

Abstract
Dihydrolipoamide succinyltransferase (DLST) is a subunit enzyme of the α-ketoglutarate dehydrogenase complex of the Krebs cycle. While studying how the DLST genotype contributes to the pathogenesis of Alzheimer’s disease (AD), we found a novel mRNA that is transcribed starting from intron 7 in the DLST gene. The novel mRNA level in the brain of AD patients was significantly lower than that of controls. The truncated gene product (designated MIRTD) localized to the intermembrane space of mitochondria. To investigate the function of MIRTD, we established human neuroblastoma SH-SY5Y cells expressing a maxizyme, a kind of ribozyme, that specifically digests the MIRTD mRNA. The expression of the maxizyme specifically eliminated the MIRTD protein and the resultant MIRTD-deficient cells exhibited a marked decrease in the amounts of subunits of complexes I and IV of the mitochondrial respiratory chain, resulting in a decline of activity. A pulse-label experiment revealed that the loss of the subunits is a post-translational event. Thus, the DLST gene is bifunctional and MIRTD transcribed from the gene contributes to the biogenesis of the mitochondrial respiratory complexes.
Keywords: Alzheimer’s disease/cytochrome c oxidase/dihydrolipoamide succinyltransferase/mitochondria/oxidative stress
Introduction
α-ketoglutarate dehydrogenase complex is a rate-limiting enzyme of the Krebs cycle in the mitochondrial matrix. The complex is composed of three major protein subunits, one of which is the structural core protein dihydrolipo amide succinyltransferase (DLST). The human DLST gene is located on chromosome 14 at q24.2–q24.3 (Nakano et al., 1993) and consists of 15 exons (Nakano et al., 1994). A genetic association has been reported between polymorphisms of the DLST gene and Alzheimer’s disease (AD) in both Japanese and Caucasian populations (Nakano et al., 1997; Sheu et al., 1999a,b). Interestingly, the polymorphisms are also seen in Swedish patients with familial AD (Sheu et al., 1998). However, the association remains controversial (Kunugi et al., 1998; Matsushita et al., 2001), perhaps because the risk is not great and, additionally, because controls and AD patients were not matched for age and gender. In any case, the polymorphisms do not change amino acid residues of the DLST protein. Thus, the reason why the nucleotide substitutions in the DLST gene contribute to the pathogenesis of AD is not clear. Therefore, it is important to elucidate the role of the DLST gene when investigating the molecular mechanism involved in AD.
AD is a complex neurodegenerative disease caused by multiple genetic and environmental factors. The accumulation of amyloid β protein is considered a main cause of neuronal cell death. Additionally, mitochondrial dysfunction may be associated with the pathogenesis of AD (Bonilla et al., 1999; Schon and Manfredi, 2003). In particular, a deficiency of cytochrome c oxidase of the mitochondrial respiratory chain in the brains of AD patients has been reported from several laboratories (Kish et al., 1992; Parker and Parks, 1995; Cottrell et al., 2001). On the other hand, amyloid β protein inhibits the activity of cytochrome c oxidase and other key enzymes of mitochondria (Caseley et al., 2002). Damage to mitochondria causes a decline in ATP synthesis and an increase in the generation of reactive oxygen species (ROS). ROS damage various molecules, including DNA, protein and lipid, and induce apoptosis. Evidence has recently emerged that oxidative stress is involved in the pathogenesis of various neurodegenerative diseases such as AD and Parkinson’s disease (Schulz et al., 2000; Smith et al., 2000; Ohsawa et al., 2003). Moreover, it has been shown that amyloid β protein generates hydrogen peroxide (Behl et al., 1994; Harris et al., 1995).
In the present study, we found a novel truncated mRNA that is transcribed starting from intron 7 of the DLST gene, in addition to full-length mRNA. The amount of this truncated mRNA is significantly reduced in the brain of patients with AD. In addition, selective digestion of the truncated mRNA lowered the steady-state level of the subunits of complexes I and IV of the mitochondrial respiratory chain. These results strongly suggest that the truncated DLST protein transcribed from the bifunctional DLST gene contributes to the biogenesis of the respiratory chain.
Results
Haplotype analysis of the DLST gene
We have reported that polymorphisms at nucleotide 19116 in intron 13 and nucleotide 19183 at the third codon base in exon 14 of the DLST gene are associated with AD (Figure 1; Nakano et al., 1997). The gene was classified into three haplotypes, designated ac, at and gc depending on the nucleotides 19116 (A or G) and 19183 (C or T), in which the homozygous ac genotype was associated with AD (Nakano et al., 1997). Since these polymorphisms do not change any amino acid residues, we searched for pathogenic mutations that are in linkage disequilibrium with the haplotype.

Fig. 1. Schematic presentation of the structure of the DLST gene. Nucleotide residues are numbered on the basis of the revised sequence starting from the transcription initiation site of DLST mRNA at position 1 (Nakano et al., 1994). Haplotypes are designated by two nucleotide variants at 19116 (A or G) and at 19183 (C or T). Nucleotides specific to haplotype ac are listed. The transcription initiation site of MIRTD mRNA is at 10555.
The nucleotide sequence of the entire DLST gene including ∼1000 bp of the upstream region was directly determined in six volunteers, two each with the homozygous ac, at and gc genotypes, by using PCR products from total DNA isolated from peripheral blood. On comparing the three haplotypes, we found an additional six polymorphisms at 10578, 11043, 11103, 13844, 15455 and 17106 in linkage disequilibrium specific to haplotype ac (19116A and 19183C haplotype) (Figure 1). A11043G lies at the third codon base in exon 8, while the other polymorphisms lie in introns. As a result, we found no polymorphism that leads to a change in amino acid in the haplotype ac.
Truncated transcript from intron 7 of the DLST gene with poly(A)+ RNA
We have reported the existence of a truncated DLST protein in muscle (Matuda et al., 1997). We hypothesized that this kind of unusual protein derived from the DLST gene contributes to cell death, that mRNA transcribed from an intron generates a truncated protein and that a polymorphism in the intron contributes to the expression of the truncated mRNA. Therefore, we tried to find mRNA that is transcribed from an intron of the DLST gene. We extensively performed PCR coupled with reverse transcription (RT) of poly(A)+ RNA from a human neuroblastoma cell line SH-SY5Y using various sets of primers, one of which corresponds to the sequence of an intron and the other to that of an exon. The primer sets covered most regions of the DLST gene. When a pair of primers corresponding to intron 7 and exon 9 was used (Figure 2A), a fragment was clearly amplified (Figure 2B, lane 1). The length was ∼240 bp, which agrees with a fragment length covering intron 7 to exons 8 and 9. The amplified RT–PCR product was sequenced to confirm that the fragment indeed consisted of intron 7, exon 8 and exon 9 of the DLST gene. The absence of the sequence of intron 8 strongly suggests that the RNA is derived from mRNA.

Fig. 2. Existence of a truncated mRNA transcribed from intron 7. (A) Schematic diagram of the primers (a–d) and TaqMan probe (e) used in this study. Arrows represent positions of primers. To detect the truncated mRNA (MIRTD mRNA), PCR was performed by preheating for 10 min at 95°C, followed by 35 cycles of 15 s at 95°C and 1 min at 57°C. Primers a, b, c and d represent the MIRTD forward primer 5′-ctcatttcagacagtgccagtg-3′ (intron 7), MIRTD reverse primer 5′-tggctcagctagtggtggg-3′ (exon 9), DLST forward primer 5′-acctacagcagcggcagt-3′ (exon 8) and DLST reverse primer 5′-ctcctggctcagctagtggt-3′ (exon 9), respectively. (B) Poly(A)+ RNA was isolated from the SH-SY5Y cells. Complementary DNA was synthesized with oligo(dT) primer and then PCR was carried out using primers a and b (lane 1) or primers c and d (lane 2). The poly(A)+ RNA fraction was untreated with reverse transcriptase, but subjected to PCR (lane 3). Lane 4 shows a marker of φX174 DNA digested with HincII. Arrows indicate each amplified fragment. (C) Initiation site of MIRTD mRNA was determined by an oligonucleotide-capping method (Yamabe et al., 1998) with Cap Site cDNA™ of human brain (Nippon Gene, Toyama, Japan) as described in Materials and methods.
Next, its transcription initiation site was determined by an oligonucleotide capping method (Yamabe et al., 1998). This method confirmed the presence of the cap structure at the initiation site, and revealed the initiation site to be A at 10555 (Figure 2C). Thus we concluded the existence of a novel mRNA, with the cap structure, which is transcribed from intron 7 of the DLST gene. We designated the truncated protein derived from the novel mRNA as MIRTD (MItochondrial Respiration Generator of Truncated DLST) in consideration of its function as described later.
Expression of MIRTD mRNA is significantly reduced in the brains of patients with AD
As a first step, we examined whether the truncated mRNA is associated with AD. Relative amounts of MIRTD mRNA in AD and control brains were compared using poly(A)+ RNA prepared from cortex. The amounts of MIRTD mRNA were measured using real-time quantitative RT–PCR and normalized with the amounts of DLST or glyceraldehyde-3-phospate dehydrogenase (GAPDH) mRNA. When mRNA of DLST was not detected, we omitted the samples from analysis. The ratio of MIRTD mRNA to DLST mRNA was not correlated with age (correlation coefficient 0.097) or post-mortem period (correlation coefficient 0.22) of the control brains.
In control brains, the ratios of MIRTD mRNA to DLST and GAPDH mRNAs were (0.0621 ± 0.0505) and (1.62 ± 1.47) × 10–4, respectively. In the AD brains, they were (0.0248 ± 0.0368) and (0.713 ± 1.15) × 10–4, respectively (Figure 3). The differences were apparently significant (p = 0.004 for MIRTD/DLST mRNA and p = 0.018 for MIRTD/GAPDH mRNA by Student’s t-test). Strikingly, MIRTD mRNA was not detected in half of the AD brains (Figure 3). In addition, the ratio of MIRTD mRNA to DLST mRNA from carriers with the ac haplotype tended to be lower than that from non-carriers irrespective of the clinical diagnosis (0.041 ± 0.040 versus 0.055 ± 0.047), despite showing no statistical significance (p = 0.23). Thus, these results suggest that a decrease in MIRTD mRNA is associated with AD.

Fig. 3. Decreased amounts of MIRTD mRNA in brains of patients with AD. Amounts of MIRTD mRNA in the brains of AD patients or controls were measured by real-time quantitative RT–PCR and normalized against those of (A) DLST or (B) GAPDH mRNA as described in Materials and methods. Numbers of samples, mean values with standard deviations (± SD) and p values as judged by Student’s t-test are given in the figure.
Regulation of expression of MIRTD mRNA by haplotypes of the DLST gene
Around the initiation site of MIRTD mRNA, there is a polymorphism (10578T) that is specific to haplotype ac (Figure 1). Therefore, we investigated whether haplotypes of the DLST gene influence the expression of MIRTD mRNA in vitro. For this purpose, we used the rat cell line PC12, because the nucleotide sequence of intron 7 of the rat DLST gene is distinguishable from the human counterpart. We isolated two independent cosmid clones carrying the full-length human DLST genes of each haplotype (the ac and gc haplotypes) and then transfected the human genes into PC12, where the endogenous rat DLST gene is present. The amount of exogenous human MIRTD mRNA was measured by real-time quantitative RT–PCR and normalized to that of human DLST mRNA (Figure 4). Each primer set specific to the human DLST gene was selected to detect human DLST mRNA and human MIRTD mRNA. In fact, only exogenous human DLST and MIRTD mRNAs, and no endogenous mRNAs derived from the host, were detected (Figure 4A and B). The ratios of human MIRTD mRNA to DLST mRNA in the cells carrying 10578T (haplotype ac) were 3.6 ± 1.9% and 4.6 ± 2.3%, compared with 8.7 ± 2.4% and 9.7 ± 3.2% in the cells carrying 10578A (haplotype gc) in each of two independent clones (Figure 4C). The difference was statistically significant, as shown in the figure legend. Therefore, expression of human MIRTD mRNA depends on the haplotype of the DLST gene.
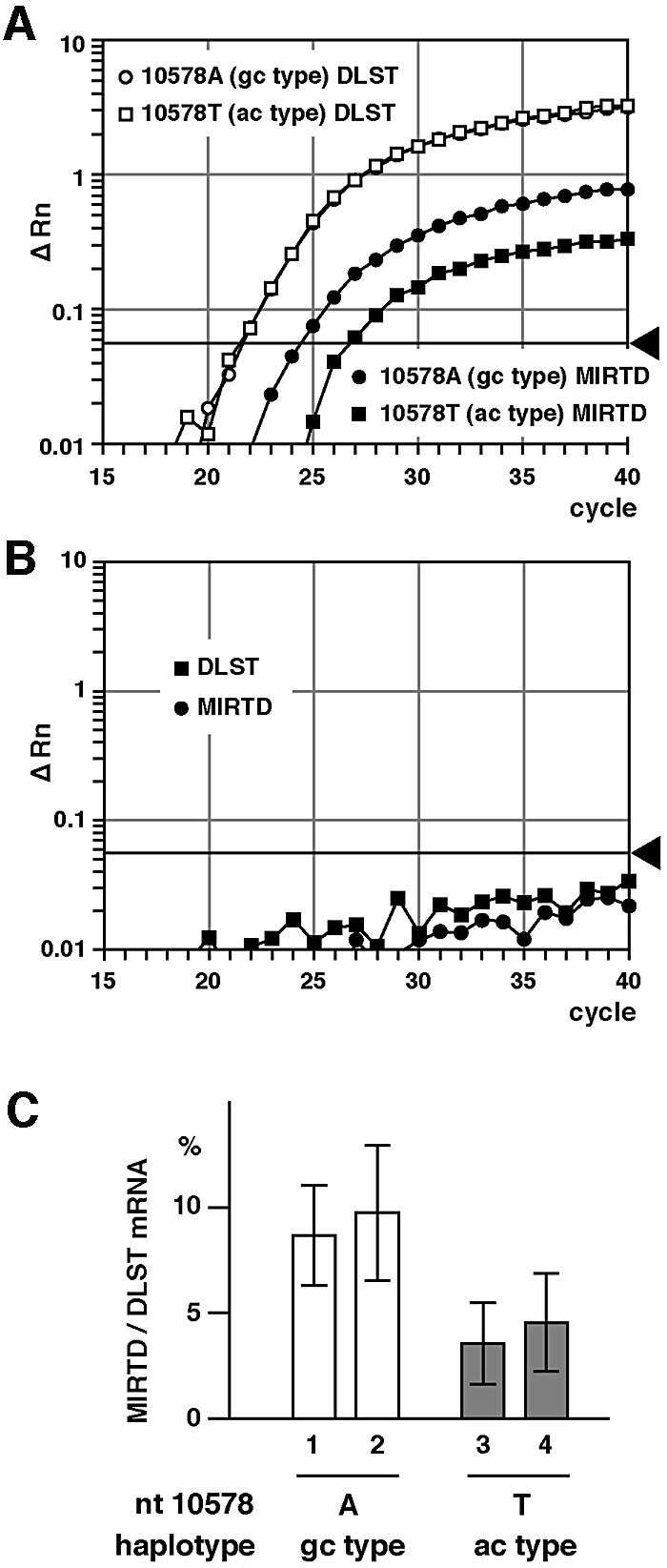
Fig. 4. Decreased expression of human MIRTD in rat PC12 cells carrying the human DLST gene with haplotype ac. PC12 cells were cotransfected with the cosmid containing the ac- or gc-type DLST gene and the geneticin-resistance gene using Lipofectin (Invitrogen). Transfectants were enriched by selecting geneticin-resistant cells for 14 days and then poly(A)+ RNA was prepared from the transfectant pools. Amounts of human MIRTD and DLST mRNAs were measured by real-time quantitative RT–PCR. (A) Profiles of the real-time quantitative RT–PCR showing increases of PCR products derived from human DLST and MIRTD mRNAs. (B) Profiles of the real-time quantitative RT–PCR using poly(A)+ RNA from untransfected PC12 cells, detecting neither DLST nor MIRTD mRNA. (C) Relative values of MIRTD to DLST mRNA are shown with the mean ± SD obtained from four independent experiments. The difference was significant for 1 versus 3 (p = 0.039), 1 versus 4 (p = 0.049), 2 versus 3 (p = 0.016) and 2 versus 4 (p = 0.016).
Location of the MIRTD protein
To obtain evidence for the existence of the MIRTD protein, its location was determined using rat liver, which is more suitable for isolating intact organella for a subcellular fractionation experiment. Since a short RNA corresponding to the MIRTD mRNA was detected in the total rat liver RNA fraction by northern blotting (Figure 5A), there should be MIRTD protein in rat liver.

Fig. 5. Identification and location of the MIRTD protein. (A) Total RNA (50 µg) isolated from rat liver was analyzed by northern blotting. DLST and MIRTD mRNAs were detected with 32P-labeled rat DLST cDNA. The positions of 28S and 18S for rRNAs are indicated on the right. Arrowheads indicate mRNAs corresponding to DLST and MIRTD. (B) Fractionation of rat liver was carried out as described in Materials and methods. Post-nuclear supernatant was centrifuged at 10 000 g. The pellet fraction (P10 000) contained most of the mitochondria. The supernatant was centrifuged at 100 000 g. The pellet (P100 000) and the supernatant (S100 000) contained microsomes and cytosol, respectively. Samples were analyzed by western blotting with anti-DLST monoclonal antibody. (C) Isolated rat liver mitochondria were diluted with isotonic (lanes 1–3) or hypotonic (lanes 4–6) buffer to prepare mitoplast. Samples were divided into three aliquots and treated with 0 (lanes 1, 4), 10 (lanes 2, 5) or 50 µg/ml (lanes 3, 6) proteinase K. Samples were analyzed by western blotting with anti-DLST monoclonal, anti-Tom40 polyclonal or Tom20 polyclonal antibody. Asterisk, degraded product of DLST; PK, proteinase K; M, mitochondria; MP, mitoplast.
To find the location of the MIRTD protein, rat liver was fractionated as usual and then western blotting was performed (Figure 5B). In the P10 000 fraction (the mitochondrial fraction), a 55 kDa product and an additional 30 kDa product were detected (Figure 5B, lane 1). The 55 kDa product corresponds to the full-length DLST protein. If the 30 kDa polypeptide is translated from the first AUG codon located in exon 8 of the DLST gene through the same reading frame with the full-length DLST protein, the molecular weight is estimated as 29.7 kDa, which corresponds to the apparent molecular weight of 30 kDa of the product. MIRTD was detected mainly in the P10 000 fraction (Figure 5B, lanes 2 and 3), suggesting that the protein belonged to the mitochondrial fraction.
To determine the location of MIRTD more precisely, mitochondria isolated from rat liver were treated with external proteinase K. Tom20, Tom40 and the full-length DLST (a matrix protein) were used as internal controls. Tom40 and Tom20 are components of the translocation complex for transporting the nuclear-encoded mitochondrial precursor proteins. Tom40 is exposed to the intermembrane side, while Tom20 is exposed outside mitochondria (Pfanner and Geissler, 2001). Tom20 was easily digested with 10 µg/ml proteinase K, whereas Tom40, DLST and the MIRTD protein were resistant (Figure 5C, lanes 1–3). On the other hand, when mitochondria were converted to mitoplast, the MIRTD band disappeared (Figure 5C, lane 4), whereas Tom40 and Tom20 remained in the mitoplast. These results indicate that the MIRTD protein is located in the intermembrane space of mitochondria.
Construction of cell lines for digesting the MIRTD mRNA
To investigate the function of MIRTD, we tried to eliminate MIRTD from cultured cells. In order to reduce the amount of specific mRNA in general, antisense oligonucleotides covering initiation codons are introduced into the cell. However, in this case, the sequence of MIRTD mRNA is identical to that of the full-length DLST mRNA. If the antisense oligonucleotides for the MIRTD mRNA are introduced into the cell, the full-length DLST mRNA should be reduced together with the MIRTD mRNA. Thus, we used maxizyme technology to digest the MIRTD mRNA with high specificity. Recently, a new type of ribozyme that specifically digests a target RNA has been developed (Kuwabara et al., 1998). Maxizyme is a heterodimeric ribozyme and has sensor arms that can recognize target sequences. In the presence of a specific target sequence, it can form a cavity that can capture catalytically indispensable magnesium ions. To express the maxizyme under the control of a strong promoter in vivo, we embedded each monomeric unit downstream of the sequence of a human tRNAVal promoter (Kawasaki et al., 1996) that is recognized by RNA polymerase III (Perriman and de Feyter, 1997) to generate MzL (maxizyme left) and MzR (maxizyme right). The specific design of the tRNAVal constructs was based on previous success in attaching a ribozyme sequence to the 3′-modified side of the tRNAVal portion of the human gene to be exported into cytosol (Koseki et al., 1999). In order to obtain good substrate activity, we designed sequences of the maxizyme that can adopt an active conformation only in the presence of the intron 7–exon 8 junction of the DLST gene (Figure 6A). Indeed, the maxizyme digested the MIRTD mRNA with high specificity, but not the DLST mRNA in vitro as had been reported (Tanabe et al., 2000).

Fig. 6. Decreased amounts of MIRTD mRNA by introducing the maxizyme. (A) The sequences of MIRTD mRNA and the maxizyme heterodimer MzR and MzL are shown. The maxizyme heterodimer can form an active conformation only in the presence of the junction between intron 7 and exon 8 in the DLST gene. A triangle indicates the site of cleavage by the maxizyme. (B) Neuroblastoma SH-SY5Y cells were cotransfected with DNA of the maxizyme and the geneticin-resistance gene. Independent clones stably expressing the maxizyme (Z5, Z7 and Z21) or monomeric controls (L1 and R2) were isolated and then relative values of MIRTD mRNA to DLST mRNA were obtained as described in Materials and methods. (C) Upper panel: protein (50 µg) from each sample was subjected to SDS–PAGE followed by western blotting using antibody against the C-terminal region of the DLST protein. Lower panel: the amount of the MIRTD protein was quantified and normalized to that of the DLST protein. Results are shown as the average ± SD of three experiments.
The DNA fragments that code MzL and MzR for MIRTD mRNA were transfected into human neuroblastoma SH-SY5Y cells and then the cells expressing the maxizyme were selected. Poly(A)+ RNA was isolated from the transfectants and the amount of MIRTD mRNA was measured by real-time quantitative RT–PCR. As expected, amounts of MIRTD mRNA were markedly reduced in the maxizyme-expressing cells (Figure 6B). The DLST and MIRTD proteins were semiquantified by western blotting (Figure 6C). Amounts of the DLST protein were not affected by the introduction of the maxizyme, whereas the MIRTD protein markedly decreased (Figure 6C, lanes Z5 and Z21). Thus, we concluded that the maxizyme specifically digested the MIRTD mRNA and that the 30 kDa protein is the product translated from the MIRTD mRNA.
Phenotypes of the MIRTD-deficient cell lines
To elucidate the role of MIRTD, we tried to find phenotypes that appeared on specific digestion of the MIRTD mRNA. First, we exposed the cells expressing the maxizyme or monomer (inactive form) to hydrogen peroxide, stained their nuclei with a mixture of fluorescent dyes [Hoechst 33342 (blue) and propidium iodide (PI) (pink) as indicators of living and dead cells, respectively] and then counted the cells stained with each dye to calculate viability. Cell viability against hydrogen peroxide was significantly reduced compared with the controls in a dose- and time-dependent manner (Figure 7A and B). When the transfectants expressing the maxizyme and monomeric controls differentiated into putative neurons on treatment with retinoic acid, the transfectants with the maxizyme were also more sensitive to hydrogen peroxide than the controls (Figure 7C).

Fig. 7. Sensitivity to oxidative stress in the MIRTD-deficient cells. (A) Maxizyme-expressing cells (Z5, Z7 and Z21) and control cells (L1 and R2) were exposed to medium containing 0.2, 0.4 or 0.6 mM H2O2. After 24 h, the cells were stained with Hoechst 33342 (blue, dead and living cells) and propidium iodide (pink, dead cells). Cell viability was obtained by counting 200 cells in each clone exposed to various concentrations of H2O2. (B) Z5, Z21, L1 and R2 cells were exposed to 0.4 mM H2O2 for the periods indicated. Cell viability was calculated as above. (C) The transfectants (L1, R2, Z7 and Z21) were induced to differentiate into putative neurons with 10 µM retinotic acid in the presence of 1% FBS for 5 days and then exposed to various concentration of H2O2 for 12 h in the presence of 1% FBS. Cell viability was assessed as described above. (D) Total protein was prepared from the L1 and Z21 cells exposed to 0.2 mM H2O2 at 0, 6, 12 and 24 h. Protein (50 µg) from each sample was subjected to SDS–PAGE and western blotting with antibody against the C-terminal region of the DLST protein. Arrowheads indicate MIRTD and the full-length DLST protein. (E) Poly(A)+ RNA was isolated from L1 and Z21 cells exposed to 0.2 mM H2O2 at 0, 3, 6, 12 or 24 h. The amounts of MIRTD mRNA were determined by real-time quantitative RT–PCR and normalized to those of DLST mRNA. (F) The amounts of the MIRTD protein were quantified by western blotting of (D) and normalized to those of DLST. (G) Z5, Z21, L1 and R2 cells were exposed to the medium containing menadione at the indicated concentration. After 24 h, cell viability was measured as described in (A). (H) Z21 and L1 cells were incubated in the presence or absence of 10 µM menadione for 3 h and then treated with CM-H2DCFDA (Molecular Probes) (5 µM) for 1 h. The cells were trypsinized and subjected to flow cytometry to measure the fluorescent signal of DCF with an EPICS ELITE flow cytometer (Beckman Coulter). Twenty thousand cells were measured in each experiment and mean values were obtained. Results in (A–C) and (E–H) are shown as the average ± SD obtained from three or four experiments.
To confirm the contribution of MIRTD to vulnerability, the amounts of the mRNA and protein of MIRTD were monitored by real-time RT–PCR (Figure 7E) and western blotting (Figure 7D and F), respectively, in the time course during exposure to hydrogen peroxide. The MIRTD protein and mRNA slightly increased and then gradually decreased in the control transfectant, whereas they were maintained at low level in the transfectants expressing the maxizyme (Figure 7D–F). Therefore, these results showed that the reduction of MIRTD made the cells more sensitive to external hydrogen peroxide.
Next, we tested sensitivity against another reagent that should cause internal oxidative stress. The cells expressing the maxizyme were exposed to menadione, a quinone derivative that generates, from the mitochondrial respiratory chain, superoxide anion that is promptly converted into hydrogen peroxide. Unexpectedly, the cells expressing the maxizyme (Z5, Z21) became more resistant to the reagent than the control cells (Figure 7G). Then, hydrogen peroxide production with menadione was tested by flow cytometry with a fluorescent dye [5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescin diacetate acetyl ester (CM-H2DCFDA)] that is specific to hydrogen peroxide. The control clone L1 actively generated hydrogen peroxide on treatment with menadione, whereas the maxizyme-expressing clone Z21 did not (Figure 7H). Therefore, the MIRTD-deficient cells generated less superoxide anion, resulting in resistance against menadione (Figure 7G).
Respiration deficiency in the cells expressing the maxizyme
To clarify why the MIRTD-deficient cells are insensitive to menadione, we investigated the mitochondrial respiratory chain in the maxizyme-expressing cells. First, we measured the rate of oxygen consumption using whole cells with an oxygen electrode and found that maxizyme-expressing clones Z5 and Z21 showed almost no respiration, while the control clones (L1 and R2) did respire (Figure 8A). Then, we measured the enzymatic activity of each complex of the respiratory chain using mitochondria isolated from each clone (Figure 8B). Interestingly, the activity of complex IV (cytochrome c oxidase) was not detected in mitochondria isolated from the MIRTD-deficient Z5 and Z21 clones (there is no bar, indicating no activity of complex IV). In clones Z5 and Z21, the activity of NADH-cytochrome c oxidoreductase (complex I + III) was also markedly decreased, but that of succinate-cytochrome c oxidoreductase (complex II + III) was not. Thus, it is concluded that the activity of cytochrome c oxidase is completely lost, whereas that of complex I (NADH–ubiquinone oxidoreductase) is significantly but not completely affected, leading to loss of respiration in the MIRTD-deficient cells.

Fig. 8. Decrease in the activity and amounts of the mitochondrial respiratory chain complexes. (A) Semiconfluent cells were trypsinized and harvested. The harvested cells were suspended at (0.7–1.5) × 107 cells/ml in the culture medium and placed into a chamber (50 µl) of an oxygen electrode. The rate of oxygen consumption by cells was measured at 37°C using a Clark-type electrode (Strathkelvin Instruments, Glasgow). Left panel: traces of oxygen consumption by cells at 1 × 107 cells/ml. Right panel: average values with SD normalized to cell number from three independent experiments. (B) The activities of NADH–cytochrome c oxidoreductase (I + III), succinate–cytochrome c oxidoreductase (II + III), ubiquinol–cytochrome c oxidoreductase (III) and cytochrome c oxidase (IV) were measured as described in Materials and methods. Results are shown with the relative activities. The level of activity in mitochondria isolated from SH-SY5Y cells was set to 100. Note that no complex IV activity in the mitochondria isolated from Z5 and Z21 clones was detected. Average values with the SD from three independent experiments are shown. (C) Proteins (10 µg) from the mitochondrial fraction prepared from each clone were subjected to western blotting using each antibody against the subunits of the respiratory complexes and ATP synthase. CO I and CO II are mitochondrial-encoded proteins, while the others are encoded by nuclear genes. F1β, F1-ATPase β subunit. (D) Mitochondrial translation products labeled with [35S]methionine in vivo were analyzed as described (Hayashi et al., 1993). Briefly, semiconfluent cells were labeled with [35S]methionine in the presence of emetine (0.2 mg/ml) for 60 min at 37°C. Proteins of the mitochondrial fraction were separated by SDS–PAGE (15–25% gradient gel). The translation products were analyzed with a bioimaging analyzer FLA2000 (Fuji Film, Tokyo). ND 1–5, subunits of complex I (NADH–ubiquinone oxidoreductase); CO I–III, subunits of cytochrome c oxidase (complex IV); cyt b, the cytochrome b subunit of ubiquinol–cytochrome c oxidoreductase (complex III); ATP 6 and ATP 8, subunits of ATP synthase (complex V). (E) Poly(A)+ RNA from each clone was converted into cDNA and CO IV mRNA was quantified by real-time RT–PCR as described in Materials and methods. Average values with the SD from three experiments are shown after normalization to GAPDH cDNA.
To investigate the mechanism behind the respiratory deficiency, steady-state levels of subunits involved in the respiratory chain were examined by western blotting (Figure 8C). Amounts of the nuclear-encoded subunits of complexes II, III and V were independent of the MIRTD deficiency. In contrast, the amount of the nuclear-encoded 15K subunit of complex I was slightly decreased in the MIRTD-deficient cells. More striking, amounts of the mitochondrial-encoded subunits of complex IV (CO I and CO II) were severely reduced. Additionally, a nucleus-encoded subunit of complex IV (CO IV) was also severely affected. These declines were in good agreement with the loss of enzymatic activity. To examine whether the mitochondrial translation is responsible for the decline in the mitochondrial-encoded subunits (CO I and CO II), translation in mitochondria was examined by pulse-labeling the gene products with [35S]methionine in the presence of emetine, an inhibitor of cytosolic translation. The labeled translational products were separated by SDS–PAGE followed by autoradiography (Figure 8D). The strength of the signals did not change enough to explain the declines in the steady-state levels of CO I and CO II in each clone. Thus, the decrease in the steady-state level of the complex IV subunits took place post-translationally. In addition, amounts of mRNA of CO IV, a nuclear-encoded subunit of complex IV, were independent of MIRTD (Figure 8E). These findings strongly suggest that the MIRTD deficiency affects the assembly of complex IV, and to a slight extent that of complex I, to abolish the activities, and that the unassembled subunits are easily digested by mitochondrial proteinases.
Discussion
In the present study, we found an unusual mRNA transcribed from intron 7 of the DLST gene in addition to the full-length form and designated the translated protein MIRTD. We detected the novel mRNA by the RT–PCR method and confirmed its existence based on: (i) a lack of the sequence of intron 8 in the PCR product covering exon 8 and exon 9 (Figure 2A and B); (ii) the presence of the cap structure in the MIRTD mRNA (Figure 2C), confirming that the transcription was indeed initiated from intron 7; (iii) the presence of a short mRNA that corresponds to MIRTD mRNA (Figure 5A); (iv) the presence of a protein that corresponds to the MIRTD protein, as judged by the apparent molecular weight and immunoreactivity (Figure 5B and C); (v) a decrease in MIRTD mRNA on expressing a maxizyme that was designed to digest the MIRTD mRNA (Figure 6B); and (vi) a decrease in MIRTD protein accompanied by that of MIRTD mRNA (Figure 6C).
Amounts of MIRTD mRNA in the brain varied among individuals, suggesting that the expression or degradation of the mRNA was regulated depending upon genetic background and various factors. Even under these complicated conditions, the amount of MIRTD mRNA was significantly associated with AD. Haplotype ac of the DLST gene seemed to show only a trend in terms of reduced amounts in the brain. However, this would be reasonable when the number of samples is not sufficient for a statistical analysis and when the risk is not great. Thus, this moderate genetic background might be statistically overcome by other risk factors in the brain. On the other hand, in an in vitro experiment, the expression of MIRTD was dependent upon the haplotype of the DLST gene (Figure 4). Therefore, it is suggested that haplotypes of the DLST gene influence the expression of MIRTD. Although further analyses in specific regions of the brain might have supported the association more strongly, the amount of MIRTD mRNA in the brain was shown to be associated with AD. In particular, the inability to detect MIRTD mRNA in half of the AD patients suggests an association with AD.
The MIRTD protein is located in the mitochondrial intermembrane space, despite lacking the putative mitochondrial targeting sequence in the N-terminal region, whereas the full-length DLST is found in the matrix. It is unknown how the MIRTD protein targets mitochondria and reaches this specific region.
We used maxizyme technology to find the physiological role of the MIRTD protein. When MIRTD mRNA was eliminated using this technique, the cells became sensitive to hydrogen peroxide and showed strikingly reduced steady-state levels of the subunits of cytochrome c oxidase, leading to complete loss of enzymatic activity. Cytochrome c oxidase is composed of 13 different subunits, of which three are mitochondrial encoded and the rest are nuclear encoded (Barrientos et al., 2002a). Although the molecular mechanism of its assembly is not fully understood, factors essential for assembly have been identified. For example, SURF-1, whose mutation is responsible for a mitochondrial disease (Leigh’s syndrome), contributes to the assembly of cytochrome c oxidase. As a result of mutation, the enzymatic activity of cytochrome c oxidase declines in patients (Zhu et al., 1998; Tiranti et al., 1999; Barrientos et al., 2002b). The following model has been proposed for the cytochrome c oxidase assembly: CO I acts as the core of the complex assembly, CO IV binds to the core in the initial step, and CO II and the other 10 subunits join to form the complex (Nijtmans et al., 1998). In Figure 8C, the extent of decrease in CO II is most severe and the amount of CO IV is less than that of CO I in the MIRTD-deficient cells. This finding agrees with the assembly model, strongly suggesting that MIRTD contributes to the assembly of the cytochrome c oxidase complex. Since the activity of complex I is significantly decreased, MIRTD should also contribute to its assembly. MIRTD probably aids in the assemblies by acting from the intermembrane space side. The molecular mechanism of the function of MIRTD will be investigated in detail in the future.
Even when mitochondrial respiration is completely abolished, cultured mammalian cells are often able to grow (King and Attardi, 1989). We have previously isolated HeLa cell lines completely lacking mitochondrial DNA (Hayashi et al., 1991). Thus, it is not surprising that cell lines deficient in respiration have been established by digesting MIRTD mRNA. HeLa cells lacking mitochondrial DNA grow in conventional media, but are susceptible to various toxic treatments including exposure to hydrogen peroxide (S.Ohta, unpublished results). Similarly, the MIRTD-deficient cells may become more sensitive to hydrogen peroxide because of the respiration defect. Alternatively, the secondary change in the expression of several factors involved in oxidative stress may totally influence the sensitivity to hydrogen peroxide.
In conclusion, the MIRTD-deficient cells exhibit a decline in respiration because the assembly of the respiratory complexes is disturbed. If these findings can be applied to results obtained in the brains of AD patients lacking MIRTD mRNA, they would suggest that the MIRTD-deficient brain cells of AD patients would be less active in respiration and more sensitive to hydrogen peroxide. In fact, several laboratories have reported a correlation between AD and defects of cytochrome c oxidase (e.g. Kish et al., 1992; Parker and Parks, 1995; Cottrell et al., 2001). Moreover, it has been shown that amyloid β protein generates hydrogen peroxide (Behl et al., 1994; Harris et al., 1995), which may be more toxic in the MIRTD-deficient cells. It will be necessary to investigate the close correlation of the MIRTD protein with cytochrome c oxidase in the brains of AD patients.
Taken together, the results of this study explain why a genotype of the DLST gene is associated with AD without a change of amino acid residues in the DLST protein: through a decrease in the expression of MIRTD mRNA and the function of MIRTD. Although it remains unclear just how much MIRTD actually contributes to the pathogenesis of AD, the present study has revealed that the DLST gene is bifunctionally involved in mitochondrial energy metabolism, the full-length DLST protein is involved in the Krebs cycle and the truncated version, MIRTD, contributes to the biogenesis of the respiratory chain.
Materials and methods
Cell culture
The human neuroblastoma cell line SH-SY5Y was cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 15% fetal bovine serum (FBS), 2 mM glutamine, 20 IU/ml penicillin and 20 µg/ml streptomycin in 5% CO2 at 37°C. The rat pheochromocytoma cell line PC12 was cultured in DMEM supplemented with 5% FBS, 5% immobilized horse serum, 100 IU/ml penicillin and 100 µg/ml streptomycin.
Brain samples
The brain samples of patients with AD were kindly provided by Dr Colin L.Masters (Department of Pathology, University of Melbourne, Melbourne, Australia), Dr Dennis J.Selkoe (Department of Neurology, Harvard Medical School, USA), Center for Neurologic Diseases, Brigham, USA, Women’s Hospital, Hatsuishi Hospital, Hatsuishi, Chiba, Japan and Tokyo Metropolitan Geriatric Medical Center, Itabashi, Tokyo, Japan. Control samples were obtained from patients who died of malignant neoplasms in the Gunma Cancer Center. Cortexes of both patients and controls had been obtained with informed consent from family members. Post-mortem periods ranged from 40 to 660 min (255.0 ± 174.9 min). The mean age of the AD patients was 77.7 ± 6.5 years (range 60–86 years), while that of the controls was 65.9 ± 9.0 years (range 48–79 years). Frequencies of carriers of the ac haplotype were 32 and 24% in the AD and control groups, respectively.
Quantitative RT–PCR
Total RNA was isolated from samples of human brain and cultured cells using RNeasy Mini Kit (Qiagen) or ISOGEN (Wako) according to the manufacturer’s protocol. Poly(A)+ RNA was purified from total RNA using a QuickPrep mRNA Purification Kit (Amersham Bioscience). First-stranded cDNA was synthesized with SUPERSCRIPT II RT (Invitrogen) using oligo(dT) primer (Invitrogen).
Amounts of mRNA were quantified by real-time quantitative RT–PCR using the ABI PRISM 7700 Sequence Detector System (Applied Biosystems) (Holland et al., 1991; Livak et al., 1995). The measurements were repeated at least three times and the average values were obtained. Primers and probes, which had been designed with the Primer Express software program (Applied Biosystems), were as follows: DLST forward primer, 5′-acctacagcagcggcagt-3′ (exon 8); DLST reverse primer, 5′-ctcctggctcagctagtggt-3′ (exon 9); MIRTD forward primer, 5′-ctcatttcagacagtgccagtg-3′ (intron 7); MIRTD reverse primer, 5′-tggctcagctagtggtggg-3′ (exon 9); TaqMan probe for both DLST and MIRTD, 5′-cctccttctggcaaacctgtgtct-3′ (covering exons 8 and 9); GAPDH forward primer, 5′-gggaaggtgaaggtcgga-3′; GAPDH reverse primer, 5′-gcagccctggtgaccag-3′; TaqMan probe for GAPDH, 5′-caacggatttggtcgtattgggcg-3′. For measurement of the amounts of mRNA of subunit IV of cytochrome c oxidase, the forward primer 5′-tgtgtgtgtacgagctcatgaaag-3′, reverse primer 5′-gtggtcacgccgatccat-3′ and TaqMan probe 5′-ttgtgaagagcgaagacttttcgctccc-3′ were used. The values were normalized with respect to GAPDH cDNA. TaqMan probes contain fluorophores FAM as a reporter at the 5′ end and TAMRA as a quencher at the 3′ end.
Determination of the transcriptional start site of MIRTD mRNA
The transcriptional start site of MIRTD mRNA was determined by an oligonucleotide-capping method (Yamabe et al., 1998) with Cap Site cDNA™ of human brain (Nippon Gene, Toyama, Japan) according to the manufacturer’s protocol. The first PCR was performed using 5′-caaggtacgccacagcgtatg-3′ (1RC, Nippon Gene) and 5′-aagtaacgaatgtctactgggtatcagca-3′. Samples were amplified for 40 cycles under the following conditions: 94°C for 30 s, 55°C for 30 s and 72°C for 45 s. Aliquots of the first reaction product were used as the template in the second PCR. The second PCR (nested PCR) was performed using 5′-gtacgccacagcgtatgatgc-3′ (2RC2; Nippon Gene) and 5′-actgggtatcagcaatatgcaaag-3′, and was followed by DNA sequencing.
Western blotting
For total cell lysate, cultured cells were washed once with PBS and scraped in PBS. The cells were collected by centrifugation and lysed with 2% SDS, 20 mM Tris–HCl pH 6.8 and 1 mM phenylmethylsulfonyl fluoride (PMSF). The protein concentration was determined by BCA assay (Pierce). Samples with 2-mercaptoethanol were subjected to SDS–PAGE and then western blotting was carried out using antibody.
Preparation of anti-DLST monoclonal antibody and the other antibodies
The C-terminal region of DLST, which contained the region downstream from methionine in exon 8, was expressed with glutathione S-transferase in Escherichia coli. Inclusion bodies containing the fusion protein were isolated and subjected to preparative SDS–PAGE. The fusion protein was eluted from the excised gel, concentrated and dialyzed with phosphate-buffered saline (PBS). Spleen cells were isolated from immunized BALB/c mice and fused with PAI myeloma cells (Ohta and Schatz, 1984). Antibody-producing hybridoma cells were screened by solid-phase enzyme-linked immunosorbent assay using the purified recombinant C-terminal region of DLST with His6. Positive cells were cloned by limited dilution.
Antibodies against mitochondrial Tom40 and Tom20 were kind gifts from Professor K.Mihara (Kyushu University, Fukuoka, Japan). All antibodies against the subunits of the respiratory chain complex were purchased from Molecular Probes.
Construction of PC12 cells carrying the human DLST gene
Genomic DNA was isolated from peripheral blood and partially digested with Sau3AI. The digested DNA was inserted into the BamHI site in the cosmid vector SuperCos 1 (Stratagene). The ligated DNA was packaged using Gigapack III Gold packaging extract (Stratagene) according to the manufacturer’s directions. Positive clones were selected by colony hybridization using a PCR fragment corresponding to the promoter region in the DLST gene (Nakano et al., 1994). The selected cosmid was confirmed to contain the full length by amplifying the upstream region, exons 1, 8 and 14, and the downstream regions by PCR. PC12 cells were cotransfected with the cosmid containing the ac- or gc-type DLST gene and the geneticin-resistance gene using Lipofectin (Invitrogen). Selecting geneticin-resistant cells for 14 days enriched the transfectants.
Sub-organellar fractionation of rat liver mitochondria
To isolate intact mitochondria, fresh rat liver was washed in precooled breaking buffer (0.22 M mannitol, 0.07 M sucrose, 20 mM HEPES–KOH pH 7.4 and 1 mM EDTA) and weighed. It was minced with scissors and suspended in the breaking buffer. The suspension was homogenized in a Potter–Elvehjem glass homogenizer using a Teflon pestle. The homogenate was loaded onto 0.34 M sucrose and centrifuged at 1000 g for 10 min. The upper phase (post-nuclear supernatant) was centrifuged at 10 000 g for 15 min and the pellet (P10 000 fraction) was isolated. The resulting supernatant was centrifuged at 100 000 g for 2 h and divided into a pellet (P100 000) and supernatant (S100 000). To prepare mitoplast, mitochondria (10 mg/ml) were diluted 10-fold with 20 mM HEPES–KOH pH 7.4 and 1 mM EDTA. Then samples of mitochondria and mitoplast were divided into three aliquots and treated with 0, 10 or 50 µg/ml proteinase K for 30 min on ice. Mitochondria or mitoplasts were re-isolated by centrifugation and analyzed by SDS–PAGE and western blotting.
Establishment of cells stably expressing the maxizyme
Chemically synthesized oligonucleotides encoding the maxizyme for the MIRTD mRNA and pol III termination sequence were converted to double-stranded sequences by PCR. After digestion with Csp45I and SalI, each appropriate fragment was cloned downstream of the tRNAVal promoter of pUC-dt, which contained the chemically synthesized promoter for a human gene for tRNAVal between the EcoRI and SalI sites of pUC19 (Kuwabara et al., 1999). The sequences of the constructs were confirmed by direct sequencing. SH-SY5Y cells were cotransfected with the maxizyme and the geneticin-resistance gene using Effectene reagent (Qiagen) according to the manufacturer’s protocol. Several independent clones stably expressing the maxizyme or monomeric controls were isolated by selection with geneticin (Invitrogen).
Assay of the respiratory chain complex activity
The activity of each mitochondrial respiratory chain complex was spectrophotometrically determined at 30°C as described (Trounce et al., 1996; Birch-Machin and Turnbull, 2001). For the assay of NADH-cytochrome c oxidoreductase (complex I + III) activity, frozen–thawed mitochondria (15 µg protein/ml) were preincubated in 25 mM KPi pH 7.2, 5 mM MgCl2 and 2.5 mg/ml bovine serum albumin (BSA) containing 2 mM KCN at 30°C for 3 min. Then, cytochrome c (III) (50 µM) and NADH (130 µM) were added and the change between absorbance of cytochrome c at 550 nm and that at 580 nm (extinction coefficient 19 mM–1 cm–1) was measured. After 2 min, rotenone (2 µg/ml) was added and the rotenone-insensitive rate was measured. For the assay of succinate–cytochrome c oxidoreductase (complex II + III) activity, frozen–thawed mitochondria (15 µg/ml) were preincubated in 25 mM KPi pH 7.2, 5 mM MgCl2 and 2.5 mg/ml BSA containing 20 mM sodium succinate, 2 mM KCN and 2 µg/ml rotenone at 30°C for 10 min. Then, cytochrome c (III) (30 µM) was added and the change in absorbance was measured. For the assay of ubiquinol–cytochrome c oxidoreductase (complex III) activity, 35 µM ubiquinol and cytochrome c (III) were added to 25 mM KPi pH 7.2, 5 mM MgCl2 and 2.5 mg/ml BSA containing 2 mM KCN, 2 µg/ml rotenone and 0.6 mM n-dodecyl β-d-maltoside, and the non-enzymatic rate was measured. Then, mitochondria (10 µg protein/ml) were added and the change in absorbance of cytochrome c was measured. Cytochrome c oxidase (complex IV) activity was measured by following the change between absorbance of cytochrome c at 550 nm and that at 580 nm (extinction coefficient 19 mM–1 cm–1). Namely, reduced cytochrome c (15 µM) was added to 20 mM KPi pH 7.2 and 0.45 mM n-dodecyl β-d-maltoside, and the non-enzymatic rate was measured. Then, mitochondria (10 µg protein/ml) were added and the change in absorbance of cytochrome c was measured.
Acknowledgments
Acknowledgements
We are grateful to Professor Katsuyoshi Mihara (Kushu University) for providing anti-Tom40 and anti-Tom20 antisera.
References
- Barrientos A., Barros,M.H., Valnot,I., Rotig,A., Rustin,P. and Tzagoloff,A. (2002a) Cytochrome oxidase in health and disease. Gene, 286, 53–63. [DOI] [PubMed] [Google Scholar]
- Barrientos A., Korr,D. and Tzagoloff,A. (2002b) Shy1p is necessary for full expression of mitochondrial COX1 in the yeast model of Leigh’s syndrome. EMBO J., 21, 43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behl C., Davis,J.B., Lesley,R. and Schubert,D. (1994) Hydrogen peroxide mediates amyloid β protein toxicity. Cell, 77, 817–827. [DOI] [PubMed] [Google Scholar]
- Birch-Machin M.A. and Turnbull,D.M. (2001) Assaying mitochondrial respiratory complex activity in mitochondria isolated from human cells and tissues. Methods Cell Biol., 65, 97–117. [DOI] [PubMed] [Google Scholar]
- Bonilla E., Tanji,K., Hirano,M., Vu,TH., DiMauro,S. and Schon,EA. (1999) Mitochondrial involvement in Alzheimer’s disease. Biochim. Biophys. Acta, 1410, 171–182. [DOI] [PubMed] [Google Scholar]
- Caseley C.S., Canevari,L., Land,J.M., Clark,J.B. and Sharpe,M.A. (2002) β-amyloid inhibits integrated mitochondrial respiration and key enzyme activity. J. Neurochem., 80, 91–100. [DOI] [PubMed] [Google Scholar]
- Cottrell D.A., Borthwick,G.M., Johnson,M.A., Ince,P.G. and Turnbull,D.M. (2001) Cytochrome c oxidase deficient hippocampal neurons and choroids epithelial cells are increased in Alzheimer’s disease. Neurology, 57, 260–264.11468310 [Google Scholar]
- Harris M.E., Hensley,K., Butterfield,D.A., Leedle,R.A. and Carney,J.M. (1995) Direct evidence of oxidative injury produced by the Alzheimer’s β-amyloid peptide (1–40) in cultured hippocampal neurons. Exp. Neurol., 131, 193–202. [DOI] [PubMed] [Google Scholar]
- Hayashi J., Ohta,S., Kikuchi,A., Takemitsu,M., Goto,Y. and Nonaka,I. (1991) Introduction of disease-related mitochondrial DNA deletions into HeLa cells lacking mitochondrial DNA results in mitochondrial dysfunction. Proc. Natl Acad. Sci. USA, 88, 10614–10618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi J., Ohta,S., Takai,D., Miyabayashi,S., Sakuta,R., Goto,Y. and Nonaka,I. (1993) Accumulation of mtDNA with a mutation at position 3271 in tRNA(Leu)(UUR) gene introduced from a MELAS patient to HeLa cells lacking mtDNA results in progressive inhibition of mitochondrial respiratory function. Biochem. Biophys. Res. Commun., 197, 1049–1055. [DOI] [PubMed] [Google Scholar]
- Holland P.M., Abramson,R.D., Watson,R. and Gelfand,D.H. (1991) Detection of specific polymerase chain reaction product by utilizing the 5′-3′ exonuclease activity of Thermus aquaticus DNA polymerase. Proc. Natl Acad. Sci. USA, 88, 7276–7280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki H., Ohkawa,J., Tanishige,N., Yoshinari,K., Murata,K., Yokoyama,K.K. and Taira,K. (1996) Selection of the best target site for ribozyme-mediated cleavage within a fusion gene for adenovirus E1A-associated 300 kDa protein (p300) and luciferase. Nucleic Acids Res., 24, 3010–3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King MP. and Attardi,G. (1989) Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science, 246, 500–503. [DOI] [PubMed] [Google Scholar]
- Kish S.J., Bergeron,C., Rajput,A., Dozic,S., Mastrogiacomo,F., Chang,L.J., Wilson,J.M., DiStefano,L.M. and Nobrega,J.N. (1992) Brain cytochrome oxidase in Alzheimer’s disease. J. Neurochem., 59, 776–779. [DOI] [PubMed] [Google Scholar]
- Koseki S., Tanabe,T., Tani,K., Asano,S., Shioda,T., Nagai,Y., Shimada,T., Ohkawa,J. and Taira,K. (1999) Factors governing the activity in vivo of ribozymes transcribed by RNA polymerase III. J. Virol., 73, 1868–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunugi H., Nanko,S., Ueki,A., Isse,K. and Hirasawa,H. (1998) DLST gene and Alzheimer’s disease. Lancet, 351, 1584. [DOI] [PubMed] [Google Scholar]
- Kuwabara T., Warashina,M., Tanabe,T., Tani,K., Asano,S. and Taira,K. (1998) A novel allosterically trans-activated ribozyme, the maxizyme, with exceptional specificity in vitro and in vivo. Mol. Cell, 2, 617–627. [DOI] [PubMed] [Google Scholar]
- Kuwabara T., Warashina,M., Nakayama,A., Ohkawa,J. and Taira,K. (1999) tRNAVal-heterodimeric maxizymes with high potential as gene-inactivating agents: Simultaneous cleavage at two sites in HIV-1 tat mRNA in cultured cells. Proc. Natl Acad. Sci. USA, 96, 1886–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak K.J., Flood,S.J., Marmaro,J., Giusti,W. and Deetz,K. (1995) Oligonucleotides with fluorescent dyes at opposite ends provide a quenched probe system useful for detecting PCR product and nucleic acid hybridization. PCR Methods Appl., 4, 357–362. [DOI] [PubMed] [Google Scholar]
- Matsushita S., Arai,H., Yuzuriha,T., Kato,M., Matsui,T., Urakami,K. and Higuchi,S. (2001) No association between DLST gene and Alzheimer’s disease or Wernicke–Korsakoff syndrome. Neurobiol. Aging, 22, 569–574. [DOI] [PubMed] [Google Scholar]
- Matuda S., Kodama,J., Goshi,N., Takase,C., Nakano,K., Nakagawa,S. and Ohta,S. (1997) A polypeptide derived from mitochondrial dihydrolipoamide succinyltransferase is located on the plasma membrane in skeletal muscle. Biochem. Biophys. Res. Commun., 241, 151–156. [DOI] [PubMed] [Google Scholar]
- Nakano K. et al. (1993) Human dihydrolipoamide succinyltransferase: cDNA cloning and localization on chromosome 14q24.2-q24.3. Biochim. Biophys. Acta, 1216, 360–368. [DOI] [PubMed] [Google Scholar]
- Nakano K., Takase,C., Sakamoto,T., Nakagawa,S, Inazawa,J., Ohta,S. and Matuda,S. (1994) Isolation, characterization and structural organization of the gene and pseudogene for the dihydrolipoamide succinyltransferase component of the human 2-oxoglutarate dehydrogenase complex. Eur. J. Biochem., 224, 179–189. [DOI] [PubMed] [Google Scholar]
- Nakano K., Ohta,S., Nishimaki,K., Miki,T. and Matuda,S. (1997) Alzheimer’s disease and DLST genotype. Lancet, 350, 1367–1368. [DOI] [PubMed] [Google Scholar]
- Nijtmans L.G.J., Taanman,J-W., Muijsers,A.O., Speijer,D. and van den Bogert,C. (1998) Assembly of cytochrome-c oxidase in cultured human cells. Eur. J. Biochem., 254, 389–394. [DOI] [PubMed] [Google Scholar]
- Ohsawa I., Nishimaki,K., Yasuda,C., Kamino,K. and Ohta,S. (2003) Deficiency in a mitochondrial aldehyde dehydrogenase increases vulnerability to oxidative stress in PC12 cells. J. Neurochem., 84, 1110–1117. [DOI] [PubMed] [Google Scholar]
- Ohta S. and Schatz,G. (1984) A purified precursor polypeptide requires a cytosolic protein fraction for import into mitochondria. EMBO J., 3, 651–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker W.D. and Parks,J.K. (1995) Cytochrome c oxidase in Alzheimer’s disease brain: Purification and characterization. Neurology, 45, 482–486. [DOI] [PubMed] [Google Scholar]
- Perriman R. and de Feyter,R. (1997) tRNA-delivery systems for ribozymes. In Turner,P.C. (ed.), Methods in Molecular Biology, Ribozyme Protocols. Humana Press, Totowa, NJ, pp. 393–402. [DOI] [PubMed]
- Pfanner N. and Geissler,A. (2001) Versatility of the mitochondrial protein import machinery. Nat. Rev. Mol. Cell Biol., 2, 339–349. [DOI] [PubMed] [Google Scholar]
- Schon E.A. and Manfredi,G. (2003) Neuronal degeneration and mitochondrial dysfunction. J. Clin. Invest, 111, 303–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz J.B., Lindenau,J., Seyfried,J. and Dichgans,J. (2000) Glutathione, oxidative stress and neurodegeneration. Eur. J. Biochem., 267, 4904–4911. [DOI] [PubMed] [Google Scholar]
- Sheu K.F. et al. (1998) Polymorphisms of the DLST gene associate with late-onset and with familial Alzheimer’s disease. Neurobiol. Aging, 19, S293. [Google Scholar]
- Sheu K.F. et al. (1999a) Modulation by DLST of the genetic risk of Alzheimer’s disease in a very elderly population. Ann. Neurol., 45, 48–53. [PubMed] [Google Scholar]
- Sheu K.F., Brown,A.M., Kristal,B.S., Kalaria,R.N., Lilius,L., Lannfelt,L. and Blass,J.P. (1999b) A DLST genotype associated with reduced risk for Alzheimer’s disease. Neurology, 52, 1505–1507. [DOI] [PubMed] [Google Scholar]
- Smith MA., Rottkamp,C.A., Nunomura,A., Raina,A.K. and Perry,G. (2000) Oxidative stress in Alzheimer’s disease. Biochim. Biophys. Acta, 1502, 139–144. [DOI] [PubMed] [Google Scholar]
- Tanabe T., Takata,I., Kuwanbara,T., Warashina,M., Kawasaki,H., Tani,K., Ohta,S., Asano,S. and Taira,K. (2000) Maxizymes, novel allosterically controlable ribozymes, can be designed to cleave various substrates. Biomacromolecules, 1, 108–117. [DOI] [PubMed] [Google Scholar]
- Tiranti V., Galimberti,C., Hijtmans,L., Bovolenta,S., Perini,M.P. and Zeviani,M. (1999) Characterization of SURF-1 expression and Surf-1p function in normal and disease conditions. Hum. Mol. Genet., 8, 2533–2540. [DOI] [PubMed] [Google Scholar]
- Trounce I.A., Kim,Y.L., Jun,A.S. and Wallace,D.C. (1996) Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts and transmitochondrial cell lines. Methods Enzymol., 264, 484–509. [DOI] [PubMed] [Google Scholar]
- Yamabe Y., Shimamoto,A., Goto,M., Yokota,J., Sugawara,M. and Furuichi,Y. (1998) Sp1-mediated transcription of the Werner helicase gene is modulated by Rb and p53. Mol. Cell. Biol., 18, 6191–6200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z. et al. (1998) SURF1, encoding a factor involved in the biogenesis of cytochrome c oxidase, is mutated in Leigh syndrome. Nat. Genet., 20, 337–343. [DOI] [PubMed] [Google Scholar]