

Abstract
A method is described to study the precise positioning of transmembrane peptides in a phospholipid bilayer combining hydrogen/deuterium (H/D) exchange and nanoelectrospray ionization mass spectrometry. The method was tested by using model systems consisting of designed α-helical transmembrane peptides [acetylGW2(LA)5W2Aethanolamine (WALP16) and acetyl-(GA)3W2(LA)5W2(AG)3ethanolamine (WALP16(+10))] incorporated in large unilamellar vesicles of 1,2-dimyristoyl-sn-glycero-3-phosphocholine. Both peptides consist of an alternating leucine/alanine hydrophobic core sequence flanked by tryptophan residues as interfacial anchor residues. In the case of WALP16(+10), this sequence is extended at both ends by 5-aa glycine/alanine tails extending into the aqueous phase surrounding the bilayer. H/D exchange of labile hydrogens in these peptides was monitored in time after dilution of the vesicles in buffered deuterium oxide. It was found that the peptides can be measured by direct introduction of the proteoliposome suspension into the mass spectrometer. Several distinct H/D exchange rates were observed (corresponding to half-life values varying from ≤2 to ≈2 × 104 min). Fast exchange rates were assigned to the water-exposed tails of WALP16(+10). For both WALP16 and WALP16(+10), intermediate exchange rates were assigned to the residues close to the membrane/water interface, and the slow exchange rates to the membrane-embedded hydrophobic core. These assignments were confirmed by results from collision-induced dissociation tandem mass spectrometry experiments, which allowed analysis of exchange of individual peptide amide linkages. This proteoliposome nanoelectrospray ionization mass spectrometry technique is shown to be an extremely sensitive and powerful tool for revealing site-specific information on peptide–membrane interactions.
Membrane proteins are biologically important because they are responsible for crucial functions in the cell, such as signal transduction, hormone reception, and transport of proteins, nutrients, and ions across cell membranes. The way in which membrane proteins are embedded in the lipid bilayer and interact with surrounding lipids is of fundamental significance for membrane protein structure and function. Insight into the exact positioning of the transmembrane segment(s) of such proteins with respect to the membrane–water interface and their structural and dynamic properties is hereby essential.
The analysis of hydrogen/deuterium (H/D) exchange kinetics of protein backbone amide protons has long been used as a source of structural and dynamic information (1–3). The H/D exchange process of amide hydrogens with solvent deuteriums can take place only when these hydrogens are exposed to the solvent. For a membrane-incorporated peptide, the rate of H/D exchange is influenced by the extent of solvent permeation to the site of exchange in different regions of the bilayer, as well as by the participation of amide hydrogens in the hydrogen-bonding network that defines secondary and tertiary structure (2).
Until now, H/D exchange in membrane peptides and proteins has been studied mostly by using Fourier-transformed IR (FTIR) and NMR spectroscopic techniques. Although both techniques have given useful insight into the structure and dynamics of peptides in membranes, both exhibit some disadvantages. FTIR spectroscopy can determine the total amount of deuterium uptake but does not give information about specific sites of deuterium exchange (4–6). Usually, such studies on membrane peptides require bilayers that are not hydrated in excess water, and the use of buffers is not trivial. In contrast, high-resolution NMR techniques can determine exchange rates of specific amide hydrogens. However, such NMR studies typically use micellar systems, which are not ideal alternatives for membrane systems. Moreover, relatively large amounts of material, long measuring times, and a low pH are required. The latter may not be physiologically relevant. Nevertheless, H/D exchange rates have been measured for individual assigned amide protons of various membrane-associated peptides and proteins in detergent solutions (6–9). Another NMR approach makes use of trapping techniques and subsequent transfer of the peptide from an aqueous membrane environment to isotropic media (10). Also, solid-state NMR techniques have been used to study H/D exchange kinetics of individual amide backbone hydrogens of a helical transmembrane peptide (11). However, the limited sensitivity is a disadvantage of all NMR methods.
In recent years, several studies have been reported in which mass spectrometry (MS) was used to investigate H/D exchange behavior of water-soluble peptides and proteins (12–16). From the observed H/D exchange patterns, information can be extracted on protein folding (13, 15) and noncovalent complex formation (14). Information on the exchange of hydrogens at individual peptide amide linkages may be obtained by MS by using limited proteolysis (16) or collision-induced dissociation tandem MS (MS/MS), as has been shown for the α-helical peptide melittin (17) as well as for several peptic fragments of cytochrome c (18). The results of these latter studies indicate that H/D exchange measured by MS/MS may in some cases be an appropriate tool to investigate the amount of secondary structure in a peptide.
In this study, we apply nanoelectrospray ionization MS (nano-ESI-MS) as a tool to measure H/D exchange in water-insoluble transmembrane peptides. In this approach, the peptides are reconstituted in fully hydrated dispersed phospholipid bilayers, which are then directly introduced into the mass spectrometer. We tested this technique by using well-defined model transmembrane peptides, acetylGW2(LA)5W2Aethanolamine (WALP16) and acetyl(GA)3W2(LA)5W2(AG)3ethanolamine [WALP16(+10)] (Table 1). The WALP16 peptide consists of an alternating Leu/Ala hydrophobic core sequence flanked by tryptophan residues, which are found in many membrane proteins near the membrane/water interfacial region and are assumed to interact in a specific way with the membrane interface (19–21). Moreover, it forms a transmembrane α-helix in phosphatidylcholine bilayers (20), and therefore it may resemble a consensus transmembrane α-helical segment of intrinsic membrane proteins. The length of WALP16 [22.5 Å, (22)] is similar to the hydrophobic thickness of a 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) bilayer in the fluid state [≈23 Å, (23)], which implies that in this lipid system, WALP16 is completely embedded in the hydrophobic region of the bilayer. The WALP16(+10) peptide has alternating Gly/Ala extensions at both termini of WALP16. These tails were designed to have sufficient hydrophilicity to penetrate into the water phase surrounding the bilayer and to adopt no secondary structure because of the α-helix breaking Gly residues. Because the tail hydrogens therefore should be accessible to the aqueous phase and not involved in hydrogen bonding, they are expected to exchange much more rapidly than those in the hydrophobic core of the peptide. The membrane incorporation of both peptides is schematically depicted in Fig. 1.
Table 1.
Amino acid sequences of the peptides used, their molecular masses, and their numbers of exchangeable hydrogen atoms
| Peptide | Amino acid sequence | Monoisotopic molecular mass, amu | No. of exchangeable hydrogens |
|---|---|---|---|
| WALP16 | a-GWWLALALALALAWWA-e | 1,896.05 | 22 |
| WALP16(+10) | a-GAGAGAWWLALALALALAWWAGAGAG-e | 2,536.35 | 32 |
Figure 1.

Schematic representation of the incorporation of (A) WALP16 and (B) WALP16(+10) in a DMPC bilayer. The cylinder implies the α-helical transmembrane region; the Gly/Ala extensions of WALP16(+10) are denoted by randomly ordered strings.
The results of the experiments described here show that our proteoliposome nano-ESI-MS technique may be an extremely powerful and sensitive tool to analyze the exact positioning of membrane peptides in a lipid bilayer and to reveal site-specific information on peptide–membrane interactions. We believe that this method could be equally well applied to study the positioning of larger transmembrane proteins in model or biological membranes.
Materials and Methods
Chemicals.
Trifluoroacetic acid and 1,4-dioxane (p.A.) were obtained from Merck (Darmstadt, Germany) and 2,2,2-trifluoroethanol (TFE) from Sigma. Deuterium oxide (>99.9% D) was obtained from Aldrich. D2O was stored under nitrogen at 4°C. Acetonitrile (HPLC gradient grade) was obtained from Biosolve (Valkenswaard, The Netherlands). Sodium iodide was from OPG Farma (Utrecht, The Netherlands). Ammonium acetate was from Fluka. The phospholipid DMPC was obtained from Avanti Polar Lipids. The peptide WALP16 was synthesized as described by Killian et al. (20). WALP16(+10) was synthesized from fluorenylmethoxycarbonyl-Gly Sasrin resin and cleaved with 10% ethanolamine in dichloromethane at 24°C for 48 hr. Details will be published elsewhere (D. V. Greathouse and R. Goforth, personal communication). The peptides were tested for purity by electrospray MS and found to be essentially pure.
Procedure for Peptide Incorporation into Phospholipid Vesicles.
Peptides were first dissolved in a small volume of trifluoroacetic acid (TFA) (10 μl per mg of peptide) and dried under a nitrogen stream. To remove residual TFA, the peptides were subsequently dissolved in TFE (1 mg/ml) followed by evaporation of the solvent in a rotavapor. Peptides were then again dissolved in TFE to a final concentration of 1 mg/ml. Dry mixed films of WALP16 or WALP16(+10) and DMPC (peptide to lipid ratio 1:25) were prepared as follows. Peptide solutions in TFE (1 ml; 0.46 mM) were added to DMPC solutions in methanol (1 ml; 12 mM) and vigorously vortexed. The solvent was removed by evaporation in a rotavapor. The mixed films were then dried for 24 hr under vacuum. The films were hydrated at about 40°C, well above the gel-to-liquid crystalline phase transition temperature of the phospholipid [24°C (24)] in 0.5 ml 10 mM ammonium acetate buffer (pH 7.5). This results in the formation of extended bilayers, as was confirmed by 31P NMR measurements, performed as described previously (20). Large unilamellar vesicles (LUVETs) were prepared by extrusion through a 400-nm filter at room temperature and kept at 4°C until use.
Before the start of H/D exchange, LUVETs were preincubated at 30°C for at least 30 min. LUVETs suspensions were then diluted 50 times in deuterated ammonium acetate buffer at 30°C (10 mM, pH 7.5) containing approximately 0.1 mM NaI. At selected time points, 2 μl of this diluted DMPC/peptide suspension was transferred into a gold-coated glass capillary, and the measurement was started as quickly as possible. The deadtime between dilution and measurement was at a minimum 1.5 min.
MS Measurements.
MS measurements were performed on a quadrupole time-of-flight (Q-Tof) instrument (Micromass, Manchester, U.K.) operating in positive ion mode, equipped with a Z-spray nanoelectrospray source. Nanoelectrospray needles were made from borosilicate glass capillaries (Kwik-Fil, World Precision Instruments, Sarasota, FL) on a P-97 puller (Sutter Instruments, Novato, CA). A special procedure was developed to pull needles with a relatively large tip opening (several tens of micrometers), which resulted in higher flow rates than typical for nano-ESI-MS. The needles were coated with a thin gold layer (approximately 500 Å) by using an Edwards Scancoat (Edwards Laboratories, Milpitas, CA) six Pirani 501 (at 40 mV, 1 kV, for 200 sec). The nano-ES needle was positioned approximately 5 mm before the orifice of the mass spectrometer. For MS experiments the quadrupole was set in the radio-frequency-only mode to act simply as an ion guide to efficiently link the electrospray ion source with the high sensitivity reflectron time-of-flight analyzer. In MS/MS mode, the quadrupole was used to select precursor ions, which were fragmented in the hexapole collision cell, generating product ions that were subsequently mass analyzed by the orthogonal time-of-flight mass analyzer. The potential between the nanospray needle and the orifice of the mass spectrometer was typically set to 1,800 V; the cone voltage was 140 V. The nanospray needle was constantly kept at approximately 30°C. For MS/MS measurements, the collision energy was set to 135 V. Argon was used as collision gas. The quadrupole mass resolution parameters were set to a relatively large mass window to select the entire isotope envelope of the precursor ions. The reflectron time-of-flight parameters were set such that the fragment ions were detected at more than unit mass resolution, as required to obtain isotopically resolved H/D profiles. Increases in deuterium content (in Da) were calculated by using the average mass-to-charge (m/z) values of the isotope clusters of the undeuterated peptide and the (partly) deuterated peptides.
Results and Discussion
ESI-MS.
By using the proteoliposome nano-ESI-MS technique as described in this paper, high-quality spectra can be obtained of peptides in model membranes. Figs. 2 A and B show typical positive ion spectra of LUVETs of WALP16 and WALP16(+10), respectively, reconstituted in DMPC bilayers dispersed in 10 mM ammonium acetate buffer. In both cases the monomers, dimers, trimers, and tetramers of DMPC are observed as [M+H]+ as well as [M+Na]+ ions (22 mass units higher). Also the peptide ion peaks are clearly visible, in spite of the relatively low peptide/lipid ratio. The [M+Na]+ ions of WALP16 as well as of WALP16(+10) are more abundant than the corresponding [M+H]+ ions. The spectrum of the WALP/DMPC LUVETs indicates that the vesicles are destabilized during the ionization process in such a way that only smaller aggregates of phospholipid molecules are observed. The largest singly charged multimer was found to be the [16M+H]+ ion of DMPC (not shown), with an intensity of about four orders of magnitude lower than the intensity of the [M+H]+ ion of DMPC.
Figure 2.

Nano-ESI-MS spectrum of LUVETs of (A) WALP16/DMPC (1:25) and (B) WALP16(+10)/DMPC (1:25). The [M+Na]+ ions of the DMPC monomer, dimer and trimer are most abundant; the isotope envelopes of the [M+Na]+ ions of the WALP16 and WALP16(+10) peptides are observed around m/z 1919 (A) and m/z 2559 (B); the [M+H]+ ions are around m/z 1897 (A) and m/z 2537 (B), respectively. For the recording of each spectrum, ≈200 nl of a vesicle suspension containing 20 μM peptide was used.
Fig. 3 shows the overall deuterium content in time of WALP16 and WALP16(+10) reconstituted in DMPC bilayers upon dilution in buffered deuterium oxide. As shown in Fig. 3A, within the deadtime of the experiment, approximately six hydrogens have exchanged for WALP16 and 15 for WALP16(+10). From the first time point at 2 min to approximately 100 min, the exchange is slower, as can also be observed in Fig. 3B, in which the exchange is monitored during much longer incubation times. At longer incubation times, exchange becomes very slow. From the total number of exchangeable hydrogens (22 for WALP16 and 32 for WALP16(+10); see Table 1), it can be concluded that even after several days, not all exchangeable hydrogens have exchanged completely. For both peptides, approximately 10 hydrogens remain protected from exchange. This appears to be in good agreement with the model shown in Fig. 1, in which WALP16 is incorporated in the DMPC bilayer with its hydrophobic core embedded inside the phospholipid bilayer, whereas the 5-aa tails of WALP16(+10) at both sides extend into the aqueous phase surrounding the bilayer.
Figure 3.

Deuterium content of WALP16 (●) and WALP16(+10) (▴) incorporated in DMPC bilayers as a function of incubation time. A shows the time envelope from 0 to 15 min. The dotted line between 0 and 2 min represents an estimated extrapolation of the curve, because no data points were available in this region. B shows the time envelope from 0 to 5 × 103 min.
The various exchange rates can be quantified by using the expression (25):
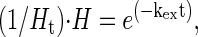 |
1 |
where H is the amount of protected hydrogens, Ht is the total amount of exchangeable hydrogens, kex is the H/D exchange rate constant, and t is the time the peptide is incubated in D2O. From Eq. 1, it follows that in plots of ln(H) vs. t, the slope is inversely proportional to kex, and populations of hydrogens having different rate constants can thus be distinguished. In this way, from plots over the entire time interval, roughly three populations of exchangeable hydrogens could be discerned. These are summarized in Table 2 and will be discussed below.
Table 2.
H/D exchange half-life values (in min) of populations of hydrogens of WALP16 and WALP16(+10), calculated from logarithmic plots of the data in Fig. 3 (see text for details)
| WALP16
|
WALP16(+10)
|
|||
|---|---|---|---|---|
| No. of hydrogens | Half-life, min | No. of hydrogens | Half-life, min | |
| Fast | 6 | ≤3 | 15 | ≤2 |
| Intermediate | 1.5 | 1.4 × 101 | 2 | 1.9 × 101 |
| 2 | 2.4 × 102 | 2 | 7.0 × 101 | |
| 2 | 3.4 × 103 | 2 | 3.5 × 102 | |
| Slow | 10 | 1.8 × 104 | 10 | 2.3 × 104 |
Fast exchange was defined as between 0 and 2 min, intermediate exchange from 2–1,000 min and slow exchange starting from about 1,000 min. The number of hydrogens in each population is indicated (estimated errors in these numbers are in all cases 0.5).
Both peptides contain a number of fast-exchanging hydrogens (half-lives ≤3 min). These hydrogens have already exchanged completely at the time of the first measurements. They are most likely unprotected by hydrogen bonding and readily accessible to the solvent. As shown in Table 2, WALP16 has about six and WALP16(+10) about 15 fast-exchanging hydrogens. Therefore, these hydrogens comprise most likely the two terminal backbone hydrogens of WALP16 and the terminal extensions of WALP16(+10), corresponding to approximately 10 additional fast-exchanging hydrogens. Also, the four indole hydrogens of the tryptophans in both peptides may exchange very fast. For both the WALP16 and WALP16(+10) hydrogens, it is not possible to obtain the exact half-life values for these populations, because no data points are available at these very early times.
The population of hydrogens that display intermediate exchange rates (half-lives from ≈101 to ≈103 min) comprises five to six hydrogens for both peptides. In this intermediate exchange region, it was even possible to distinguish subpopulations of hydrogens with different exchange kinetics. This is illustrated in Fig. 4 A and B, which show H/D exchange data of WALP16 and WALP16(+10) in the time interval of 2 to 100 min, respectively. For WALP16, the subpopulations with the shortest half-life values correspond to lines 1 and 2 in Fig. 4A. The third subpopulation has a slightly longer half-life (not visible on the timescale of Fig. 4A). An analogous set of subpopulations was observed for WALP16(+10) (represented by lines 1, 2, and 3 in Fig. 4B). These hydrogens most likely represent the backbone hydrogens of the tryptophan residues and residues near the ends of the transmembrane helices, i.e., Gly-1 and Ala-16 for WALP16, and Ala-6 and Ala-21 for WALP16(+10). Differences between the calculated half-life values in the two peptides are ascribed to small differences in local environment at these positions.
Figure 4.

Kinetics of H/D exchange of (A) WALP16 and (B) WALP16(+10). On the y-axis, the natural logarithm of the amount of protected hydrogens in the peptide [ln(H)] is shown. The time envelopes from 2–100 min for both peptides are indicated. From the slopes of the curves in the plot, the half-lives of specific hydrogen populations in this time period were calculated, and from the intercepts with the y-axis, the number of hydrogens involved. For WALP16, the intermediate exchange data are represented by the lines (1) y = −4.9x 10−2x + 2.81 (r = 0.95), and (2) y = −2.9x 10−3x + 2.66 (r = 0.99); for the intermediate exchange of WALP16(+10) (1) y = −3.7 × 10−2x + 2.81 (r = 0.97), (2) y = −9.9 × 10−3x + 2.68 (r = 0.99), and (3) y = −2.0 × 10−3x + 2.49 (r = 0.99).
The slowest exchanging hydrogen populations (half-lives ≈104 min) comprise approximately 10 hydrogens in both peptides and are assigned to the backbone amide hydrogens of the Leu/Ala core of the peptides. Such a protection of exchange by incorporation into a membrane is in agreement with exchange studies on other transmembrane peptides and proteins (7–9).
ESI MS/MS.
From the results of the single-stage MS experiments, information with regard to the accessibility of exchangeable hydrogens of membrane-buried peptides could be obtained. However, from such data it is not possible to directly determine which sites have actually been exchanged. In principle this should be possible by fragmentation studies on the partly deuterated peptides. To investigate this possibility, MS/MS experiments were performed on the [M+Na]+ ions of WALP16 and WALP16(+10). Experiments were performed on the undeuterated peptides and partly deuterated peptides when about 12 of 22 (WALP16) and 20 of 32 [WALP16(+10)] hydrogens had been exchanged. Fig. 5 shows part of a typical MS/MS spectrum of partly deuterated WALP16(+10) in which series of A and Y′ fragment ions can be observed, together covering almost the entire peptide. The MS/MS spectra of the sodiated WALP16 and WALP16(+10) ions showed predominantly characteristic A and Y′ fragment ions, corresponding to the N- and C-terminal fragments, respectively (26).
Figure 5.

Part of a nano-ESI MS/MS spectrum of the [M+Na]+ ion of WALP16(+10) incorporated in a DMPC bilayer 100 min after incubation in a deuterated ammonium acetate buffer. The sodiated partly deuterated peptide parent ions as well as parts of the A fragment ion series and Y′ fragment ion series are indicated [Y′ is defined as [Y′′ −H + Na] (26)]. (Inset) Fragmentation pattern; A ions correspond to N-terminal fragments and Y′ ions correspond to C-terminal fragments.
As an illustration of the quality of the MS/MS data, isotope envelopes of A10, A18, and A23 fragment ions of WALP16(+10) are shown in Fig. 6. Fragment ions of WALP16(+10) in DMPC bilayers in undeuterated buffer (Fig. 6A) are compared with those in deuterated buffer when on average 20 of 32 exchangeable hydrogens had been exchanged (Fig. 6B). The numbers in bold denote the differences between the average m/z values of the undeuterated and the deuterated fragment ions. The difference in deuterium content between the A10 and A18 fragment ions is much smaller than the difference between the A18 and A23 fragment ions, indicating that the deuterium labels were not randomly distributed over the WALP16(+10) peptide. Instead, it seems that in the partly exchanged WALP16(+10) precursor ions, much less deuterium was present in the Leu/Ala core than in the other parts of the peptide.
Figure 6.

Isotope envelopes of A10, A18, and A23 fragment ions of WALP16(+10) incorporated in DMPC bilayers in undeuterated buffer (A) as opposed to the analogous fragment ions after incubation in deuterated buffer (B). The arrows and numbers indicate the average m/z values for the fragment ions. The differences between the average m/z values of the undeuterated and the deuterated fragment ions are printed in boldface in parentheses. These spectra were recorded when about 20 of 32 hydrogens had exchanged. The amino acid sequence (Upper) indicates how the A-fragment ions are formed (a, acetyl; e, ethanolamine).
The deuterium content of all A and Y′ fragment ions as derived from the MS/MS data is shown in Fig. 7 A and B for WALP16 and WALP16(+10), respectively (solid bars). The solid lines depicted in Fig. 7 are theoretical curves using a simplified model, in which the exchanged hydrogens in the partly deuterated peptides would originate exclusively from the outermost exchangeable hydrogens. For WALP16(+10), these would be: the C-terminal and N-terminal Gly/Ala extensions, including the two exchangeable hydrogen atoms of the ethanolamine moiety (14 hydrogens), the side chains of the tryptophans (4 hydrogens), and the backbone hydrogens of the outermost tryptophans (Trp-7 and Trp-20) at each side of the peptide (2 hydrogens). The data collection of WALP16(+10) (Fig. 7B) shows a remarkable resemblance to this hypothesized exchange kinetics.
Figure 7.

Bar diagrams representing the average deuterium content of the partly deuterated A and Y′ fragment ions (26) (A) of WALP16 and (B) of WALP16(+10), as derived from nano-ESI-MS/MS measurements. The An fragment comprises the amide and side chain deuteriums of the N-terminal n amino acid residues; the Y′n fragment comprises the amide and side chain deuteriums of the C-terminal n amino acid residues plus the ethanolamine terminal group. The MS/MS spectra of the [M+Na]+ ions of WALP16 and WALP16(+10) were recorded when 12 of 22 and 20 of 32 hydrogens were exchanged, respectively. The deuterium content of the fragments as based on a simplified model (see text for explanation) is indicated by the black circles connected by a line (-●-). Data points for the fragments for which no bar is shown could not be obtained because of poor signal-to-noise ratio.
Also, WALP16 follows such a hypothetical pattern rather well. However, the observed patterns suggest in both cases the following deviations from the model. First, there appears to be a small gradient in exchange rate toward the ends of the transmembrane segments. This can be attributed to local dynamic fraying as is generally observed near the termini of α-helices (27). Second, for both peptides the N-terminal fragments contain more deuterium than the C-terminal fragments. A possible explanation is that the C termini are more protected from exchange, because in a right-handed α-helical conformation, amide hydrogens at the C terminus can form hydrogen bonds with backbone carbonyl oxygens in the preceding turn, whereas the N termini cannot form such hydrogen bonds.
It is important to note here that we cannot exclude the possibility that the measured deuterium content as depicted in Fig. 7 A and B has been affected by gas-phase deuterium scrambling. It is known that proton mobility within smaller protonated gas-phase peptide ions can often be sufficiently rapid to scramble deuterium labeling (28). However, recent findings of Anderegg et al. (17) and Deng et al. (18) have shown that under certain conditions, hydrogen exchange at individual peptide amide linkages can be determined by collision-induced dissociation MS. The results obtained in the present study are fully consistent with the expected exchange properties of the peptides based on the model in Fig. 1. Therefore, it is suggested that MS/MS as used under the conditions described here can give a reliable indication about the deuterium content in different regions of the peptide, and that in our case the extent of scrambling is limited. We believe that the fact that the investigated peptides are sodiated may hamper proton randomization.
Conclusions.
In this paper, we have described a direct approach to measure hydrogen/deuterium kinetics of model transmembrane peptides reconstituted in fully hydrated dispersed phospholipid bilayers by using nano-ESI-MS and MS/MS. The uniqueness of this proteoliposome nano-ESI-MS technique is that the peptide-membrane system is directly introduced into the mass spectrometer without use of any membrane-mimicking detergents, “isotropic” media, or exchange trapping techniques. The results of the experiments described here show that this method is very convenient to perform studies on the exact positioning of membrane peptides (such as the WALP peptides) in a lipid bilayer.
The method has many general advantages. It is both fast and sensitive, and it allows a large flexibility in choice of environmental conditions, such as pH and ionic strength. It also allows variation in lipid as well as peptide composition, as we observed in pilot experiments with biological peptides in negatively charged phospholipid bilayers. Another advantage is the possibility of simultaneously incorporating different peptides into the membrane and subsequently monitoring them independently of each other. Therefore, there is also no stringent requirement for high purity of the peptides. We propose that this relatively simple method could be equally well applied to study the positioning of larger transmembrane proteins in model or even biological membranes.
We note that other potential applications of our direct proteoliposome nano-ESI-MS technique lie in the field of proteomics of membrane proteins (29), which are difficult to characterize because of their inherent insolubility in aqueous solutions as well as the fact that membrane protein-solubilizing detergents cannot easily be used in nano-ESI-MS. Moreover, this technique could be exploited in the investigation of lipid membrane structures by direct introduction of, for instance, plasma membrane vesicles or detergent-resistant membrane vesicles (30).
Acknowledgments
We thank Kees Versluis for technical assistance, Dr. Denise V. Greathouse for synthesis of the peptides, and Dr. Ben de Kruijff for fruitful discussions and helpful suggestions on the manuscript. This project was supported by the Netherlands Organization for Scientific Research and by National Institutes of Health Grant GM 34968 (to R.E.K.).
Abbreviations
- DMPC
1,2-dimyristoyl-sn-glycero-3-phosphocholine
- MS
mass spectrometry
- nano-ESI-MS
nanoelectrospray ionization MS
- TFE
2,2,2-trifluoroethanol
- H/D
hydrogen/deuterium
- LUVETs
large unilamellar vesicles prepared by extrusion
- MS/MS
collision-induced dissociation tandem MS
- m/z
mass-to-charge ratio
- WALP16
acetylGW2-(LA)5W2Aethanolamine
- WALP16(+10)
acetyl(GA)3W2(LA)5W2(AG)3ethanolamine
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.050444797.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.050444797
References
- 1.Hvidt A, Nielsen S O. Adv Protein Sci. 1966;21:287–386. doi: 10.1016/s0065-3233(08)60129-1. [DOI] [PubMed] [Google Scholar]
- 2.Englander S W, Sosnick T R, Englander J J, Mayne L. Curr Opin Struct Biol. 1996;6:18–23. doi: 10.1016/s0959-440x(96)80090-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clarke J, Itzhaki L S. Curr Opin Struct Biol. 1998;8:112–118. doi: 10.1016/s0959-440x(98)80018-3. [DOI] [PubMed] [Google Scholar]
- 4.Goormaghtigh E, Raussens V, Ruysschaert J M. Biochim Biophys Acta Rev Biomemb. 1999;1422:105–185. doi: 10.1016/s0304-4157(99)00004-0. [DOI] [PubMed] [Google Scholar]
- 5.Sturgis J, Robert B, Goormaghtigh E. Biophys J. 1998;74:988–994. doi: 10.1016/S0006-3495(98)74022-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang Y P, Lewis R N, Henry G D, Sykes B D, Hodges R S, McElhaney R N. Biochemistry. 1995;34:2348–2361. doi: 10.1021/bi00007a031. [DOI] [PubMed] [Google Scholar]
- 7.Henry G D, Weiner J H, Sykes B D. Biochemistry. 1987;26:3626–3634. doi: 10.1021/bi00386a056. [DOI] [PubMed] [Google Scholar]
- 8.Chupin V, Killian J A, Breg J, de Jongh H H, Boelens R, Kaptein R, de Kruijff B. Biochemistry. 1995;34:11617–11624. doi: 10.1021/bi00036a038. [DOI] [PubMed] [Google Scholar]
- 9.Pervushin K V, Orekhov V, Popov A I, Musina L, Arseniev A S. Eur J Biochem. 1994;219:571–583. doi: 10.1111/j.1432-1033.1994.tb19973.x. [DOI] [PubMed] [Google Scholar]
- 10.Dempsey C E, Butler G S. Biochemistry. 1992;31:11973–11977. doi: 10.1021/bi00163a003. [DOI] [PubMed] [Google Scholar]
- 11.Cotten M, Fu R, Cross T A. Biophys J. 1999;76:1179–1189. doi: 10.1016/S0006-3495(99)77282-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Katta V, Chait B T. Rapid Commun Mass Spectrom. 1991;5:214–217. doi: 10.1002/rcm.1290050415. [DOI] [PubMed] [Google Scholar]
- 13.Bai Y, Sosnick T R, Mayne L, Englander S W. Science. 1995;269:192–197. doi: 10.1126/science.7618079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benjamin D R, Robinson C V, Hendrick J P, Hartl F U, Dobson C M. Proc Natl Acad Sci USA. 1998;95:7391–7395. doi: 10.1073/pnas.95.13.7391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miranker A, Robinson C V, Radford S E, Aplin R T, Dobson C M. Science. 1993;262:896–900. doi: 10.1126/science.8235611. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Z, Smith D L. Protein Sci. 1996;5:1282–1289. doi: 10.1002/pro.5560050707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anderegg R J, Wagner D S, Stevenson C L, Borchardt R T. J Am Soc Mass Spectrom. 1994;5:425–433. doi: 10.1016/1044-0305(94)85058-5. [DOI] [PubMed] [Google Scholar]
- 18.Deng Y Z, Pan H, Smith D L. J Am Chem Soc. 1999;121:1966–1967. [Google Scholar]
- 19.Wimley W C, White S H. Nat Struct Biol. 1996;3:842–848. doi: 10.1038/nsb1096-842. [DOI] [PubMed] [Google Scholar]
- 20.Killian J A, Salemink I, de Planque M R, Lindblom G, Koeppe R E, II, Greathouse D V. Biochemistry. 1996;35:1037–1045. doi: 10.1021/bi9519258. [DOI] [PubMed] [Google Scholar]
- 21.de Planque M R R, Kruijtzer J A W, Liskamp R M J, Marsh D, Greathouse D V, Koeppe R E, II, de Kruijff B, Killian J A. J Biol Chem. 1999;274:20839–20846. doi: 10.1074/jbc.274.30.20839. [DOI] [PubMed] [Google Scholar]
- 22.de Planque M R R, Greathouse D V, Koeppe R E, II, Schafer H, Marsh D, Killian J A. Biochemistry. 1998;37:9333–9345. doi: 10.1021/bi980233r. [DOI] [PubMed] [Google Scholar]
- 23.Lewis B A, Engelman D M. J Mol Biol. 1983;166:211–217. doi: 10.1016/s0022-2836(83)80007-2. [DOI] [PubMed] [Google Scholar]
- 24.Blume A. Biochemistry. 1983;22:5436–5442. [Google Scholar]
- 25.Liu Y Q, Smith D L. J Am Soc Mass Spectrom. 1994;5:19–28. doi: 10.1016/1044-0305(94)85080-1. [DOI] [PubMed] [Google Scholar]
- 26.Roepstorff P, Fohlman J. Biomed Mass Spectrom. 1984;11:601. doi: 10.1002/bms.1200111109. [DOI] [PubMed] [Google Scholar]
- 27.Wand A J, Roder H, Englander S W. Biochemistry. 1986;25:1107–1114. doi: 10.1021/bi00353a025. [DOI] [PubMed] [Google Scholar]
- 28.Johnson R S. J Am Soc Mass Spectrom. 1996;7:515–521. doi: 10.1016/1044-0305(96)00009-8. [DOI] [PubMed] [Google Scholar]
- 29.Whitelegge J P, le Coutre J, Lee J C, Engel C K, Prive G G, Faull K F, Kaback H R. Proc Natl Acad Sci USA. 1999;96:10695–10698. doi: 10.1073/pnas.96.19.10695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fridriksson E K, Shipkova P A, Sheets E D, Holowka D, Baird B, McLafferty F W. Biochemistry. 1999;38:8056–8063. doi: 10.1021/bi9828324. [DOI] [PubMed] [Google Scholar]