

Abstract
The distance that separates αβ protomers of the Na+,K+-ATPase in microsomes and in purified membranes prepared from duck nasal salt glands was estimated by measuring fluorescence resonance energy transfer between anthroylouabain bound to a population of αβ protomers and either N-[7-nitrobenz-2-oxa-1,3-diazol-4-yl]-6-aminohexyl ouabain or 5-(and-6)-carboxyfluorescein-6-aminohexyl ouabain bound to the rest. Energy transfer between probes bound in the microsomal preparation was less than in the purified membranes. The efficiency of energy transfer between anthroylouabain and N-[7-nitrobenz-2-oxa-1,3-diazol-4-yl]-6-aminohexyl ouabain was 29.2% in the microsomes compared with 62.6% in the purified preparation. Similar results were obtained with 5-(and-6)-carboxyfluorescein-6-aminohexyl ouabain as acceptor. We calculate that either the protomer bound probes were on the average 13 Å farther apart in the microsomes than in the purified membranes, or that 53% of the protomers are monomeric in the microsome preparation. Microsomes prepared in the presence of phalloidin (a toxin that binds to F actin and stabilizes the actin-based cytoskeleton) showed less quench than those prepared in its absence. The data support the hypothesis that protomers are kept apart by their association with the cytoskeleton. The turnover rate while hydrolyzing ATP is the same in the microsomal and purified preparations; higher oligomer formation has no significant effect on the enzyme reaction mechanism.
Na+,K+-ATPase (adenosine triphosphatase, 3.6.1.3) is the biochemical manifestation of the Na+,K+ pump, which maintains gradients of Na+ and K+ across cell membranes. Its basic structural unit is an αβ heterodimer consisting of an α chain that contains the functionally important ion binding sites, the nucleotide binding site, the phosphorylation site, a ouabain binding site, and a β chain of uncertain function. Although there is abundant evidence that monomeric αβ protomers derived from Na+,K+-ATPase solubilized by detergents are able to carry out all the functions of the enzyme (1–5), models of the reaction mechanism that require protein interaction within a dimer remain popular (5). Much of this popularity is accounted for by evidence derived from chemical and physical methods applied to membrane particles enriched in Na+,K+ ATPase. These studies for the most part are interpreted as indicating that pumps are close enough in these preparations to form dimers.
Electron micrographs of purified membrane preparations that reveal αβ protomers arranged in closely packed clumps and strands with large empty areas of membrane between them have been published frequently (6), and the arrangement is illustrated in Fig. 1. There is no apparent order in the clumps or strands. Pump density in a purified preparation was reported as 12,000 μm−2, and the minimum distance between particle centers was found to be close to 5 nm (7), about the diameter of an αβ protomer. Measurements such as cross-linking, radiation inactivation, and fluorescence resonance energy transfer (FRET) confirm that αβ protomers are close together in such preparations, but provide no information about whether they are structural dimers or αβ protomers in casual contact in a crowded environment.
Figure 1.

Electron micrograph of purified Na+,K+-ATPase preparation. A Na+,K+-ATPase preparation suspended in Tris, pH 7.5 buffer was adhered to a formvar-coated copper grid, negatively stained with 1% uranyl acetate and viewed under an 80 KV beam at ×200,000 magnification. Na+,K+-ATPase molecules in this negatively stained preparation appear as white dots. (Scale bar = 0.1 μm.)
Microsomes from nasal glands of salt-adapted ducks have a pump density of 3,400 μm−2 (8) so that, if they were randomly distributed in the membrane, they would be separated by an average distance of about 19 nm. However, if they are free to diffuse in the cell membrane, rough calculation from the estimate (5 × 105 M−1–1.1 × 107 M−1) of the KA for self-association of α chains in detergent solutions (5) leads to the conclusion that αβ protomers must be aggregated in microsomes just as they are in purified membrane preparations. But, αβ protomers cannot diffuse freely; diffusion is limited by their association with the actin-based cytoskeleton (9). In endothelial cells, α chains bind to ankyrin by means of a high-affinity ankyrin binding site, and ankyrin in turn binds to the actin-based cytoskeleton (10). The fraction of α chains that self-associate should be a function of the concentration of α chains, the concentration of ankyrin, the KA for self-association of α chains, and the KA for the association of α chains with ankyrin:
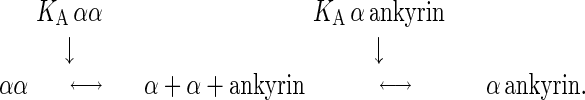 |
Because KA for α self-association, given above (5), is lower than KA for association of α with ankyrin (108 M−1; ref. 11), the fraction of α chains that self-associate will be determined largely by the relative amounts of α chains and ankyrin molecules.
The rationale for our experiments can be developed with the help of Fig. 2. In endothelial cells most αβ protomers are attached to the cytoskeleton, and only unattached protomers are free to self-associate. In purified membrane preparations, the cytoskeleton is completely removed and all αβ protomers are free to diffuse so that all self-associate. In microsomes, the cytoskeleton has been disrupted by homogenization of the gland, so that only a fraction of the αβ protomers are restricted from diffusing, and therefore the fraction of nonaggregated protomers should be intermediate between that in the purified membranes and that in the intact microsomes. We also performed some homogenizations in the presence of the mushroom toxin phalloidin, which binds to F actin and stabilizes actin filaments (12), in the hope that the cytoskeleton of the microsomes so formed would be somewhat protected against disaggregation.
Figure 2.

Model of the interaction of Na+ pumps and cytoskeleton in different membrane preparations.
We measured the distance between αβ protomers by measuring FRET between a subset of protomers labeled with anthroylouabain (AO) (13) and the remainder of the protomers labeled with N-[7-nitrobenz-2-oxa-1,3-diazol-4-yl]-6-aminohexyl ouabain (NBDO) or 5-(and-6)-carboxyfluorescein-6-aminohexyl ouabain (FO). We found that the signal from AO was quenched less in the microsomes than in the purified membranes, and less in the microsomes prepared with phalloidin than in those prepared without it. In all groups, the turnover rate of the enzyme while hydrolyzing ATP was identical.
Experimental Procedures
Materials.
Reagents.
AO was purchased from Sigma. Phalloidin was purchased from Molecular Probes. All other reagents were the highest grade available.
Synthesis of ouabain derivatives.
FO and NBDO were synthesized by the Chemical Synthesis Center at the State University of New York at Stony Brook. All reagents were from Sigma or Molecular Probes and were used without further purification. Pyridine was dried by refluxing over and then distilling from calcium hydride. Fluorescein and nitrobenz-2-oxa-1,3-diazol-4-yl (NBD) groups were covalently linked to ouabain by using a common intermediate that had a short amino terminated six-carbon chain attached to the rhamnose sugar hydroxyl groups.
N-carbobenzyloxy (CBZ) 6-aminohexanoic acid was converted to the acid chloride by using a 2-fold excess of oxalyl chloride in methylene chloride at room temperature (14). Ouabain was reacted with the acid chloride (1.2 mol equivalent) in pyridine at room temperature in the dark for 5 days. Reaction occurred primarily at the 2- and 3-hydroxyl groups of the rhamnose to form the ester. The crude material was purified by column chromatography on silica gel. No attempt was made to separate the two isomeric esters. The CBZ-protecting group was removed by catalytic hydrogenolysis at room temperature by using 5% Pd/C in ethanol and a hydrogen pressure of 45 psi. Under these conditions the unsaturated lactone is unaffected (15). The 6-aminohexyl ouabain (1 mol equivalent) was reacted with 1 mole equivalent of either 5(6)-carboxyfluorescein or NBD chloride in dimethylformamide to yield the fluorescent derivatives that were purified by preparative TLC on silica gel. Structures were confirmed by 1H- and 13C-NMR.
Na+,K+-ATPase preparations.
Microsomes and purified Na+,K+-ATPase were prepared from nasal salt glands of salt-adapted ducks as described (16). Phalloidin-treated microsomes were prepared by conducting all steps of microsome preparation and isolation in the presence of 1 μm phalloidin.
Methods.
P-nitrophenyl phophastase (PNPPase) activity.
PNPPase activity was measured at 37°C in assay media containing 5 mM EGTA, 40 mM KCl, 5 mM MgCl2, 10 mM p-nitrophenyl phosphate, and 10 mM Tris⋅HCl, pH 7.5. The reaction was initiated by adding Na+,K+-ATPase (usually at 1 μg/ml) to a temperature-equilibrated cuvette, and the change in absorbance at 410 nm was recorded with time by using a Hewlett–Packard 8452A diode array spectrophotometer.
ATPase activity, phosphorylation, and turnover rate.
Na+,K+-ATPase activity, phosphorylation from 32Pi, and calculation of ATP hydrolysis turnover rate were determined as described (16).
FRET.
Free AO in solution has an excitation maximum of about 363 nm. AO when bound to the Na+,K+-ATPase can be excited at 290 nm by energy transfer from tryptophan that produces a substantially higher emission compared with excitation at 363 nm (13). By exciting at 290 nm, background emission from nonspecifically associated or free AO is substantially reduced, and the resultant emission at 470 nm increases proportionally above background with increasing occupancy of ouabain sites by AO. Briefly, the energy transfer was measured as follows. AO (fluorescent donor) was bound to Na+,K+-ATPase at low levels such that 20–35% of the ouabain binding sites were occupied. Subsequently, the remaining ouabain binding sites were occupied with saturating concentrations of NBDO (or FO), which served as the fluorescent acceptor. The quench of the AO emission after the binding of acceptor was used to calculate the energy transfer between donor and acceptor fluorophores.
The above scheme was executed by measuring the fluorescence of a series of six incubation conditions conducted in parallel. The experimental design is summarized in Table 1 and given in detail below. The final conditions were obtained after three stages of incubation. The common medium for all incubations was 5 mM MgCl2, 10 mM Tris-phosphate, and 10 mM Tris⋅HCl, pH 7.5. During the first stage, ATPase (1 mg/ml purified ATPase or 3.5 mg/ml microsomes) was incubated under three conditions: condition A = 70 μM ouabain (saturating); condition B = 70 μM AO (saturating); condition C produced the subsaturating AO occupancy of ouabain binding sites (20–35%). Because of the relatively high ATPase concentration (>6 μM), the subsaturating AO concentration had to be determined empirically and varied between ATPase preparations, but generally ranged from 0.25 to 0.75 μM. The first stage was incubated 30 min at room temperature. During the second stage, the incubation conditions A, B, and C were used to establish the six final conditions: condition 1 = condition A + an equal volume of 70 μM AO; condition 2 = condition A + an equal volume of AO at the subsaturating concentration used in condition C; condition 3 = condition B diluted in ice-cold common media to yield the final dilution described below and transferred to an ice bath; condition 4 = condition C + an equal volume of 70 μM ouabain; condition 5 = condition C + an equal volume of 70 μM NBDO (or FO); condition 6 = condition C + an equal volume of 70 μM ouabain. Conditions 1, 2, 4, and 5 were incubated 20 min at room temperature. Condition 6 was incubated 10 min at room temperature and then received the same moles of NBDO (or FO) as condition 5 followed by 10 min at room temperature. Control studies demonstrated that lengthier incubations for condition 6 were unnecessary. During the third stage, conditions 1, 2, 4, 5, and 6 were diluted in ice-cold common media and were transferred to an ice bath. This final step resulted in a 70-fold dilution of all ATPase, ouabain, and ouabain derivatives such that the concentration of all free fluorophores was ≤ 1 μM and ATPase concentration was ≤ 0.1 μM. Studies involving phalloidin-treated microsomes maintained a phalloidin concentration of 1 μM at all stages of the procedure. The fluorescent emissions of the resultant incubates were recorded by using an Aminco SPF-500 Spectrofluorometer set at an excitation wavelength of 290 nm, bandpass 10 nm and an emission wavelength of 470 nm, bandpass 10 nm. The cuvette holder was cooled to 2°C, and the sample compartment was purged with dry N2. During each experimental set-up the six final conditions were formed in quadruplicate and the mean and SD were determined. These experiments required extreme care because, generally speaking, experiments in which the SD was > 7% of the mean of the quadruplicates yielded unacceptable results.
Table 1.
Experimental design
| Condition | Sequential addition of reactants |
|---|---|
| 1 | ATPase + Sat [O] + Sat [AO] |
| 2 | ATPase + Sat [O] + SubSat [AO] |
| 3 | ATPase + Sat [AO] |
| 4 | ATPase + SubSat [AO] + Sat [O] |
| 5 | ATPase + SubSat [AO] + Sat [NBDO] |
| 6 | ATPase + SubSat [AO] + Sat [O] + Sat [NBDO] |
Reactants were added sequentially and incubated at room temperature after each addition. ATPase was incubated with the first reactants for 30 min. The next incubations with ouabain and derivatives were for 20 min. After the final incubation the incubates were diluted in ice-cold common buffer and kept on ice until read in a spectrofluorometer. Sat = saturating; SubSat = sub-saturating. Experiments involving FO substituted FO for NBDO. See Methods for details.
The levels of AO binding and fluorescence from bound AO were determined from the mean fluorescent emissions of the quadruplicates as follows:
Total AO bound (FT) = condition 3 − condition 1;
Subsaturating AO bound = fluorescence of donor (FD) = condition 4 − condition 2;
Fluorescence of donor after addition of acceptor (FDA′) = condition 5 − condition 2;
Fluorescence decrease because of nonspecific quench (FNSQ) = condition 4 − condition 6.
The results of the above determinations then were used to calculate the FRET as follows:
Fraction of ouabain sites occupied by AO (FB) = FD/FT;
Apparent fluorescence of donor after energy transfer to acceptor (FDA") = FDA′ + FNSQ;
Apparent relative fluorescence ratio = FDA"/FD.
Control studies indicated that the value of FB agreed well with the fractional level of inhibition of PNPPase activity induced by the subsaturating AO.
We used a dimer model as the minimal structural unit that would permit energy transfer between α subunits. Accordingly, at any level of fractional occupancy of ouabain sites by AO, some dimers would have both ouabain binding sites occupied by AO and therefore no energy transfer would occur in those dimers after addition of acceptor. The observed reduction in fluorescence after addition of acceptor results from energy transfer within those dimers, which initially had one ouabain site occupied with AO, and had the other occupied with acceptor during stage 2 of the above incubations. To complete the FRET calculation, we determined the quench resulting in the dimers that bound one AO and one acceptor (NBDO or FO). If binding of AO is equally probable for all available ouabain binding sites, the fraction of assumed α-α dimers that have only one ouabain binding site occupied by AO can be calculated by use of the binomial theorem. In a population of N dimers, there will be FB⋅2N molecules of AO bound at a fractional occupancy of FB. The number of AO molecules bound to both sites of individual dimers (i.e., unquenchable) will be (FB)2⋅2N. The fraction of bound AO that is unquenchable is therefore equal to (FB)2⋅2N/FB⋅2N = FB. The fraction of bound AO that is quenchable is (1−FB). The measured donor fluorescence before quench (FD) can be defined as:
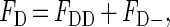 |
where FDD is the fluorescence from the unquenchable dimers and FD− is the fluorescence from the quenchable dimers. We determined above that FDD = FB⋅FD and FD− = (1−FB)⋅FD. The fluorescence measured after adding the acceptor (FDA′′) can be defined as:
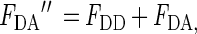 |
where FDA is the fluorescence from the quenched dimers with bound donor and acceptor. A simple calculation is then sufficient to determine what value of FDA from the quenchable units would give rise to the observed apparent FDA′′. Rearrangement and substitution of the equations involved gives rise to the following expression to determine the fluorescence ratio of quenchable bound AO (FDA/FD−):
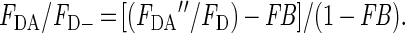 |
The energy transfer efficiency (E), which is the quench caused by FRET, can then be calculated:
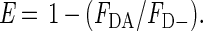 |
The distance between probes can be calculated by using the Förster relationship (17)
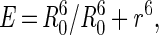 |
where Ro is the Förster distance (the distance for 50% transfer efficiency) for a particular donor-acceptor pair and r is the distance between probes.
The level of nonspecific quench (FNSQ) varied between preparations. Nonspecific quench using NBDO as acceptor was minimal with purified Na+,K+-ATPase resulting in FDA′/FDA′′ ≥ 0.9. Nonspecific quench was greater and more variable with microsomal preparations where FDA′/FDA′′ ≈ 0.81 ± 0.09. When FO was the acceptor, nonspecific quench was somewhat higher and more variable with the microsomal preparations, FDA′/FDA′′ ≈ 0.75 ± 0.17. Correction for nonspecific quench did not qualitatively change the results of the study, but it did improve the precision of the data.
Results
The high pump density of the microsomes prepared from salt-adapted ducks allowed us to obtain a sufficiently strong signal to permit good data reproduction. Attempts to use microsomes prepared from nonsalt-adapted ducks or membranes from duck red blood cells resulted in an unsatisfactory signal/noise ratio, which we assume reflects the lower pump densities in those preparations.
Each of the probes used in this study had a different effective affinity for the ouabain binding site. Fig. 3 shows K-dependent PNPPase activity of purified Na+,K+-ATPase as a function of the concentration of the ouabain derivatives. The order of affinity for the probes was ouabain>AO>NBDO>FO. During the course of this study we determined that when free probe concentrations exceeded 1 μM, fluorescence self-absorbance phenomena became significant, which was particularly true with studies involving FO. The experimental protocol was designed to result in a final probe concentration of ≤ 1 μM to avoid these effects. However, as shown in Fig. 3, 1 μM is below the saturating concentration for FO, which was not a problem because initial binding of FO was at 70 μM and the 1 μM concentration was reached only after lowering the incubation temperature to ≈ 2°C in an ice bath. At this temperature, dissociation of bound ouabain and derivatives is very slow and should have been minimal during the course of the fluorescence readings. However, as noted above, nonspecific quench effects were also greater and more variable when FO was the acceptor. Taking these limitations into account, we present in this paper only the data obtained by using NBDO as the acceptor; however, the same qualitative conclusions could be drawn from the FO data.
Figure 3.

Effects of varied concentrations of ouabain and ouabain derivatives on PNPPase activity. Purified Na+,K+-ATPase was incubated at 20 μg/ml with varied concentrations of ouabain, AO, FO, and NBDO for 30 min at room temperature, and the PNPPase activity was measured as described in Methods. The data were fitted to a sigmoidal dose-response curve by using prizm 2.01 (GraphPad, San Diego). The fitted parameters and symbols are: ○ = ouabain, EC50 = 6.4 nM; ● = AO, EC50 = 64 nM; □ = NBDO, EC50 = 210 nM; ■ = FO, EC50 = 860 nM.
The results of our measurements are summarized in Table 2. The first data column in the table shows the energy transfer efficiency measured in 10 experiments with purified Na+,K+-ATPase and 19 experiments with salt gland microsomes. Energy transfer in the purified preparation is much greater than that in microsomes. We also have broken down the microsome experiments into those prepared in the absence and presence of phalloidin. Microsomes that were prepared in the presence of phalloidin showed a significantly lower efficiency of energy transfer. Microsomes that were prepared in the absence of phalloidin and subsequently were incubated with phalloidin during the FRET experiments had the same energy transfer efficiency as microsomes without phalloidin (not shown).
Table 2.
Energy transfer measurements and calculated values
| Samples* | Quench (E) | P values† | Separation, Å | % Monomers‡ | Turnover, min−1§ |
|---|---|---|---|---|---|
| Purified ATPase (10) | 0.6260 ± 0.0315 | 49.6 ± 1.1 | 0 | 9,690 ± 340 | |
| Microsomes (19) | 0.2822 ± 0.0287 | <0.0001 | 63.1 ± 1.2 | 53.3 ± 4.6 | 9,473 ± 397 |
| Microsomes, no phalloidin (10) | 0.3378 ± 0.0338 | 60.4 ± 1.1 | 47.6 ± 5.4 | 9,476 ± 420 | |
| Microsomes + phalloidin (9) | 0.2202 ± 0.0397 | 0.0183 | 66.7 ± 2.1 | 61.6 ± 6.3 | 9,470 ± 363 |
*Numbers in parentheses are number of FRET experiments. Values given in the table are the means ± standard error of the mean.
†The P values were determined by using an unpaired, one-tailed t-test.
‡ % Monomers were determined by assuming the purified ATPase was 100% dimer and dividing the energy transfer efficiency (E) of the microsomes by E (0.626) of the purified ATPase.
§Turnover values for purified ATPase are the mean ± SD from ref. 16. Turnover values for microsomes are the mean ± SD from three experiments ± phalloidin.
Discussion
The major finding of this study is that fluorescence of AO bound to a fraction of αβ protomers is quenched more by NBDO or FO bound to the rest of the αβ protomers in purified membrane preparations than it is in the microsomes from which they are derived. The results can be interpreted in either of two ways: either the average distance between αβ protomers is greater in the microsomes than in the purified membranes, or the microsomes contain a population of αβ protomers packed as closely as they are in purified preparations and another population of protomers that are so far apart that fluorescence resonance energy transfer is not possible, i.e., monomers.
We calculated the average separation between ouabain derivatives bound to αβ protomers by means of the Förster relation, which assumes that differences in energy transfer reflect differences in the separation of the probes. Because the measurements were made with the same probes in the purified membranes and in the microsomes, and because the Na+, K+ pumps were the same in the two preparations, we reckoned that orientation differences would not occur. We were interested only in the relative distances between protomers, so we used a value for Ro (54 Å) in the Förster relationship for the pair AO and NBDO and 45 Å for the pair AO and fluorescein mercuric acetate given by Fortes and coworkers (18, 19) rather than calculate them ourselves. These results are collected in the fourth column of Table 2. Using the AO-NBDO pair we obtained a value of 50 Å for the average distance between protomers in purified membranes, about the same as others have reported for this measurement (20). The average separation in microsomes is 63 Å, about 13 Å greater. Because the radius of an αβ protomer is 25 Å, and because the average separation in purified membranes calculated from the quench data is 50 Å, most protomers in purified membranes must exist as diprotomers. The average separation in microsomes is 63 Å so that the separation between protomers could in all cases be 63 Å or could vary between 50 Å and a much larger value as long as the average separation is 63 Å. Dimer structure requires a fixed protomer separation, because the distance between protomers in the two preparations varies; diprotomers in the purified preparation must result from casual association in a crowded environment rather then from stable, fixed structural contacts.
Perhaps a more realistic view is that αβ protomers in the microsomes are a mixture of a population packed as closely as the diprotomers in purified membranes and a population of monomers. We assumed that all αβ protomers in the purified preparations exist as diprotomers (which is supported by the separation data) and used the average quench of the purified membranes (0.626) as the value for the quench of a diprotomer pair. We then divided the quench measured in the microsomes by this value to determine the percentage of protomers that must be diprotomers in microsomes to produce the observed quench. The remainder of the protomers in the microsomes must be monomers. The monomer compositions are given in the fifth column of Table 2. We calculated that monomers must make up 53% of the total number of protomers in microsomes. Similar results were obtained by using the AO-FO pair.
In either case, protomers in the microsomes must be mostly monomers. Although the protomers in purified membranes are close enough to be dimers, there is no reason to conclude from these studies that they are, in fact, structural or functional dimers.
In microsomes prepared with phalloidin, αβ protomers are either farther apart than in the untreated microsomes, or the fraction of αβ protomers that are monomers is greater in the treated microsomes. This is the prediction from our hypothesis that protomers tend to be monomers in microsomes because they are prevented from aggregating by their association with the actin-based cytoskeleton. But the observation that protomers in microsomes are largely monomeric does not depend on whether or not the hypothesis is credible. Calculations using KA = 5 × 105 M−1 for self-association of α chains obtained with detergent solubilized preparations lead to the conclusion that 97% of the protomers in purified membranes must be present as diprotomers (5). Using the same value for KA, we calculated that if αβ protomers in microsomes are free to diffuse, then 94% should form diprotomers. The apparent KA for self-association of α chains in a membrane is likely to be higher than the KA found in detergent solutions because, within the membrane, protomers are subject to molecular crowding, reduction of diffusional motion to two dimensions, limitation of rotation to a single axis, etc. If attachment of αβ protomers to the cytoskeleton is not responsible for keeping them apart, some other mechanism must be found.
The last column in Table 2 gives the turnover rate for the hydrolysis of ATP in the various preparations. The turnover rate is identical in the purified membranes and the microsomal preparations. When αβ protomers are close enough to form diprotomers, there is no significant effect on the turnover rate. The reaction mechanism does not require protomer interaction, nor is it altered when protomers are closely packed.
These results are not unprecedented. Studies with detergent solubilized purified membranes in which solubilized enzyme was quantitatively cross-linked showed that both αβ protomers and diprotomers are able to hydrolyze ATP (1). Analytical centrifugation of detergent-solubilized enzyme found that ATPase activity migrated with the mobility of an αβ protomer (2). The specific activity of the αβ protomers was the same as the specific activity of protomers in the membrane preparation from which they were derived, in which protomers presumably are closely packed. In a subsequent study (3), it was found that solubilized monomers are capable of occluding Rb and Na. In an active enzyme chromatography study (5), ATPase activity migrated with both αβ monomers and (αβ)2 diprotomers. The specific activity was the same for both the monomers and dimers. Finally, an active enzyme centrifugation study (4) showed that ATPase activity migrated with αβ protomers. The studies with solubilized enzyme are entirely consistent with ours using natural membranes in which the turnover rate is the same in purified membranes in which αβ protomers are closely crowded and in microsomes in which closely packed protomers and monomeric protomers coexist.
Most studies of the oligomeric state of αβ protomers in natural membranes have been performed with purified membrane preparations, and, as expected, have shown that αβ protomers in such preparations are closely crowded. Human red blood cells contain about 500 αβ protomers per cell, or about 3 μm−2, and about 1.2 × 106 copies of the anion transporter, band 3, or about 7,800 μm−2. Cu and orthophenanthroline cross-link α chains in purified membrane preparations in a conformationally dependent manner (21). We found that, in red cell membranes, Cu and orthophenanthroline cross-link α chains to band 3 but not to α chains in a conformationally dependent fashion (22). Similar results were obtained with another cross-linking reagent, bis-(4-fluoro-3-nitrophenyl)-sulfone, which cross-links by a different chemical mechanism. We concluded that, in purified membrane preparations, α chains are close to each other either because they are organized as dimers, or because they are closely packed. In red cell membranes, however, α chains are not close to each other (i.e., are not organized as dimers), but are closer to the predominant band 3 molecules. We mentioned a possibility that we thought remote: that α chains are, in fact, organized as dimers both in purified membrane preparations and in red cell membranes, but that cross-links form only between the portion of an α chain not involved in dimer formation and its nearest neighbor, and not between α chains in dimers. The experiments with FRET reported here support the interpretation that αβ protomers are not organized as dimers in red cell membranes.
Monomeric αβ protomers, therefore, are capable of carrying out the entire reaction mechanism of the Na+,K+ pump including cation transport. Because monomeric protomers have the same turnover rate as closely packed αβ protomers, dimerization, if it occurs, makes no notable change in the overall reaction mechanism. However, interaction of αβ protomers in a closely packed environment may in some way modify a property of the enzyme. Many such effects have been described but rarely with any independent evidence that protomer interaction is involved. Functional evidence for oligomerization in the absence of physical evidence is at best uncertain. In one case, however, an effect of protein interaction has been credibly shown. Scatchard plots of the level of ATP or ADP binding to αβ protomers are curvilinear in the presence of K+, but are converted to linear when most of the protomers are inhibited by ouabain (23). This is strong evidence of cooperativity between protomers. Physical evidence supported this conclusion; detergent solubilization of the enzyme converted the nonlinear plot to a linear one (24). The protomer interaction changed the dissociation constant for nucleotides, which is less than 1 μM, by less than 2-fold. It is inconceivable that such a small difference could have a significant effect on the reaction mechanism.
αβ protomers of the Na+,K+ ATPase must exist in natural membranes as a mixture of diprotomers and monomers and the conclusion that subunit interaction has no significant role in the reaction mechanism of the Na+,K+ pump is inescapable. In natural membranes, αβ protomers probably are prevented from aggregating by their connection to the actin cytoskeleton.
Acknowledgments
We are grateful to Cheryl Martin for excellent technical assistance. This work was supported by U.S. Public Health Service Grant DK-19185.
Abbreviations
- AO
anthroylouabain
- FO
5-(and-6)-carboxyfluorescein-6-aminohexyl ouabain
- NBDO
N-[7-nitrobenz-2-oxa-1,3-diazol-4-yl]-6-aminohexyl ouabain
- FRET
fluorescence resonance energy transfer
- PNPPase
p-nitrophenyl phophastase
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.050558397.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.050558397
References
- 1.Craig W S. Biochemistry. 1982;21:5707–5717. doi: 10.1021/bi00265a049. [DOI] [PubMed] [Google Scholar]
- 2.Brotherus J R, Jacobsen L, Jørgensen P L. Biochem Biophys Acta. 1983;731:290–303. doi: 10.1016/0005-2736(83)90021-4. [DOI] [PubMed] [Google Scholar]
- 3.Vilsen B, Andersen J P, Petersen J, Jørgensen P L. J Biol Chem. 1987;262:10511–10517. [PubMed] [Google Scholar]
- 4.Ward D G, Cavieres J D. Proc Natl Acad Sci USA. 1993;90:5332–5336. doi: 10.1073/pnas.90.11.5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hayashi Y, Mimura K, Matsui H, Takagi T. Biochim Biophys Acta. 1989;983:217–229. doi: 10.1016/0005-2736(89)90237-x. [DOI] [PubMed] [Google Scholar]
- 6.Yokoyama T, Kaya S, Abe K, Taniguchi K, Katoh T, Zazawa M, Hayashi Y, Mårdh S. J Biol Chem. 1999;274:31792–31796. doi: 10.1074/jbc.274.45.31792. [DOI] [PubMed] [Google Scholar]
- 7.Deguchi N, Jørgensen P L, Maunsbach A B. J Cell Biol. 1977;75:619–634. doi: 10.1083/jcb.75.3.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Merchant J L, Papermaster D S, Barrnett R J. J Cell Sci. 1985;78:233–246. doi: 10.1242/jcs.78.1.233. [DOI] [PubMed] [Google Scholar]
- 9.Paller M S. J Membr Biol. 1994;142:127–135. doi: 10.1007/BF00233390. [DOI] [PubMed] [Google Scholar]
- 10.Nelson W J, Hammerton R W, McNeil H. In: The Sodium Pump: Structure, Mechanism, and Regulation. Kaplan J H, DeWeer P, editors. New York: Rockefeller Univ. Press; 1990. pp. 77–87. [Google Scholar]
- 11.Davis J Q, Bennett V. J Biol Chem. 1990;265:17252–17256. [PubMed] [Google Scholar]
- 12.Estes J E, Selden L A, Gershman L C. Biochemistry. 1981;20:708–712. doi: 10.1021/bi00507a006. [DOI] [PubMed] [Google Scholar]
- 13.Fortes P A G. Biochemistry. 1977;16:531–540. doi: 10.1021/bi00622a030. [DOI] [PubMed] [Google Scholar]
- 14.Adams R, Ulrich L H. J Am Chem Soc. 1920;42:599–611. [Google Scholar]
- 15.Baran J S. J Org Chem. 1964;29:527–536. [Google Scholar]
- 16.Martin D W, Sachs J R. Biochemistry. 1999;38:7485–7497. doi: 10.1021/bi983019b. [DOI] [PubMed] [Google Scholar]
- 17.Förster T. Ann Phys (Leipaig) 1948;2:55–75. [Google Scholar]
- 18.Chong P L G, Fortes P A G, Jameson D M. J Biol Chem. 1985;260:14484–14490. [PubMed] [Google Scholar]
- 19.Jesartis A J, Fortes P A G. J Biol Chem. 1980;255:459–467. [PubMed] [Google Scholar]
- 20.Amler E, Abbott A, Ball W J. Biophys J. 1992;61:553–568. doi: 10.1016/S0006-3495(92)81859-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Periyasamy S M, Huang W- H, Askari A. J Biol Chem. 1983;258:9878–9885. [PubMed] [Google Scholar]
- 22.Martin D W, Sachs J R. J Biol Chem. 1992;267:23922–23929. [PubMed] [Google Scholar]
- 23.Ottolenghi P, Jensen J. Biochim Biophys Acta. 1983;727:89–100. doi: 10.1016/0005-2736(83)90372-3. [DOI] [PubMed] [Google Scholar]
- 24.Jensen J, Ottolenghi P. Biochim Biophys Acta. 1983;731:282–289. doi: 10.1016/0005-2736(83)90020-2. [DOI] [PubMed] [Google Scholar]