

Abstract
Protease-activated receptors (PARs) 1 and 2 modulate the gastric and intestinal smooth muscle motility in vitro. In the present study, we examined if activation of PAR-2 and PAR-1 could alter gastrointestinal transit in mice.
Intraperitoneal administration of the PAR-2-activating peptide SLIGRL-NH2, but not the inactive control LSIGRL-NH2, at 1–5 μmol kg−1, in combination with the aminopeptidase inhibitor amastatin at 2.5 μmol kg−1, facilitated gastrointestinal transit in a dose-dependent manner. The human PAR-1-derived peptide SFLLR-NH2 and the specific PAR-1 agonist TFLLR-NH2, but not the inactive control FSLLR-NH2, at 2.5–10 μmol kg−1, in combination with amastatin, also promoted gastrointestinal transit.
The Ca2+-activated, small conductance K+ channel inhibitor apamin at 0.01 μmol kg−1 significantly potentiated the actions of SLIGRL-NH2 and TFLLR-NH2 at subeffective doses.
The increased gastrointestinal transit exerted by either SLIGRL-NH2 at 5 μmol kg−1 or TFLLR-NH2 at 10 μmol kg−1 was completely abolished by the L-type Ca2+ channel inhibitor verapamil at 61.6 μmol kg−1. In contrast, the tyrosine kinase inhibitor genistein at 18.5 μmol kg−1 failed to modify the effects of the agonists for PAR-2 or PAR-1.
These findings demonstrate that PAR-1 and PAR-2 modulate gastrointestinal transit in mice in vivo. Our data also suggest that the PAR-1-and PAR-2-mediated effects are modulated by apamin-sensitive K+ channels and are dependent on activation of L-type Ca2+ channels, but independent of tyrosine kinase. Our study thus provides novel evidence for the physiological and/or pathophysiological roles of PARs 1 and 2 in the digestive systems, most probably during inflammation.
Keywords: Protease (proteinase)-activated receptor (PAR), gastrointestinal motility, intestinal transit, potassium channel, calcium channel, tyrosine kinase
Introduction
Thrombin exhibits cellular actions mainly by activating protease-activated receptors (PARs) 1, 3 or 4 (Vu et al., 1991; Ishihara et al., 1997; Kahn et al., 1998; Xu et al., 1998; Nakanishi-Matsui et al., 2000), while trypsin and mast cell tryptase, do so through activation of PAR-2 (Nystedt et al., 1994; Molino et al., 1997). Activation of PARs is achieved by proteolytic unmasking of the cryptic tethered ligand present in the N-terminal peptide of the receptor (for review, see Dery et al., 1998; Hollenberg, 1999; Kawabata & Kuroda, 2000). Amongst the four members of the PAR family, PARs 1 and 2 are now recognized as physiologically important molecules, especially in the early stages of inflammation, tissue-injury or haemorrhage, with protective and/or aggravating effects (Cirino et al., 1996; Kawabata et al., 1998; 1999a; Hollenberg, 1999; Vergnolle et al., 1999a, 1999b; Kawabata & Kuroda, 2000; Cocks & Moffatt, 2000).
The alimentary systems are, in particular, abundant in PARs 1 and 2 (Hollenberg, 1999; Kawabata & Kuroda, 2000). PAR-2 or PAR-2-like receptors are involved in salivary and pancreatic exocrine secretion (Bohm et al., 1996; Nguyen et al., 1999; Kawabata et al., 2000c, 2000d), and in intestinal ion transport (Vergnolle et al., 1998). There is also in vitro evidence that PARs 1, 2 and 4 modulate smooth muscle motility throughout the gastrointestinal tract including the oesophagus (Hollenberg et al., 1993; 1997; 1999; Corvera et al., 1997; Zheng et al., 1998; Cocks et al., 1999; Kawabata et al., 1999b; 2000a, 2000b). Agonists for PARs 1, 2 and 4 produce contraction of the gastric longitudinal smooth muscle from rats or guinea-pigs (Hollenberg et al., 1993; 1997; 1999; Saifeddine et al., 1996; Zheng et al., 1998), whereas activation of PARs 1 and 2 elicits relaxation of the precontracted smooth muscle of mouse gastric fundus or guinea-pig taenia coli (Cocks et al., 1999). Further, PAR-1 agonists exhibit a dual action, relaxation followed by contraction, in the rat duodenal smooth muscle, while PAR-2 agonists elicit only contraction and PAR-4 agonists have no effect in the same tissue (Kawabata et al., 1999b; 2000b). The signal transduction mechanisms for the contraction and/or relaxation of smooth muscle due to activation of PARs may include activation of L-type Ca2+ channels, tyrosine kinase and apamin-sensitive K+ channels in some tissues, but do not necessarily appear to be common in distinct tissues (Hollenberg et al., 1993; 1997; 1999; Corvera et al., 1997; Zheng et al., 1998; Cocks et al., 1999; Kawabata et al., 1999b; 2000a, 2000b). Questions that have yet to be answered are: (1) what happens in vivo as an outcome of activation of PAR-1 and PAR-2 in the gastrointestinal tract? and (2) can the mechanisms suggested in previous in vitro studies as described above be demonstrated in vivo? In the present study, therefore, we examined if agonists for PAR-1 and PAR-2 could modulate gastrointestinal transit in the mouse in vivo, and attempted to characterize the mechanisms of the in vivo modulation of gastrointestinal transit through PARs.
Methods
Animals employed
Male ddY mice weighing 25–35 g (Japan SLC. Inc., Japan) were used with approval from the Kinki University Faculty of Pharmaceutical Sciences' Committee for the Care and Use of Laboratory Animals. After 1 week of acclimatization (temperature 24±1°C; humidity 60%), food was withheld for 16–20 h before experiments, but animals had free access to tap water.
Assessment of gastrointestinal transit
As described elsewhere (Izzo et al., 2000), 10% charcoal suspension in 5% gum arabic was administered orally to conscious mice, and the mice were killed by cervical dislocation after 20 min. The gastrointestinal tract was removed, and the length of the small intestine traversed by the marker was measured. Data are expressed as percentages of the total length of the small intestine of each mouse.
Drug administration schedules
PAR-related peptides used were: the PAR-2 agonist SLIGRL-NH2 (based on murine and rat PAR-2), the PAR-2-inactive control peptide LSIGRL-NH2, the PAR-1 agonist SFLLR-NH2 (based on human PAR-1) and TFLLR-NH2, and the PAR-1-inactive control peptide FSLLR-NH2. These peptides at various doses were administered intraperitoneally (i.p.) to the mouse 1 min after i.p. amastatin at 2.5 μmol kg−1 (Kawabata et al., 2000d), and followed immediately by the charcoal meal administered orally. In some experiments, carbachol at 0.055 or 0.55 μmol kg−1 was administered i.p. without amastatin. The Ca2+-dependent, small conductance K+ channel inhibitor apamin at 0.01 μmol kg−1 was given i.p. 6 min before i.p. administration of the agonist peptides, SLIGRL-NH2 at 1 μmol kg−1 or TFLLR-NH2 at 2.5 μmol kg−1 (5 min before amastatin); the dose of apamin was decided on the basis of our previous in vitro study where apamin at 0.1 μM completely abolished the relaxation of duodenal smooth muscle in response to TFLLR-NH2 at 50 μM (Kawabata et al., 1999b). The L-type Ca2+ channel inhibitor verapamil at 61 μmol kg−1 (30 mg kg−1) (Rupniak et al., 1993) and the tyrosine kinase inhibitor genistein at 18.5 μmol kg−1 (5 mg kg−1) (Campos et al., 1999; Deodato et al., 1999) were administered 16 min before SLIGRL-NH2 at 5 μmol kg−1 or TFLLR-NH2 at 10 μmol kg−1 (15 min before amastatin). In control experiments, each vehicle was administered in the same manner.
Drugs employed
All peptides were prepared by a standard solid-phase synthesis method by ourselves. The concentration, purity and composition of the peptides were determined by high-performance liquid chromatography, mass spectrometry and quantitative amino acid analysis. Amastatin was purchased from Peptide Institute Inc. (Japan), and genistein, apamin, verapamil hydrochloride and carbachol were from Sigma (U.S.A.). Genistein was dissolved in 5% Tween 80 solution. All other chemicals were dissolved in phosphate-buffered saline or saline.
Statistical analysis
The results are represented as mean±s.e.mean. Statistical significance was analysed by Newman-Keuls' multiple comparison test, and was set at a P<0.05 level.
Results
Effects of receptor-activating peptides for PAR-2 and PAR-1 on gastrointestinal transit in mice
The murine PAR-2-derived receptor-activating peptide SLIGRL-NH2 at 1–5 μmol kg−1, when administered i.p. in combination with i.p. amastatin, an inhibitor of aminopeptidase, a peptide degrading enzyme, at 2.5 μmol kg−1, facilitated gastrointestinal transit in a dose-dependent manner in conscious mice (Figure 1b), although without pretreatment with amastatin it failed to produce a significant effect (Figure 1a). On the other hand, the PAR-2-inactive control peptide LSIGRL-NH2 at 5 μmol kg−1, in combination with amastatin, had no effect on gastrointestinal transit (Figure 1b).
Figure 1.
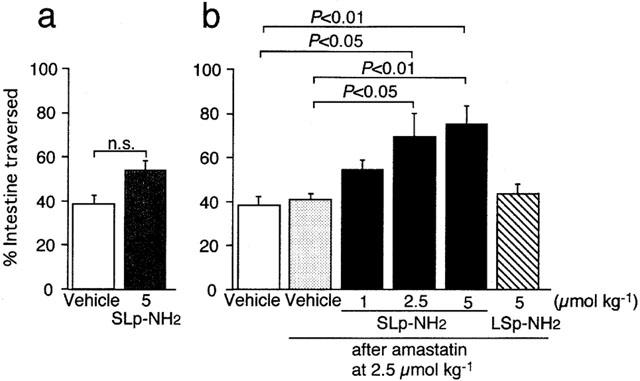
Effects of the PAR-2 agonist on gastrointestinal transit in mice. The PAR-2 agonist SLIGRL-NH2 (SLp-NH2) or the control peptide LSIGRL-NH2 (LSp-NH2), alone (a) or in combination with i.p. amastatin at 2.5 μmol kg−1 (b), were administered i.p. to the mouse. Data represent the mean±s.e.mean from 4–6 mice. n.s., not significant. Vehicle for amastatin did not modify the gastrointestinal transit.
The human PAR-1-derived receptor-activating peptide SFLLR-NH2 that also has a weak agonistic activity toward PAR-2 (Kawabata et al., 1999c), when administered i.p. at 5 and 10 μmol kg−1 in combination with amastatin, produced significant increase in gastrointestinal transit in mice (Figure 2a), although without pretreatment with amastatin it had no significant effect (data not shown). On the other hand, the PAR-1-inactive control peptide FSLLR-NH2 at 10 μmol kg−1 had no significant effect (Figure 2a). The PAR-1 agonist analogue TFLLR-NH2, known to be highly specific for PAR-1 with no PAR-2 activity, by i.p. administration in combination with amastatin, increased gastrointestinal transit in a dose-dependent manner (Figure 2b), although this peptide also required pretreatment with amastatin to produce the effect (data not shown).
Figure 2.
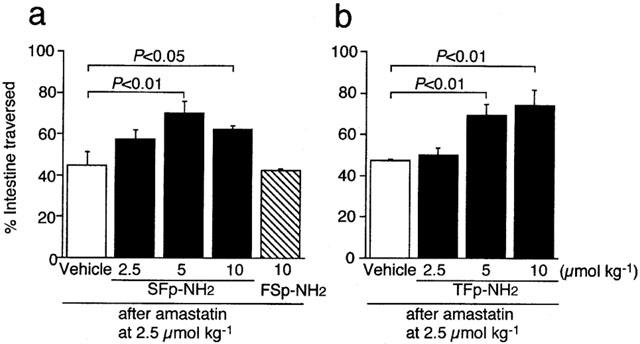
Effects of the PAR-1 agonists on gastrointestinal transit in mice. The human PAR-1-derived peptide SFLLR-NH2 (SFp-NH2) or the control peptide FSLLR-NH2 (FSp-NH2) (a) and the specific PAR-1 agonist TFLLR-NH2 (TFp-NH2) (b), in combination with i.p. amastatin at 2.5 μmol kg−1, were administered i.p. to the mouse. Data represent the mean±s.e.mean from 8–10 (a) or 4–6 (b) mice.
Potentiation by apamin of the effects of agonists for PAR-2 and PAR-1 at subeffective doses on gastrointestinal transit in mice
We tested if apamin, an inhibitor of Ca2+-activated, small-conductance K+ channels, could modify gastrointestinal transit in mice in vivo, since activation of PAR-2 and/or PAR-1 can produce relaxation of some of gastrointestinal smooth muscle preparations from mice, rats or guinea-pigs in vitro (Cocks et al., 1999; Kawabata et al., 1999b; 2000b). Apamin, when administered i.p. at 0.01 μmol kg−1, did not significantly alter gastrointestinal transit in conscious mice in vivo (Figure 3). Preadministration of apamin at the same dose significantly potentiated the effects of the PAR-2 agonist SLIGRL-NH2 and PAR-1-agonist TFLLR-NH2 at 1 and 2.5 μmol kg−1, respectively, that had no significant effect on gastrointestinal transit by themselves (Figure 3). On the other hand, the same dose of apamin failed to significantly potentiate the effect of carbachol at 0.055 μmol kg−1 that had been confirmed to be a subeffective dose in our preliminary experiments: per cent of intestine traversed was 40.4±1.9, 44.8±2.8 and 48.6±2.0 (n=4) in groups treated with vehicle plus vehicle, vehicle plus carbachol and apamin plus carbachol, respectively.
Figure 3.
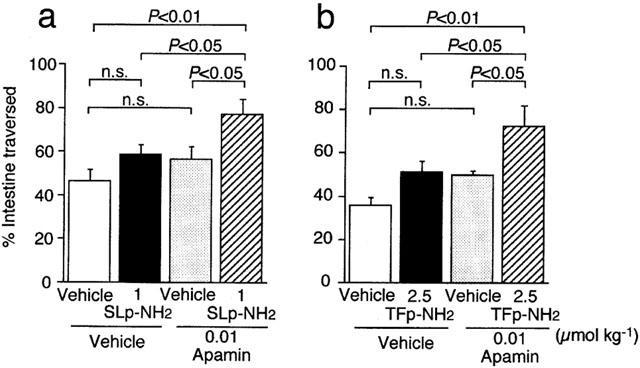
Promoting by apamin of the effects of the PAR-2 agonist SLIGRL-NH2 (SLp-NH2) (a) and the PAR-1 agonist TFLLR-NH2 (TFp-NH2) (b) at subeffective doses on gastrointestinal transit in mice. Apamin at 0.01 μmol kg−1 was administered i.p. to the mouse 6 min before i.p. injection of SLIGRL-NH2 at 1 μmol kg−1 or TFLLR-NH2 at 2.5 μmol kg−1 (5 min before amastatin at 2.5 μmol kg−1). Data represent the mean±s.e.mean from 7–8 (a) or 5–6 (b) mice. n.s., not significant.
Inhibition by verapamil, an inhibitor of voltage-dependent, L-type Ca2+ channels, of the increased gastrointestinal transit produced by agonists for PAR-2 and PAR-1 in mice
We next evaluated the effects of the L-type Ca2+ channel inhibitor verapamil on the PAR-2- or PAR-1-mediated increase in gastrointestinal transit in mice in vivo, because some in vitro studies suggested the involvement of L-type Ca2+ channels in contractile responses of gastrointestinal smooth muscle to activation of PAR-2 or PAR-1 (Saifeddine et al., 1996; Zheng et al., 1998; Kawabata et al., 1999b; 2000b). Verapamil, administered s.c. at 61.6 μmol kg−1 alone did not affect gastrointestinal transit in mice (Figure 4). The same dose of verapamil completely abolished the facilitating effects of the PAR-2 agonist SLIGRL-NH2 at 5 μmol kg−1 and the PAR-1 agonist TFLLR-NH2 at 10 μmol kg−1 on gastrointestinal transit (Figure 4). On the other hand, verapamil at the same dose significantly but only partially reduced the effect of carbachol at 0.55 μmol kg−1, a dose that had been confirmed to be submaximal in our preliminary experiments, per cent of inhibition being 43.9: per cent of intestine traversed was 36.6±2.8, 88.5±3.1 and 65.7±5.1 (n=4–6) in groups treated with vehicle plus vehicle, vehicle plus carbachol and verapamil plus carbachol, respectively.
Figure 4.
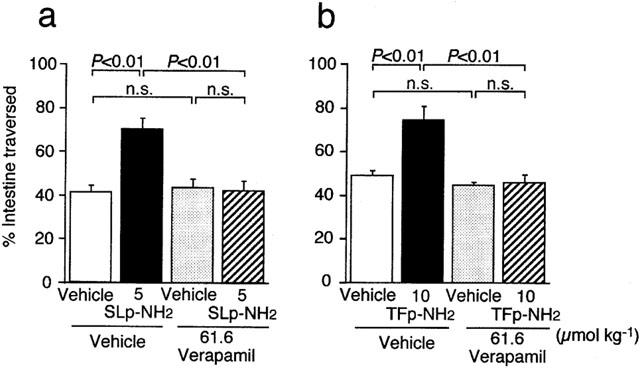
Inhibition by verapamil of the effects of the PAR-2 agonist SLIGRL-NH2 (SLp-NH2) (a) and the PAR-1 agonist TFLLR-NH2 (TFp-NH2) (b) on gastrointestinal transit in mice. Verapamil at 61.6 μmol kg−1 was administered s.c. to the mouse 16 min before i.p. injection of SLIGRL-NH2 at 5 μmol kg−1 or TFLLR-NH2 at 10 μmol kg−1 (15 min before amastatin at 2.5 μmol kg−1). Data represent the mean±s.e.mean from eight mice. n.s., not significant.
Lack of effects of genistein, an inhibitor of tyrosine kinase, on the PAR-2- and PAR-1-mediated increase in gastrointestinal transit in mice
In vitro evidence suggests that activation of tyrosine kinase might participate, at least in part, in contraction of gastrointestinal smooth muscle in response to activation of PAR-2 or PAR-1 (Saifeddine et al., 1996; Zheng et al., 1998; Kawabata et al., 1999b; 2000b). We thus finally evaluated if the tyrosine kinase inhibitor genistein could modify the effects of agonists for PAR-2 and PAR-1 on gastrointestinal transit in mice in vivo. Neither baseline values nor PAR-2- or PAR-1-mediated enhancement of gastrointestinal transit were significantly altered by s.c. preadministration of genistein at 18.5 μmol kg−1 (data not shown).
Discussion
The present study demonstrates that activation of either PAR-2 or PAR-1 increases gastrointestinal transit in mice in vivo. Our data also indicate that the augmented gastrointestinal transit via PAR-2 and PAR-1 is potentially suppressed by concomitant activation of apamin-sensitive, Ca2+-activated, small-conductance K+ channels. Furthermore, our results reveal that the effects of the agonists for PAR-2 and PAR-1 are mediated by activation of L-type Ca2+ channels, but are independent of tyrosine kinase.
Modulation by PAR-2 and PAR-1 of the smooth muscle motility in the gastrointestinal tract including the oesophagus is very complex, since both excitatory and inhibitory actions of either PAR-2 or PAR-1 agonists on isolated smooth muscle tension have been described (Hollenberg et al., 1993; 1997; Saifeddine et al., 1996; Corvera et al., 1997; Zheng et al., 1998; Cocks et al., 1999; Kawabata et al., 1999b; 2000a, 2000b). The present finding that exogenously applied agonists for PAR-2 or PAR-1 increased gastrointestinal transit suggests roles of these receptors in stimulating gastrointestinal motility. As PAR-2 and PAR-1 might be activated by agonist enzymes, i.e. trypsin, tryptase or coagulation factors VIIa and Xa for PAR-2 and thrombin for PAR-1 (Dery et al., 1998; Hollenberg, 1999; Kawabata & Kuroda, 2000; Camerer et al., 2000), in the early stage of inflammation or tissue-injury, the increased gastrointestinal transit due to activation of PAR-2 or PAR-1 might occur only in pathological conditions. A similar role of PAR-2 has been suggested in the pancreas where PAR-2 mediates pancreatic juice secretion in vivo as well as ductal secretion in vitro (Nguyen et al., 1999; Kawabata et al., 2000d). Taken together, these observations suggest that PARs might work as ‘sentries' for inflammation, as proposed elsewhere (Cocks & Moffatt, 2000). However, our hypothesis remains to be demonstrated by more detailed studies using more potent, selective non-peptide agonists and antagonists for PAR-1 and PAR-2 in future.
PAR-1, but not PAR-2, activates apamin-sensitive K+ channels, resulting in relaxation of isolated rat duodenal smooth muscle (Kawabata et al., 1999b; 2000b), and both PAR-2 and PAR-1 mediate apamin-sensitive relaxation of mouse gastric fundus precontracted with carbachol in vitro (Cocks et al., 1999). These reports predict that agonists for PAR-2 or PAR-1, given in vivo, would suppress gastrointestinal transit. The present study, however, excludes this possibility and implies that apamin-sensitive K+ channels are potentially activated depending upon activation of PAR-2 or PAR-1 in vivo, which is overcome by their excitatory effects through distinct mechanisms. The physiological significance of the potential inhibitory properties of PAR-2 and PAR-1 through apamin-sensitive K+ channels in modulation of gastrointestinal transit remains to be investigated.
That the increased gastrointestinal transit mediated by PAR-2 or PAR-1 was completely abolished by verapamil is consistent with the previous in vitro evidence that the contractile responses of gastrointestinal smooth muscle to agonists for PAR-2 or PAR-1 are largely dependent on activation of L-type Ca2+ channels (Saifeddine et al., 1996; Zheng et al., 1998; Kawabata et al., 1999b; 2000b). The finding that verapamil did not suppress the basal gastrointestinal transit (see Figure 4) is in agreement with the work by di carlo et al. (1993), while some other studies have revealed decreased gastrointestinal transit following verapamil (Shah et al., 1987; Calignano et al., 1992). The effectiveness of verapamil in the resting state might vary with experimental conditions such as the strain or size of mice employed. There is in vitro evidence that tyrosine kinase plays a role in the contraction of gastrointestinal longitudinal smooth muscle in response to activation of PAR-1 or PAR-2 (Saifeddine et al., 1996; Zheng et al., 1998; Kawabata et al., 1999b; 2000b). However, our data suggest that tyrosine kinase does not contribute to the modulation by PAR-1 and PAR-2 of gastrointestinal transit in vivo.
Our present data that PAR-2 and PAR-1 modulated gastrointestinal transit in vivo further support the importance of the roles of these receptors in the gastrointestinal systems, especially under pathophysiological conditions such as inflammation.
Acknowledgments
We are grateful to Dr Hiromasa Araki and Ms Sachiyo Nishimura (Fuso Pharmaceutical Industries Ltd.) for their assistance in synthesis of peptides.
Abbreviations
- PAR
protease-activated receptor
References
- BOHM S.K., KONG W., BROMME D., SMEEKENS S.P., ANDERSON D.C., CONNOLLY A., KAHN M., NELKEN N.A., COUGHLIN S.R., PAYAN D.G., BUNNETT N.W. Molecular cloning, expression and potential functions of the human proteinase-activated receptor-2. Biochem. J. 1996;314:1009–1016. doi: 10.1042/bj3141009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CALIGNANO A., CAPASSO A., PERSICO P., MANCUSO F., SORRENTINO L. Dexamethasone modifies morphine-, atropine-, verapamil-induced constipation in mice. Gen. Pharmacol. 1992;23:753–756. doi: 10.1016/0306-3623(92)90161-c. [DOI] [PubMed] [Google Scholar]
- CAMERER E., HUANG W., COUGHLIN S.R. Tissue factor- and factor X-dependent activation of protease-activated receptor 2 by factor VIIa. Proc. Natl. Acad. Sci. U.S.A. 2000;97:5255–5260. doi: 10.1073/pnas.97.10.5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CAMPOS M.M., SOUZA G.E.P., CALIXTO J.B. In vivo B1 kinin-receptor upregulation. Evidence for involvement of protein kinases and nuclear factor κB pathways. Br. J. Pharmacol. 1999;127:1851–1859. doi: 10.1038/sj.bjp.0702715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CIRINO G., CICALA C., BUCCI M.R., SORRENTINO L., MARAGANORE J.M., STONE S.R. Thrombin functions as an inflammatory mediator through activation of its receptor. J. Exp. Med. 1996;183:821–827. doi: 10.1084/jem.183.3.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COCKS T.M., MOFFATT J.D. Protease-activated receptors: sentries for inflammation. Trends Pharmacol. Sci. 2000;21:103–108. doi: 10.1016/s0165-6147(99)01440-6. [DOI] [PubMed] [Google Scholar]
- COCKS T.M., SOZZI V., MOFFATT J.D., SELEMIDIS S. Protease-activated receptors mediate apamin-sensitive relaxation of mouse and guinea pig gastrointestinal smooth muscle. Gastroenterology. 1999;116:586–592. doi: 10.1016/s0016-5085(99)70180-0. [DOI] [PubMed] [Google Scholar]
- CORVERA C.U., DERY O., MCCONALOGUE K., BOHM S.K., KHITIN L.M., CAUGHEY G.H., PAYAN D.G., BUNNETT N.W. Mast cell tryptase regulates rat colonic myocytes through proteinase-activated receptor 2. J. Clin. Invest. 1997;100:1383–1393. doi: 10.1172/JCI119658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEODATO B., ALTAVILLA D., SQUADRITO G., CAMPO G.M., ARLOTTA M., MINUTOLI L., SAITTA A., CUCINOTTA D., CALAPAI G., CAPUTI A.P., MIANO M., SQUADRITO F. Cardioprotection by the phytoestrogen genistein in experimental myocardial ischaemia-reperfusion injury. Br. J. Pharmacol. 1999;125:1683–1690. doi: 10.1038/sj.bjp.0702973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DERY O., CORVERA C.U., STEINHOFF M., BUNNETT N.W. Proteinase-activated receptors: novel mechanisms of signaling by serine proteases. Am. J. Physiol. 1998;274:C1429–C1452. doi: 10.1152/ajpcell.1998.274.6.C1429. [DOI] [PubMed] [Google Scholar]
- DI CARLO G., AUTORE G., IZZO A.A., MAIOLINO P., MASCOLO N., VIOLA P., DIURNO M.V., CAPASSO F. Inhibition of intestinal motility and secretion by lavonoids in mice and rats: structure-activity relationships. J. Pharm. Pharmacol. 1993;45:1054–1059. doi: 10.1111/j.2042-7158.1993.tb07180.x. [DOI] [PubMed] [Google Scholar]
- HOLLENBERG M.D. Protease-activated receptors: PAR-4 and counting: how long is the course. Trends Pharmacol. Sci. 1999;20:271–273. doi: 10.1016/s0165-6147(99)01333-4. [DOI] [PubMed] [Google Scholar]
- HOLLENBERG M.D., LANIYONU A.A., SAIFEDDINE M., MOORE G.J. Role of the amino- and carboxyl-terminal domains of thrombin receptor-derived polypeptides in biological activity in vascular endothelium and gastric smooth muscle: evidence for receptor subtypes. Mol. Pharmacol. 1993;43:921–930. [PubMed] [Google Scholar]
- HOLLENBERG M.D., SAIFEDDINE M., AL-ANI B., GUI Y. Proteinase-activated receptor 4 (PAR4): action of PAR4-activating peptides in vascular and gastric tissue and lack of cross-reactivity with PAR1 and PAR2. Can. J. Physiol. Pharmacol. 1999;77:458–464. [PubMed] [Google Scholar]
- HOLLENBERG M.D., SAIFEDDINE M., AL-ANI B., KAWABATA A. Proteinase-activated receptors: structural requirements for activity, receptor cross-reactivity, and receptor selectivity of receptor-activating peptides. Can. J. Physiol. Pharmacol. 1997;75:832–841. [PubMed] [Google Scholar]
- ISHIHARA H., CONNOLLY A.J., ZENG D., KAHN M.L., ZHENG Y.W., TIMMONS C., TRAM T., COUGHLIN S.R. Protease-activated receptor 3 is a second thrombin receptor in humans. Nature (London) 1997;386:502–506. doi: 10.1038/386502a0. [DOI] [PubMed] [Google Scholar]
- IZZO A.A., PINTO L., BORRELLI F., CAPASSO R., MASCOLO N., CAPASSO F. Central and peripheral cannabinoid modulation of gastrointestinal transit in physiological states or during the diarrhoea induced by croton oil. Br. J. Pharmacol. 2000;129:1627–1632. doi: 10.1038/sj.bjp.0703265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KAHN M.L., ZHENG Y.-W., HUANG W., BIGORNIA V., ZENG D., MOFF S., FARESE R.V., JR, TAM C., COUGHLIN S.R. A dual thrombin receptor system for platelet activation. Nature (London) 1998;394:690–694. doi: 10.1038/29325. [DOI] [PubMed] [Google Scholar]
- KAWABATA A., KURODA R. Protease-activated receptor (PAR), a novel family of G protein-coupled seven trans-membrane domain receptors: activation mechanisms and physiological roles. Jpn. J. Pharmacol. 2000;82:171–175. doi: 10.1254/jjp.82.171. [DOI] [PubMed] [Google Scholar]
- KAWABATA A., KURODA R., KUROKI N., NISHIKAWA H., KAWAI K. Dual modulation by thrombin of the motility of rat oesophageal muscularis mucosae via two distinct protease-activated receptors (PARs): a novel role for PAR-4 as opposed to PAR-1. Br. J. Pharmacol. 2000a;131:578–584. doi: 10.1038/sj.bjp.0703590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KAWABATA A., KURODA R., KUROKI N., NISHIKAWA H., KAWAI K., ARAKI H. Characterization of the protease-activated receptor-1-mediated contraction and relaxation in the rat duodenal smooth muscle. Life Sci. 2000b;67:2521–2530. doi: 10.1016/s0024-3205(00)00835-3. [DOI] [PubMed] [Google Scholar]
- KAWABATA A., KURODA R., MINAMI T., KATAOKA K., TANEDA M. Increased vascular permeability by a specific agonist of protease-activated receptor-2 in rat hindpaw. Br. J. Pharmacol. 1998;125:419–422. doi: 10.1038/sj.bjp.0702063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KAWABATA A., KURODA R., NISHIKAWA H., ASAI T., KATAOKA K., TANEDA M. Enhancement of vascular permeability by specific activation of protease-activated receptor-1 in rat hindpaw: a protective role of endogenous and exogenous nitric oxide. Br. J. Pharmacol. 1999a;126:1856–1862. doi: 10.1038/sj.bjp.0702513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KAWABATA A., KURODA R., NISHIKAWA H., KAWAI K. Modulation by protease-activated receptors of the rat duodenal motility in vitro: possible mechanisms underlying the evoked contraction and relaxation. Br. J. Pharmacol. 1999b;128:865–872. doi: 10.1038/sj.bjp.0702755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KAWABATA A., MORIMOTO N., NISHIKAWA H., KURODA R., ODA Y., KAKEHI K. Activation of protease-activated receptor-2 triggers mucin secretion in the rat sublingual gland. Biochem. Biophys. Res. Commun. 2000c;270:298–302. doi: 10.1006/bbrc.2000.2404. [DOI] [PubMed] [Google Scholar]
- KAWABATA A., NISHIKAWA H., KURODA R., KAWAI K., HOLLENBERG M.D. Proteinase-activated receptor-2 (PAR-2): regulation of salivary and pancreatic exocrine secretion in vivo in rats and mice. Br. J. Pharmacol. 2000d;129:1627–1632. doi: 10.1038/sj.bjp.0703274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KAWABATA A., SAIFEDDINE M., AL-ANI B., LEBLOND L., HOLLENBERG M.D. Evaluation of proteinase-activated receptor-1 (PAR1) agonists and antagonists using a cultured cell receptor desensitization assay: activation of PAR2 by PAR1 targeted ligands. J. Pharmacol. Exp. Ther. 1999c;228:358–370. [PubMed] [Google Scholar]
- MOLINO M., BARNATHAN E.S., NUMEROF R., CLARK J., DREYER M., CUMASHI A., HOXIE J.A., SCHECHTER N., WOOLKALIS M., BRASS L.F. Interactions of mast cell tryptase with thrombin receptors and PAR-2. J. Biol. Chem. 1997;272:4043–4049. doi: 10.1074/jbc.272.7.4043. [DOI] [PubMed] [Google Scholar]
- NAKANISHI-MATSUI M., ZHENG Y.-W., SULCINER D.J., WEISS E.J., LUDEMAN M.J., COUGHLIN S.R. PAR3 is a cofactor for PAR4 activation by thrombin. Nature. 2000;404:609–613. doi: 10.1038/35007085. [DOI] [PubMed] [Google Scholar]
- NGUYEN T.D., MOODY M.W., STEINHOFF M., OKOLO C., KOH D.-S., BUNNETT N.W. Trypsin activates pancreatic duct epithelial cell ion channels. J. Clin. Invest. 1999;103:261–269. doi: 10.1172/JCI2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NYSTEDT S., EMILSSON K., WAHLESTEDT C., SUNDELIN J. Molecular cloning of a potential proteinase activated receptor. Proc. Natl. Acad. Sci. U.S.A. 1994;91:9208–9212. doi: 10.1073/pnas.91.20.9208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RUPNIAK N.M.J., BOYE S., WILLIAMS A.R., COOK G., LONGMORE J., SEABROOK G.R., CAESER M., IVERSEN S.D., HILL R.G. Antinociceptive activity of NK1 receptor antagonists: non-specific effects of racemic RP67580. Br. J. Pharmacol. 1993;110:1607–1613. doi: 10.1111/j.1476-5381.1993.tb14008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAIFEDDINE M., AL-ANI B., CHENG C.-H., WANG L., HOLLENBERG M.D. Rat proteinase-activated receptor-2 (PAR-2): cDNA sequence and activity of receptor-derived peptides in gastric and vascular tissue. Br. J. Pharmacol. 1996;118:521–530. doi: 10.1111/j.1476-5381.1996.tb15433.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHAH M.H., DIKSHIT R.K., MANSURI S.M. The calcium channel antagonist, verapamil, potentiates the inhibitory action of morphine on intestinal and bilary motility. J. Pharm. Pharmacol. 1987;39:1037–1038. doi: 10.1111/j.2042-7158.1987.tb03157.x. [DOI] [PubMed] [Google Scholar]
- VERGNOLLE N., HOLLENBERG M.D., SHARKEY K.A., WALLACE J.L. Characterization of the inflammatory response to proteinase-activated receptor-2 (PAR2)-activating peptides in the rat paw. Br. J. Pharmacol. 1999a;127:1083–1090. doi: 10.1038/sj.bjp.0702634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VERGNOLLE N., HOLLENBERG M.D., WALLACE J.L. Pro- and anti-inflammatory actions of thrombin: a distinct role for proteinase-activated receptor-1 (PAR1) Br. J. Pharmacol. 1999b;126:1262–1268. doi: 10.1038/sj.bjp.0702408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VERGNOLLE N., MACNAUGHTON W.K., AL-ANI B., SAIFEDDINE M., WALLACE J.L., HOLLENBERG M.D. Proteinase-activated receptor 2 (PAR2)-activating peptides: identification of a receptor distinct from PAR2 that regulates intestinal transport. Proc. Natl. Acad. Sci. U.S.A. 1998;95:7766–7771. doi: 10.1073/pnas.95.13.7766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VU T.-K.H., HUNG D.T., WHEATON V.I., COUGHLIN S.R. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanisms of receptor activation. Cell. 1991;64:1057–1068. doi: 10.1016/0092-8674(91)90261-v. [DOI] [PubMed] [Google Scholar]
- XU W.-F., ANDERSEN H., WHITMORE T.E., PRESNELL S.R., YEE D.P., CHING A., GILBERT T., DAVIE E.W., GOSTER D.C. Cloning and characterization of human protease-activated receptor 4. Proc. Natl. Acad. Sci. U.S.A. 1998;95:6642–6646. doi: 10.1073/pnas.95.12.6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHENG X.-L., RENAUX B., HOLLENBERG M.D. Parallel contractile signal transduction pathways activated by receptors for thrombin and epidermal growth factor-urogastrone in guinea pig gastric smooth muscle: blockade by inhibitors of mitogen-activated protein kinase and phosphatidyl inositol-3′-kinase. J. Pharmacol. Exp. Ther. 1998;285:325–334. [PubMed] [Google Scholar]