cells due to the binding of [3H]-AEA to the wells could be determined (52±6%). This level of background binding is shown in Figure 6c as a dashed line, and was used, in conjunction with experiments undertaken in the absence of cells, to determine the degree to which the compounds at a concentration of 100 μM affected the binding of the [3H]-AEA to the wells. Although the use of a retroactive measure of binding to the wells prevents quantitative interpretation of these data, the first series of experiments indicated that several of the compounds are inactive (i.e. the homologues of palmitoylethanolamide at C⩽8 atoms) at 10 and 100 μM concentrations. Inhibition of [3H]-AEA uptake was seen at 10 μM concentrations with decanoylethanolamide, palmitoylisopropylamide, lauroylethanolamide and myristoylethanolamide, and at 100 μM concentrations with palmitoylethanolamide, palmitoylbutylamide, palmitoylcyclohexamide and oleoylethanolamide (although interpretation of the data with oleoylethanolamide is confounded by its ability to inhibit the binding of [3H]-AEA to the wells). None of the compounds affected the total uptake at concentrations of 1 μM (data not shown).
In the second series of experiments, the effects of palmitoylcyclohexamide and palmitoylisopropylamide (10 and 30 μM) were tested using both C6 and RBL-2H3 cells, and where parallel wells without cells were used to quantify the binding of the [3H]-AEA to the culture wells (Figure 6a). Propylisopropylamide reduced the specific uptake to 66 and 38% of control in the C6 cells at concentrations of 10 and 30 μM, respectively, whereas palmitoylcyclohexamide was less effective (99 and 76%, respectively). Similar results were seen in the RBL-2H3 cells (Figure 6a). Octanoyethanolamide, palmitoylethanolamide and R-palmitoyl-(2-methyl)ethanolamide at concentrations of 30 μM had no effect on the uptake of [3H]-AEA into C6 cells, whereas a small inhibition was seen with this concentration of oleoylethanolamide (Figure 6b).
Inhibition of [3H]-ethanolamine production following incubation of [3H]-AEA with intact C6 glioma cells
The six compounds investigated with respect to their effects upon the uptake of [3H]-AEA into C6 glioma cells were also tested for their abilities to inhibit [3H]-ethanolamine production following incubation of [3H]-AEA with intact C6 cells, the rationale being that such inhibition reflects the combination of effects on uptake and FAAH. The data are shown in Table 3. Oleoylethanolamide and palmitoylisopropylamide were the most potent of the compounds tested, producing >50% and >80% inhibition of [3H]-ethanolamine production at 10 and 30 μM, respectively, in both cases. AM404 was slightly less potent (40 and 68% inhibition at 10 and 30 μM, respectively), whereas palmitoylethanolamide, palmitoylcyclohexamide, R-palmitoyl-(2-methyl)ethanolamide and octanoylethanolamide produced 42, 45, 30 and 25% inhibition, respectively, at the highest concentration tested (30 μM).
Table 3.
Effect of compounds related to palmitoylethanolamide upon the hydrolysis of [3H]-AEA by intact C6 glioma cells

Discussion
The fatty acid amide palmitoylethanolamide has been found to have anti-inflammatory and anti-nociceptive properties (Aloe et al., 1993; Mazzari et al., 1996; Jaggar et al., 1998; Calignano et al., 1998). Given that this compound has a low affinity towards CB receptors in vitro and produces no significant effects at concentrations ⩽10 μM (Felder et al., 1993; Lambert et al., 1999; Ross et al., 1999; Sugiura et al., 2000), it has been suggested that palmitoylethanolamide, by preventing the metabolism of other endocannabinoids, may act as an ‘entourage' compound (Lambert & Di Marzo, 1999). The metabolism of AEA is brought about by a process of facilitated diffusion into the cell followed by intracellular FAAH-catalysed metabolism (see Mechoulam et al., 1998). In consequence, we tested both a series of palmitoylethanolamide analogues and homologues, as well as four new homologues (C3:0, C4:0, C6:0 and C8:0) and a 2-methyl analogue, for their ability to affect rat brain FAAH catalysed [3H]-AEA hydrolysis and [3H]-AEA uptake in RBL-2H3 cells. With the exception of oleoylethanolamide, none of the compounds were found at 100 μM concentrations to produce marked inhibition of the binding to [3H]-WIN 55,212-2 to CB2 receptors expressed on CHO cells. Variable effects upon CB1 receptors were seen, but for the most interesting compound emanating from this study, palmitoylisopropylamide, no dramatic interaction with this receptor subtype was found.
With respect to the ability of the compounds to inhibit the FAAH-catalyzed metabolism of [3H]-AEA, the chain length proved to be of importance for the potency at lengths of 10 carbon atoms or less. Thus, potencies of the homologues were roughly similar (given the approximate nature of the pI50 values as a result of the differences in maximum inhibition obtained) to palmitoylethanolamide as the chain length was reduced to 14 and 12 carbon atoms (or increased to 18 carbon atoms), but was slightly lower for the C10:0 compound (decanoylethanolamide). Compounds with a chain length of ⩽8 carbon atoms were inactive. This pattern of ‘stability' between C12:0 and C18:0 is also seen with saturated fatty acid sulphonyl fluoride compounds, although their potencies are greater than seen here: IC50 values of 3, 6 and 7 nM for the inhibition of rat brain AEA metabolism by lauryl- (C12), myristyl- (C14), palmityl- (C16) sulphonyl fluorides were found (Deutsch et al., 1997). In contrast, the C20:0 homologue arachidylsulphonyl fluoride is less potent (IC50 value >48 nM) (Deutsch et al., 1997).
Among the C16:0 analogues, palmitoylisopropylamide, R-palmitoyl-(2-methyl)ethanolamide and palmitoylbutylamide showed comparable potency to that of palmitoylethanolamide, although the maximal obtainable inhibition was lower. Oleoylethanolamide (C18:1) was a mixed type inhibitor of [3H]-AEA metabolism with Ki(slope) and Ki(intercept) values of 3.9 μM and 11 μM, respectively. Although the maximum observed inhibition produced by this compound was greater than seen with the saturated analogue, stearoylethanolamide, this may reflect solubility differences and the difference in pI50 values was not that great. In this respect, the potency of PTMK was very similar to that found previously for its oleoyl-analogue (pI50 value 7.24±0.077) in experiments using the same experimental conditions, (Fowler et al., 2000). The potency of PTMK found here is approximately 50 times greater than the potency of this compound towards Ca2+-independent phospholipase A2 (Ackermann et al., 1995), making it worthwhile further to investigate the potential usefulness of this compound as an FAAH inhibitor.
In theory, the palmitoylethanolamide analogues and homologues can inhibit [3H]-AEA metabolism by acting either as inhibitors or as competing substrates. Indeed, amidase activities capable of utilizing C12:0–C18:0 (but not C8:0) and C18:1 ethandamides as substrates were reported in the literature more than 16 years ago (Natarajan et al., 1984; Schmid et al., 1985). Most available studies in the literature have compared the relative activities of palmitoylethanolamide with AEA with widely varying results (see Tiger et al., 2000), although Lang et al. (1999) found a 4 fold greater rate of metabolism of 100 μM oleoylethanolamide than 100 μM stearoylethanolamide using rat brain microsomes as source of FAAH. There is more data on the ability of −amide, rather than −ethanolamide compounds to act as substrates. Thus, palmitoylamide (C16:0), myristoylamide (C14:0) and dodecanoylamide (C12:0) have been shown to act as FAAH substrates with rates of hydrolysis relative to oleamide (C18:1) of 0.72, 0.83 and 0.74, respectively (Boger et al., 2000a; see also Cravatt et al., 1996). These authors also demonstrated that introduction of a single double-bond into the 9-position of the palmitoylamide and myristoylamide molecules did not affect their rates of hydrolysis, whereas the mono-unsaturated oleamide was metabolized at a higher rate than the saturated stearamide equivalent (Boger et al., 2000a). These authors used single high concentrations of substrate (187 μM), thus presumably measuring activities near their Vmax values. Given (a) that linoleoylethanolamide and linoleoylamide are both substrates for FAAH and are metabolized with similar Vmax values (Maurelli et al., 1995); and (b) that palmitoylamide inhibits [3H]-AEA metabolism with a pI50 value (5.01±0.18, maximum inhibition attainable 51±7%, present authors, unpublished data) similar to that of palmitoylethanolamide, it is reasonable to assume that the C10:0 and C17:0 homologues of palmitoylethanolamide tested here are acting as competing substrates. The mixed-type inhibition of [3H]-AEA found here with oleoylethanolamide is perhaps surprising, given that alternate substrates should interact competitively. However, a similar mixed-type inhibition has been seen previously for the ability of 2 μM (but not 1 μM) AEA to inhibit the metabolism of [3H]-palmitoylethanolamide by rat brain FAAH (Tiger et al., 2000). Whether the ‘non-competitive component' of the inhibition in these cases represents an additional action of the compounds (such as effects secondary to membrane actions of these fatty acid amides) awaits elucidation.
For the palmitoylethanolamide analogues tested here, it is not known whether they are substrates for FAAH. One way of distinguishing inhibitors from competing substrates is to preincubate the compounds with a strong FAAH preparation prior to addition of [3H]-anandamide: for a competing substrate, the preincubation would be expected to reduce the observed potency of the compound. However, with the exception of lauroylethanolamide, no decreased potency with preincubation time was found when membranes were preincubated with the compounds for up to 3 h. This would suggest that under the conditions used, insufficient amounts of the compounds were metabolized significantly to affect the observed inhibition. Thus, further experiments are required before it can be determined whether these compounds act as substrates or as inhibitors. However, the potencies of the compounds were rather modest, and as a first step it might be more appropriate to investigate further the structure-activity relationships between palmitoylethanolamide analogues and inhibition of [3H]-AEA hydrolysis. Such studies are presently under way.
In addition to studying the effect of the compounds upon [3H]-AEA metabolism by FAAH, their ability to prevent [3H]-AEA accumulation into RBL-2H3 and C6 cells has been investigated. The cellular uptake of AEA is somewhat complicated: it is saturable but sodium- and energy- independent, and is only capable of concentrating the endocannabinoid against a modest gradient (Di Marzo et al., 1994; Beltramo et al., 1997; Hillard et al., 1997, Rakhshan et al., 2000; Deutsch et al., 2001). Thus inhibition of FAAH will result in an indirect reduction in the rate of accumulation of AEA. The compounds tested in the present study were rather modest inhibitors of [3H]-AEA accumulation into RBL-2H3 cells, and in the case of palmitoylethanolamide, the reduction in [3H]-AEA accumulation seen at the highest concentration (100 μM) is presumably a reflection of effects on [3H]-AEA metabolism. Palmitoylcyclohexamide, however, does not affect [3H]-AEA metabolism, suggesting that the ability of this compound to reduce [3H]-AEA accumulation may reflect a direct effect upon the AEA transport process. Thus, palmitoylcyclohexamide may prove useful as a template for the design of compounds that reduce the cellular accumulation of AEA without having potent effects upon either FAAH or CB2 receptors. Palmitoylisopropylamide also produces a greater inhibition of uptake than palmitoylethanolamide, despite having a similar pI50 value towards inhibition of [3H]-AEA metabolism, which also suggests a direct action on the AEA uptake process.
At first sight, the present study does not give rise to compounds that are dramatically better than palmitoylethanolamide as possible entourage compounds. However, effects on AEA metabolism in vivo will be a consequence of the combination of effects on transport and FAAH. As a first step to investigating this combination, we studied the effect of palmitoylisopropylamide, palmitoylcyclohexamide, R-palmitoyl-(2-methyl)ethanolamide and oleoylethanolamide (with palmitoylethanolamide and AM404 for comparison and octanoylethanolamide as a negative control) on the production of [3H]-ethanolamine following incubation of C6 glioma cells with [3H]-AEA. In this case, the FAAH only has access to the transported AEA, so that a combination of effects upon uptake and FAAH will produce a greater inhibition of [3H]-ethanolamine production than effects on either process alone. Such a result was indeed found: both palmitoylisopropylamide and oleoylethanolamide were more potent than either palmitoylethanolamide (which competes for FAAH but has no direct effect on AEA uptake) or palmitoylcyclohexamide (which affects AEA uptake but not FAAH). Interestingly, palmitoylisopropylamide and oleoylethanolamide were more effective than AM404. Given that AM404 potentiates endogenous AEA in vivo (Giuffrida et al., 2000), these data would suggest that palmitoylisopropylamide might be a useful entourage compound that does not have direct effects on CB receptors.
Acknowledgments
The authors would like to thank Britt Jacobsson for excellent technical assistance. This study was supported by grants from the Swedish Medical Research Foundation (Grant no. 12158), the Swedish Asthma- and Allergy Association's Research Foundation, and the Research Funds of the Medical Odontological Faculty, Umeå University. The chemical work was financially supported by the Belgian National Fund for Scientific Research and a FSR grant from the Université catholique de Louvain. The authors are grateful to the expert reviewer of this publication for constructive criticism of the paper, and for making us aware of the possibility that AM404 could block the binding of [3H]-AEA to cell culture wells.
Abbreviations
- AEA
anandamide
- FAAH
fatty acid amidohydrolase
- PTMK
palmitoyl trifluoromethyl ketone
References
- ACKERMANN E.J., CONDE-FRIBOES K., DENNIS E.A. Inhibition of macrophage Ca2+-independent phospholipase A2 by bromoenol lactone and trifluoromethyl ketones. J. Biol. Chem. 1995;270:445–450. doi: 10.1074/jbc.270.1.445. [DOI] [PubMed] [Google Scholar]
- ALOE L., LEON A., LEVI-MOLTALCINI R. A proposed autacoid mechanism controlling mastocyte behaviour. Agents Actions. 1993;39:C145–C147. doi: 10.1007/BF01972748. [DOI] [PubMed] [Google Scholar]
- BAKER D., PRYCE G., CROXFORD J.L., BROWN P., PERTWEE R.G., MAKRIYANNIS A., KHANOLKAR A., LAYWARD L., FEZZA F., BISOGNO T., DI MARZO V. Endocannabinoids control spasticity in a multiple sclerosis model. FASEB J. 2001;15:300–302. doi: 10.1096/fj.00-0399fje. [DOI] [PubMed] [Google Scholar]
- BELTRAMO M., STELLA N., CALIGNANO A., LIN S.Y., MAKRIYANNIS A., PIOMELLI D. Functional role of high-affinity anandamide transport, as revealed by selective inhibition. Science. 1997;277:1094–1097. doi: 10.1126/science.277.5329.1094. [DOI] [PubMed] [Google Scholar]
- BISOGNO T., MACCARRONE M., DE PETROCELLIS L., JARRAHIAN A., FINAZZI-AGRÒ A., HILLARD C., DI MARZO V. The uptake by cells of 2-arachidonoylglycerol, an endogenous agonist of cannabinoid receptors. Eur. J. Biochem. 2001;268:1982–1989. doi: 10.1046/j.1432-1327.2001.02072.x. [DOI] [PubMed] [Google Scholar]
- BISOGNO T., MAURELLI S., MELCK D., DE PETROCELLIS L., DI MARZO V. Biosynthesis, uptake, and degradation of anandamide and palmitoylethanolamide in leukocytes. J. Biol. Chem. 1997;272:3315–3323. doi: 10.1074/jbc.272.6.3315. [DOI] [PubMed] [Google Scholar]
- BISOGNO T., MELCK D., DE PETROCELLIS L., BOBROV M.Y., GRETSKAYA N.M., BEZUGLOV V.V., SITACHITTA N., GERWICK W.H., DI MARZO V. Arachidonoylserotonin and other novel inhibitors of fatty acid amide hydrolase. Biochem. Biophys. Res. Commun. 1998;248:515–522. doi: 10.1006/bbrc.1998.8874. [DOI] [PubMed] [Google Scholar]
- BOGER D.L., FECIK R.A., PATTERSON J.E., MIYAUCHI H., PATRICELLI M.P., CRAVATT B.F. Fatty acid amide hydrolase substrate specificity. Bioorg. Med. Chem. Lett. 2000a;10:2613–2616. doi: 10.1016/s0960-894x(00)00528-x. [DOI] [PubMed] [Google Scholar]
- BOGER D.L., SATO H., LERNER A.E., HEDRICK M.P., FECIK R.A., MIYAUCHI H., WILKIE G.D., AUSTIN B.J., PATRICELLI M.P., CRAVATT B.F. Exceptionally potent inhibitors of fatty acid amide hydrolase: The enzyme responsible for degradation of endogenous oleamide and anandamide. Proc. Natl. Acad. Sci. USA. 2000b;97:5044–5049. doi: 10.1073/pnas.97.10.5044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CALIGNANO A., KÁTONA I., DÉSARNAUD F., GIUFFRIDA A., LA RANA G., MACKIE K., FREUND T.F., PIOMELLI D. Bidirectional control of airway responsiveness by endogenous cannabinoids. Nature. 2000;408:96–101. doi: 10.1038/35040576. [DOI] [PubMed] [Google Scholar]
- CALIGNANO A., LA RANA G., GIUFFRIDA A., PIOMELLI D. Control of pain initiation by endogenous cannabinoids. Nature. 1998;394:277–281. doi: 10.1038/28393. [DOI] [PubMed] [Google Scholar]
- CHILDERS S.R., SEXTON T., ROY M.B. Effects of anandamide on cannabinoid receptors in rat brain membranes. Biochem. Pharmacol. 1994;47:711–715. doi: 10.1016/0006-2952(94)90134-1. [DOI] [PubMed] [Google Scholar]
- COMPTON D.R., MARTIN B.R. The effect of the enzyme inhibitor phenylmethylsulfonyl fluoride on the pharmacological effect of anandamide in the mouse model of cannabimimetic activity. J. Pharmacol. Exp. Ther. 1997;283:1138–1143. [PubMed] [Google Scholar]
- CRAVATT B.F., GIANG D.K., MAYFIELD S.P., BOGER D.L., LERNER R.A., GILULA N.B. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- DE PETROCELLIS L., BISOGNO T., DAVIS J.B., PERTWEE R.G., DI MARZO V. Overlap between the ligand recognition properties of the anandamide transporter and the VR1 vanilloid receptor: inhibitors of anandamide uptake with negligible capsaicin-like activity. FEBS Lett. 2000;483:52–56. doi: 10.1016/s0014-5793(00)02082-2. [DOI] [PubMed] [Google Scholar]
- DE PETROCELLIS L., MELCK D., PALMISANO A., BISOGNO T., LAEZZA C., BIFULCO M., DI MARZO V. The endogenous cannabinoid anandamide inhibits human breast cancer cell proliferation. Proc. Natl. Acad. Sci. U.S.A. 1998;95:8375–8380. doi: 10.1073/pnas.95.14.8375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEUTSCH D.G., CHIN S.A. Enzymatic synthesis and degradation of anandamide, a cannabinoid receptor agonist. Biochem. Pharmacol. 1993;46:791–796. doi: 10.1016/0006-2952(93)90486-g. [DOI] [PubMed] [Google Scholar]
- DEUTSCH D.G., GLASER S.T., HOWELL J.M., KUNZ J.S., PUFFENBARGER R.A., HILLARD C.J., ABUMRAD N. The cellular uptake of anandamide is coupled to its breakdown by fatty acid amide hydrolase (FAAH) J. Biol. Chem. 2001;276:6967–6973. doi: 10.1074/jbc.M003161200. [DOI] [PubMed] [Google Scholar]
- DEUTSCH D.G., LIN S., HILL W.A., MORSE K.L., SALEHANI D., ARREAZA G., OMEIR R.L., MAKRIYANNIS A. Fatty acid sulfonyl fluorides inhibit anandamide metabolism and bind to the cannabinoid receptor. Biochem. Biophys. Res. Commun. 1997;231:217–221. doi: 10.1006/bbrc.1997.6072. [DOI] [PubMed] [Google Scholar]
- DI MARZO V., FONTANA A., CADAS H., SCHINELLI S., CIMINO G., SCHWARTZ J.C., PIOMELLI D. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature. 1994;372:686–691. doi: 10.1038/372686a0. [DOI] [PubMed] [Google Scholar]
- EISENTHAL R., CORNISH-BOWDEN A. The direct linear plot. A new graphical procedure for estimating enzyme kinetic parameters. Biochem. J. 1974;139:715–720. doi: 10.1042/bj1390715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FELDER C.C., BRILEY E.M., AXELROD J., SIMPSON J.T., MACKIE K., DEVANE W.A. Anandamide, an endogenous cannabimimetic eicosanoid, binds to the cloned human cannabinoid receptor and stimulates receptor-mediated signal transduction. Proc. Natl. Acad. Sci. U.S.A. 1993;90:7656–7660. doi: 10.1073/pnas.90.16.7656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FOWLER C.J., TIGER G., STENSTRÖM A. Ibuprofen inhibits rat brain deamidation of anandamide at pharmacologically relevant concentrations. Mode of inhibition and structure-activity relationship. J. Pharmacol. Exp. Ther. 1997;283:729–734. [PubMed] [Google Scholar]
- FOWLER C.J., BÖRJESSON M., TIGER G. Differences in the pharmacological properties of rat and chicken brain fatty acid amidohydrolase. Br. J. Pharmacol. 2000;131:498–504. doi: 10.1038/sj.bjp.0703569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GAULDIE S.D., MCQUEEN D.S., PERTWEE R., CHESSELL I.P. Anandamide activates peripheral nociceptors in normal and arthritic knee joints. Br. J. Pharmacol. 2001;132:617–621. doi: 10.1038/sj.bjp.0703890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GIFFORD A.N., BRUNEUS M., LIN S., GOUTOPOULOS A., MAKRIYANNIS A., VOLKOW N.D., GATLEY S.J. Potentiation of the action of anandamide on hippocampal slices by the fatty acid amide hydrolase inhibitor, palmitylsulphonyl fluoride (AM 374) Eur. J. Pharmacol. 1999;383:9–14. doi: 10.1016/s0014-2999(99)00609-3. [DOI] [PubMed] [Google Scholar]
- GIUFFRIDA A., RODRIGUEZ DE FONSECA F., NAVA F., LOUBET-LESCOULIÉ P., PIOMELLI D. Elevated circulating levels of anandamide after administration of the transport inhibitor, AM404. Eur. J. Pharmacol. 2000;408:161–168. doi: 10.1016/s0014-2999(00)00786-x. [DOI] [PubMed] [Google Scholar]
- HARRINGTON C.R. Lowry protein assay containing sodium dodecyl sulfate in microtiter plates for protein determination on fractions from brain tissue. Analyt. Biochem. 1990;186:285–287. doi: 10.1016/0003-2697(90)90081-j. [DOI] [PubMed] [Google Scholar]
- HILLARD C.J., EDGEMOND W.S., JARRAHIAN A., CAMPBELL W.B. Accumulation of N-arachidonoylethanolamine (anandamide) into cerebellar granule cells occurs via facilitated diffusion. J. Neurochem. 1997;69:631–638. doi: 10.1046/j.1471-4159.1997.69020631.x. [DOI] [PubMed] [Google Scholar]
- JACOBSSON S.O.P., FOWLER C.J. Characterization of palmitoylethanolamide transport in mouse Neuro-2a neuroblastoma and rat RBL-2H3 basophilic leukaemia cells: comparison with anandamide. Br. J. Pharmacol. 2001;132:1743–1754. doi: 10.1038/sj.bjp.0704029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JAGGAR S.I., HASNIE F.S., SELLATURAY S., RICE A.S.C. The anti-hyperalgesic actions of the cannabinoid anandamide and the putative CB2 receptor agonist palmitoylethanolamide in visceral and somatic inflammatory pain. Pain. 1998;76:189–199. doi: 10.1016/s0304-3959(98)00041-4. [DOI] [PubMed] [Google Scholar]
- JARRAHIAN A., MANNA S., EDGEMOND W.S., CAMPBELL W.B., HILLARD C.J. Structure-activity relationships among N-arachidonoylethanolamine (anandamide) head group analogues for the anandamide transporter. J. Neurochem. 2000;74:2597–2606. doi: 10.1046/j.1471-4159.2000.0742597.x. [DOI] [PubMed] [Google Scholar]
- KOUTEK B., PRESTWICH G.D., HOWLETT A.C., CHIN S.A., SALEHANI D., AKHAVAN N., DEUTSCH D.G. Inhibitors of arachidonoyl ethanolamide hydrolysis. J. Biol. Chem. 1994;269:22937–22940. [PubMed] [Google Scholar]
- LAMBERT D.M., DI MARZO V. The palmitoylethanolamide and oleamide enigmas: are these two fatty acid amides cannabimimetic. Curr. Med. Chem. 1999;6:757–773. [PubMed] [Google Scholar]
- LAMBERT D.M., DIPAOLO F.G., SONVEAUX P., KANYONYO M., GOVAERTS S.J., HERMANS E., BUEB J., DELZENNE N.M., TSCHIRHART E.J. Analogues and homologues of N-palmitoylethanolamide, a putative endogenous CB2 cannabinoid, as potential ligands for the cannabinoid receptors. Biochim. Biophys. Acta. 1999;1440:266–274. doi: 10.1016/s1388-1981(99)00132-8. [DOI] [PubMed] [Google Scholar]
- LANG W., QIN C., LIN S., KHANOLKAR A.D., GOUTOPOULOS A., FAN P., ABOUZID K., MENG Z., BIEGEL D., MAKRIYANNIS A. Substrate specificity and stereoselectivity of rat brain microsomal anandamide amidohydrolase. J. Med. Chem. 1999;42:896–902. doi: 10.1021/jm980461j. [DOI] [PubMed] [Google Scholar]
- MACCARRONE M., LORENZON T., BARI M., MELINO G., FINAZZI-AGRÒ A. Anandamide induces apoptosis in human cells via vanilloid receptors. Evidence for a protective role of cannabinoid receptors. J. Biol. Chem. 2000;275:31938–31945. doi: 10.1074/jbc.M005722200. [DOI] [PubMed] [Google Scholar]
- MALLET P.E., BENINGER R.J. The cannabinoid CB1 receptor antagonist SR 141716A attenuates the memory impairment produced by Δ9-tetrahydro-cannabinol or anandamide. Psychopharmacology. 1998;140:11–19. doi: 10.1007/s002130050733. [DOI] [PubMed] [Google Scholar]
- MAURELLI S., BISOGNO T., DE PETROCELLIS L., DI LUCCIA A., MARINO G., DI MARZO V. Two novel classes of neuroactive fatty acid amides are substrates for mouse neuroblastoma ‘anandamide amidohydrolase'. FEBS Letts. 1995;377:82–86. doi: 10.1016/0014-5793(95)01311-3. [DOI] [PubMed] [Google Scholar]
- MAZZARI S., CANELLA R., PETRELLI L., MARCOLONGO G., LEON A. N-(2-hydroxyethyl)hexadecamide is orally active in reducing edema formation and inflammatory hyperalgesia by down-modulating mast cell activation. Eur. J. Pharmacol. 1996;300:227–236. doi: 10.1016/0014-2999(96)00015-5. [DOI] [PubMed] [Google Scholar]
- MECHOULAM R., FRIDE E., DI MARZO V. Endocannabinoids. Eur. J. Pharmacol. 1998;359:1–18. doi: 10.1016/s0014-2999(98)00649-9. [DOI] [PubMed] [Google Scholar]
- MELCK D., BISOGNO T., DE PETROCELLIS L., CHUANG H.H., JULIUS D., BIFULCO M., DI MARZO V. Unsaturated long-chain N-acyl-vanillyl-amides (N-AVAMs): vanilloid receptor ligands that inhibit anandamide-facilitated transport and bind to CB1 cannabinoid receptors. Biochem. Biophys. Res. Commun. 1999;262:275–284. doi: 10.1006/bbrc.1999.1105. [DOI] [PubMed] [Google Scholar]
- NATARAJAN V., SCHMID P.C., REDDY P.V., SCHMID H.H.O. Catabolism of N-acylethanolamine phospholipids by dog brain preparations. J. Neurochem. 1984;42:1613–1619. doi: 10.1111/j.1471-4159.1984.tb12750.x. [DOI] [PubMed] [Google Scholar]
- OMEIR R.L., CHIN S., HONG Y., AHERN D.G., DEUTSCH D.G. Arachidonoyl ethanolamide-[1,2-14C] as a substrate for anandamide amidase. Life Sci. 1995;56:1999–2005. doi: 10.1016/0024-3205(95)00181-5. [DOI] [PubMed] [Google Scholar]
- PATTERSON J.E., OLLMANN I.R., CRAVATT B.F., BOGER D.L., WONG C.-H., LERNER R.A. Inhibition of oleamide hydrolase catalyzed hydrolysis of the endogenous sleep-inducing lipid cis-9-octadecenamide. J. Am. Chem. Soc. 1996;118:5938–5945. [Google Scholar]
- PERTWEE R.G., FERNANDO S.R., GRIFFIN G., ABADJI V., MAKRIYANNIS A. Effect of phenylmethylsulphonyl fluoride on the potency of anandamide as an inhibitor of electrically evoked contractions in two isolated tissue preparations. Eur. J. Pharmacol. 1995;272:73–78. doi: 10.1016/0014-2999(94)00618-h. [DOI] [PubMed] [Google Scholar]
- PIOMELLI D., GIUFFRUDA A., CALIGNANO A., DE FONSECA F.R. The endocannabinoid system as a target for therapeutic drugs. Trends Pharmacol. Sci. 2000;21:218–224. doi: 10.1016/s0165-6147(00)01482-6. [DOI] [PubMed] [Google Scholar]
- RAFFA R.B., STONE D.J., HIPP S.J. Differential cholera-toxin sensitivity of supraspinal antinociception induced by the cannabinoid agonist Δ9-THC, WIN 55,212-2 and anandamide in mice. Neuroscience Letts. 1999;263:29–32. doi: 10.1016/s0304-3940(99)00096-8. [DOI] [PubMed] [Google Scholar]
- RAKHSHAN F., DAY T.A., BLAKELY R.D., BARKER E.L. Carrier-mediated uptake of the endogenous cannabinoid anandamide in RBL-2H3 cells. J. Pharmacol. Exp. Ther. 2000;292:960–967. [PubMed] [Google Scholar]
- RICHARDSON J.D., KILO S., HARGREAVES K.M. Cannabinoids reduce hyperalgesia and inflammation via interaction with peripheral CB1 receptors. Pain. 1998;75:111–119. doi: 10.1016/S0304-3959(97)00213-3. [DOI] [PubMed] [Google Scholar]
- ROSS R.A., MURPHY V.L., MCKAY N.G., ASHFORD M.L.J., PERTWEE R.G.Characterization of cannabinoid receptors in the spleen and in mast cells Essential Fatty Acids and Eicosanoids: Invited papers from the 4th International Congress. 1999Champaign, IL, USA, AOCS Press; 376–379.ed. Riemersma, R.A., Armstrong, R., Kelly, R.W. & Wilson, R. pp [Google Scholar]
- SARKER K.P., OBARA S., NAKATA M., KITAJIMA I., MARUYAMA I. Anandamide induces apoptosis of PC-12 cells: involvement of superoxide and caspase-3. FEBS Letts. 2000;472:39–44. doi: 10.1016/s0014-5793(00)01425-3. [DOI] [PubMed] [Google Scholar]
- SCHMID P.C., ZUZARTE-AUGUSTIN M.L., SCHMID H.H.O. Properties of rat liver N-acylethanolamine amidohydrolase. J. Biol. Chem. 1985;260:14145–14149. [PubMed] [Google Scholar]
- SUGIURA T., KONDO S., KISHIMOTO S., MIYASHITA T., NAKANE S., KODAKA T., SUHARA Y., TAKAYAMA H., WAKU K. Evidence that 2-arachidonoylglycerol but not N-palmitoylethanolamide or anandamide is the physiological ligand for the cannabinoid CB2 receptor. Comparison of the agonistic activities of various cannabinoid receptor ligands in HL-60 cells. J. Biol. Chem. 2000;275:605–612. doi: 10.1074/jbc.275.1.605. [DOI] [PubMed] [Google Scholar]
- TERRANOVA J.-P, MICHAUD J.-C, LE FUR G., SOUBRIÉ P. Inhibition of long-term potentiation in rat hippocampal slices by anandamide and WIN55212-2: reversal by SR141716 A, a selective antagonist of CB1 cannabinoid receptors. Naunyn-Schmiedebergs Arch. Pharmacol. 1995;352:576–579. doi: 10.1007/BF00169393. [DOI] [PubMed] [Google Scholar]
- TIGER G., STENSTRÖM A., FOWLER C.J. Pharmacological properties of rat brain fatty acid amidohydrolase in different subcellular fractions using palmitoylethanolamide as substrate. Biochem. Pharmacol. 2000;59:647–653. doi: 10.1016/s0006-2952(99)00373-1. [DOI] [PubMed] [Google Scholar]