

Abstract
Rho-associated kinase (ROCK), including the ROCK-I and ROCK-II isoforms, is a protein kinase involved in signaling from Rho to actin cytoskeleton. However, in vivo functions of each ROCK isoform remain largely unknown. We generated mice deficient in ROCK-II by gene targeting. ROCK-II−/− embryos were found at the expected Mendelian frequency until 13.5 days postcoitum, but approximately 90% died thereafter in utero. ROCK-II−/− mice of both genders that survived were born runts, subsequently developed without gross abnormality, and were fertile. Whole-mount staining for a knocked-in lacZ reporter gene revealed that ROCK-II was highly expressed in the labyrinth layer of the placenta. Disruption of architecture and extensive thrombus formation were found in the labyrinth layer of ROCK-II−/− mice. While no obvious alteration in actin filament structures was found in the labyrinth layer of ROCK-II−/− placenta and stress fibers were formed in cultured ROCK-II−/− trophoblasts, elevated expression of plasminogen activator inhibitor 1 was found in ROCK-II−/− placenta. These results suggest that ROCK-II is essential in inhibiting blood coagulation and maintaining blood flow in the endothelium-free labyrinth layer and that loss of ROCK-II leads to thrombus formation, placental dysfunction, intrauterine growth retardation, and fetal death.
The small GTPase Rho, including RhoA, -B, and -C, works as a molecular switch and regulates various cellular processes, such as cell adhesion, migration, cytokinesis, proliferation, and transformation (3, 28). The Rho-regulated adhesion of cell to substrate is typically seen as induction of focal adhesions and stress fibers in fibroblasts. These Rho actions are exerted by combined actions of downstream Rho effectors. A variety of Rho effectors have been identified. Of the Rho effectors, Rho-associated kinase (ROCK; also known as ROK or Rho kinase) (12, 17, 19, 22) has been studied most extensively and has been suggested to be one of the major Rho effectors mediating Rho signal to reorganize the actin cytoskeleton. Indeed, it is known that ROCK regulates several steps of actin dynamics. ROCK phosphorylates the regulatory subunit of myosin light-chain phosphatase and inhibits its activity, thereby increasing the level of phosphorylated myosin light chain and inducing actomyosin-based contraction seen in smooth muscle contraction and neurite retraction (7). ROCK also phosphorylates and activates LIM kinase. Activated LIM kinase in turn phosphorylates an actin-binding protein, coffilin/actin-depolymerizing factor (ADF), and inactivates its actin depolymerizing and severing activity, which leads to inhibition of actin filament disassembly (18). These ROCK actions on actin dynamics combined together contribute to Rho-induced actomyosin organization seen in stress fiber formation.
We previously introduced a synthetic compound, Y-27632, as a specific ROCK inhibitor (34). Since our report, this compound has been used extensively to examine not only the actions of ROCK in cultured cells but also its roles in vivo in the body. While these studies have provided significant insights into the role of ROCK in several pathological processes, such as hypertension and tumor invasion (14, 34), the involvement and significance of the ROCK-mediated pathway in development and homeostasis of the body are unknown, because Y-27632 is a relatively short-acting drug. There are also several Y-27632-sensitive and supposedly ROCK-mediated processes, such as regulation of specific genes, which appear not to be mediated through ROCK actions on actin dynamics and have to be confirmed and assessed by means other than the use of Y-27632 (36). Furthermore, because ROCK consists of two isoforms, ROCK-I and -II (12, 17, 19, 22) and Y-27632 suppresses the activity of both isoforms, the specific function of an individual isoform could not be elucidated by the use of this compound.
The mammalian placenta is the organ in which respiratory gases, nutrients, and wastes are exchanged between the maternal and fetal circulation systems. This transplacental exchange provides all the metabolic demands for fetal growth and development. Because the rate of this exchange depends primarily on blood flow, decreased blood flow in the maternal or fetal side can induce intrauterine growth retardation (IUGR) of the embryo (26). The site of the transplacental exchange is the labyrinth layer of the placenta, in which the maternal blood flow is countercurrent to the fetal capillary flow through the trophoblast cell layer. The maternal blood flows into the base of the labyrinth through a few straight canals, perfuses a dense network of sinusoids, and percolates back to the maternal side through a number of smaller canals (2). A unique feature of this maternal circulation is that, distinct from blood vessels throughout the body, the passage is not lined by endothelial cells but by specialized trophoblasts. How the homeostasis of this maternal blood circulation is maintained without endothelium is poorly understood.
To explore the specific function of mouse ROCK-II (mROCK-II), we generated ROCK-II-deficient mice by knocking in a lacZ reporter gene. Most ROCK-II-deficient mice die in utero with unusually large thrombi in the labyrinth layer of the placenta, and the survivors were born runts. This phenotype is strongly reminiscent of human IUGR. Using a whole-mount staining technique for the mROCK-II-lacZ reporter gene, we observed that ROCK-II is strongly expressed in the labyrinth layer of the placenta. Interestingly, while we found strong F-actin architecture in the labyrinth layer, there was no significant difference in the F-actin structure in the tissues of wild-type and ROCK-II−/− mice. Analysis of gene expression in the placenta revealed elevated expression of plasminogen activator inhibitor 1 (PAI-1) in ROCK-II−/− placenta, suggesting decreased fibrinolytic activity (4) that might be the primary cause of large thrombus formation in the labyrinth layer, placental dysfunction, IUGR, and fetal death of ROCK-II−/− mice.
MATERIALS AND METHODS
Generation of the targeting vector.
The multicloning site of pBluescript SK+ (Stratagene) was modified to contain restriction enzyme sites as follows: SacI-SacII-NotI-XbaI-KpnI-EcoRV-BamHI-SalI-PstI-NheI-AscI-AatII-ClaI-SalI-NcoI-AscI (pBSKV4). A 129/SvJ lambda FIXII genomic library (Stratagene) was used, and several clones containing various parts of the mROCK-II gene were isolated. The targeting vector was constructed as follows. A 2.4-kb fragment containing 34 bp of the third exon (defined in the National Center for Biotechnology Information mouse genome sequencing database), encoding part of the kinase domain and its 5′ flanking region, was generated by PCR amplification with 5′-GGG GGA TAT CTA CTC ACA TCT GAA GAT TTG G-3′ as a forward primer and 5′-GGG GAT CGA TCT GTA AAC CTC TGA TTT TTT TCA-3′ as a reverse primer. The fragment was digested with EcoRV and ClaI and subcloned into pBSKV4. The plasmid DNA was then cut open with KpnI and EcoRV, and a 0.5-kb KpnI/EcoRV 5′ fragment obtained from partial digestion of mROCK-II DNA was introduced. The resulting vector was digested with KpnI, and a 7-kb KpnI fragment of mROCK-II DNA was then introduced to generate a long arm (9.9 kb) that extends from a 5′ KpnI site to 34 bp 3′ downstream to the splicing acceptor of exon 3 of mROCK-II (Fig. 1A). This long arm was then fused with the ClaI/SalI fragment containing the β-galactosidase gene. The resulting plasmid was opened by digestion with SalI and NcoI, and the SalI/NcoI fragment containing the phosphoglycerate kinase (PGK)-thymidine kinase (TK) cassette was inserted. A 1.1-kb short arm corresponding to the 3′ sequence downstream of exon 3 was amplified by PCR with 5′-CCC CGG ATC CAT CAT AGC AGC TGA ATA GTT C-3′ as a forward primer and 5′-CCC CCC CGG GCA ACC CAT AGG GAG TAC TC-3′ as a reverse primer and inserted into a PGK-Neo cassette, which was then digested with XhoI and SmaI. The fragment was then inserted into the above vector to generate the final targeting construct.
FIG. 1.

Generation of ROCK-II-deficient mice. (A) Targeting strategy. Schematic representations of the domain structure of ROCK-II, the wild-type ROCK-II allele, targeting vector, and the targeted allele are shown. The positions of β-galactosidase (β-gal), neomycin resistance (Neo), and thymidine kinase (TK) genes, restriction sites KpnI (K), HindIII (H), PstI (P), EcoRV (E), BamHI (B), ClaI (C), and NotI (N), and exons 2 (ex2) and 3 (ex3) are shown. The positions of primers for PCR analysis are indicated by the arrowheads. The external probe is a unique 3′ genomic probe that distinguishes the wild-type 9.7-kb PstI fragment from a 5-kb PstI fragment generated by the targeted alleles. (B) Southern blot analysis of genomic DNA obtained from visceral yolk sacs at 13.5 dpc. (C) Genotyping by PCR on genomic DNA obtained from visceral yolk sacs at 13.5 dpc.
Genotyping.
Genotyping was performed by Southern blot analysis or PCR, using genomic DNA prepared from embryonic stem (ES) cells, embryonic visceral yolk sacs, or the tails of mice (Fig. 1A). PCR primers were 5′-CAG AGG TTT ACA GAT GAA AGC GGA AG-3′ (a forward primer for wild-type allele), 5′-GCT CCA GAC TGC CTT GGG AAA AGC-3′ (a forward primer for the targeted allele), and 5′-CTG TAA TCC AGC ACC TGT GAA GTG G-3′ (a reverse primer for the wild-type and targeted alleles).
Generation of ROCK-II-deficient mice.
The targeting vector linearized with NotI was electroporated into ES cells, line RW4 (Genome System). Two hundred G418-resistant clones were screened, and homologous recombination was verified in two clones by PCR genotyping. Cells of the positive clones were injected into blastocysts, and the chimeric offspring obtained were mated to C57BL/6J females. ROCK-II+/− heterozygous mice were then intercrossed to produce homozygous mutants (ROCK-II−/−). Mice were treated according to the guidelines for protection of experimental animals of Kyoto University, and all experimental procedures were approved by the Committee on Animal Research of the Kyoto University Faculty of Medicine.
Western blot analysis.
Whole embryos 13.5 days postcoitum (dpc) or whole brains of adult mice were lysed in lysis buffer containing 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% Nonidet P-40 (NP-40), 0.5% deoxycholate, and protease inhibitors (Complete tablet; Roche Diagnostics). After the protein concentrations were determined, one-third volume of 4× Laemmli buffer was added to the mixture, which was then boiled for 5 min. The lysates were subjected to sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis under reducing conditions. Separated proteins were transferred to nitrocellulose membranes and probed with rabbit anti-ROCK-II antibody raised against amino acids 775 to 860 of human ROCK-II (H-85; Santa Cruz Biotechnology), rabbit anti-ROCK-I antiserum (12), or mouse anti-β-tubulin antibody (Sigma).
Whole-mount β-galactosidase staining.
ROCK-II−/−, ROCK-II+/−, and wild-type embryos and placentas were isolated at 13.5 dpc. The samples were fixed for 30 min at 4°C with 2% paraformaldehyde in phosphate-buffered saline (PBS) containing 2 mM MgCl2, 5 mM EGTA (pH 8.0), and 0.02% NP-40. The samples were washed and stained for 16 h at 30°C for β-galactosidase in 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) solution [1-mg/ml X-Gal solution containing 5 mM K3Fe(CN)6, 5 mM K4Fe(CN)6, 2 mM MgCl2, 0.01% SDS, and 0.02% NP-40].
Histology.
Three adult ROCK-II−/− mice, one male and two females, were sacrificed, and various organs were isolated. For paraffin sections, the isolated organs, embryo, and placenta were fixed in 4% paraformaldehyde. They were then embedded in paraffin and cut into 7-μm-thick sections. Hematoxylin and eosin staining was performed using a standard protocol. Immunohistochemistry with anti-platelet-endothelial cell adhesion molecule (PECAM) antibody (MEC13.3; Pharmingen) was performed on the paraffin-wax sections as previously described (11). To stain F-actin in tissue, placentas were fixed in 2% paraformaldehyde at 4°C for 2 h and immersed in 20% sucrose for 2 h and then in PBS containing 25% sucrose overnight at 4°C. The placentas were then frozen in Tissue-Tek OCT compound in liquid nitrogen and were cut into serial 10-μm-thick sections using a cryostat. The sections were incubated with Texas red phalloidin (Molecular Probes) for 10 min at room temperature. After the sections were washed with PBS, they were examined with a Zeiss LSM 510 confocal imaging system.
Primary culture of trophoblasts from the labyrinth layer.
Cells were prepared from the labyrinth layer of the placenta at 13.5 dpc as described previously (31) with modifications. Briefly, embryos with their encapsulating decidual tissue were removed from the uterus and washed with PBS. The placenta was dissected free from decidual tissue and yolk sac and umbilical vessels, and the labyrinth layer was isolated and cut into small pieces. The tissue pieces were then treated with 0.025% trypsin and 0.5 mg of DNase I per ml in PBS for 10 min at 37°C. The pieces were gently triturated 10 times using a Pasteur pipette. Digestion was terminated by adding Dulbecco modified Eagle medium (DMEM) containing 10% heat-inactivated fetal calf serum (FCS). The mixture was then filtered and centrifuged at 100 × g for 10 min at room temperature. Cells were suspended in DMEM containing 10% FCS and plated onto a noncoated or fibronectin-coated cover glass in 35-mm-diameter dishes. After 24 h of culture, cells were washed once with PBS and fixed with 3.7% paraformaldehyde for 15 min at room temperature. The cells were then permeabilized with PBS containing 0.1% Triton X-100 for 5 min and stained for F-actin with Texas red phalloidin (Molecular Probes). Optical images were obtained with a confocal imaging system (Bio-Rad Laboratories MRC1024 or Zeiss LSM510 confocal imaging system).
DNA microarray analysis.
The placenta was excised from ROCK-II+/− and ROCK-II−/− dpc 13.5 embryos of the same litter. Total RNA was prepared by using RNeasy kit (Qiagen). Affymetrix GeneChip expression analysis using a MG-U74Av2 chip (Affymetrix Inc., Santa Clara, Calif.) was performed according to the manufacturer's protocol. Data were analyzed with the Affymetrix GeneChip Expression Analysis software (version 4.0).
RT-PCR analysis.
Total RNA (10 ng) prepared from placentas of ROCK-II+/− and ROCK-II−/− dpc 13.5 embryos was used as templates. Reverse transcription-PCR (RT-PCR) was performed by using the Superscript One-Step RT-PCR system (Invitrogen). The primers used to detect PAI-1 expression were 5′-ATG AGA TCA GTA CTG CGG ATG CCA TCT TTG-3′ (sense primer) and 5′-GCA CAG AGA CGG TGC TGC CAT CAG ACT TGT-3′ (antisense primer). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH), a housekeeping gene, was used as a control. Reaction conditions were optimized to obtain amplification within the logarithmic phase of the reaction reproducibly.
Northern blot analysis.
For Northern blot analysis, total RNA from the placentas of ROCK-II+/− and ROCK-II−/− dpc 13.5 embryos was prepared by using a RNeasy kit (Qiagen). RNA (10 μg) was separated on a 0.7% agarose gel containing 2.1% formaldehyde and transferred to a Hybond-N+ membrane (Amersham Pharmacia Biotech). A 900-bp NcoI/HindIII fragment of the mouse PAI-1 cDNA (24) was used as a probe. GAPDH was used as a control. The probe was labeled with [α-32P]dCTP using Ready-To-Go (Amersham Pharmacia), and hybridization was done in ULTRAhyb (Ambion). The filter was washed twice for 5 min in 2× SSC-0.1% SDS (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) at 42°C and twice for 15 min in 0.1× SSC-0.1% SDS at 42°C and subjected to film autoradiography.
RESULTS
Generation and embryonic lethality of mROCK-II-deficient mice.
We disrupted the mROCK-II gene by replacing most of exon 3, which encodes part of the kinase domain, with genes for β-galactosidase and neomycin resistance (Fig. 1A). Mice with the chimeric mutant allele were generated and mated with C57BL/6 females to produce heterozygous ROCK-II+/− mice, which were then intercrossed. The genotypes of the offspring were identified by Southern blot analysis and PCR on DNA obtained from the visceral yolk sacs or tails of mice (Fig. 1B and C). Disruption of the mROCK-II gene was also confirmed by Western blot analysis for ROCK-II protein. A 160-kDa band of ROCK-II protein was detected in wild-type dpc 13.5 embryo lysates and in whole-brain lysates of adult mice (Fig. 2A). The amount of ROCK-II protein in ROCK-II+/− embryo lysates decreased to half the amount in wild-type dpc 13.5 embryo lysates, and ROCK-II protein was absent in ROCK-II−/− embryos (Fig. 2A). On the other hand, there was no difference in the level of ROCK-I protein in the embryonic lysates or whole-brain lysates (Fig. 2B), indicating that the amount of ROCK-I did not increase to compensate for the loss of ROCK-II.
FIG. 2.

Western blot analysis. Wild-type and mutant dpc 13.5 embryo lysates or wild-type and mutant whole adult brain lysates of ROCK-II+/+, ROCK-II+/−, and ROCK-II−/− mice were probed with antibodies specific for ROCK-II (A), ROCK-I (B), and β-tubulin (C), respectively. The positions of molecular mass markers (in kilodaltons) are shown to the right of the blots.
Analysis of the genotype distribution in offspring from heterozygous mating by chi-square (χ2) analysis revealed that the homozygous ROCK-II−/− mice at dpc 18.5 are significantly underrepresented (P < 0.05) from the value predicted by Mendelian inheritance. We found that only 7.7% of embryos at dpc 18.5 and only 5.3% of the animals born had a homozygous ROCK-II genotype (Table 1). These results indicate that most ROCK-II−/− embryos died during the late stage of pregnancy in utero. Furthermore, it is unlikely that there is a critical period for the embryo morbidity, as the number of homozygous embryos gradually decreased.
TABLE 1.
Genotypes of offspring obtained by crossing of ROCK-II mutant micea
| Cross and stage | Total no. of offspring | No. of mice with genotype:
|
No. resorbed | χ2 value | P value (df = 2) | ||
|---|---|---|---|---|---|---|---|
| +/+ | +/− | −/− | |||||
| ♂(+/−) × ♀ (+/−) | |||||||
| 10.5 dpc | 50 | 10 | 27 | 13 | 0 | 3.35 | 0.19 |
| 12.5 dpc | 21 | 6 | 11 | 4 | 0 | 0.75 | 0.69 |
| 13.5 dpc | 110 | 23 | 45 | 22 (1)b | 10 | 0.07 | 0.97 |
| 15.5 dpc | 73 | 18 | 35 | 10 (2) | 10 | 3.58 | 0.16 |
| 18.5 dpc | 51 | 13 | 25 (2) | 4 (1) | 9 | 6.27 | 0.04 |
| Adult | 122 | 38 | 76 | 8 (2) | NAc | 23.68 | 0.00 |
| ♂ (−/−) × ♀ (+/−) | |||||||
| Adult | 24 | 0 | 23 | 1 | NA | 9.59 | 0.01 |
| ♂ (+/−) × ♀ (−/−) | |||||||
| Adult | 12 | 0 | 12 | 0 | NA | 6.00 | 0.05 |
The genotypes of the wild-type (ROCK-II+/+), ROCK-II+/−, and ROCK-II−/− mice are shown as +/+, +/−, and −/−, respectively.
The number of dead animals is shown in parentheses.
NA, not applicable.
Growth retardation was observed in the embryos that survived the late stage of pregnancy (Fig. 3A), and the ROCK-II−/− mice that survived were always runts. Most of the surviving ROCK-II−/− mice subsequently developed normally and were apparently healthy. One mouse lived 11 months, and two males and two females tested were fertile (Table 1). However, two ROCK-II−/− mice that displayed severe growth retardation (were about 40 to 50% the size of the control mice) died after 3 to 4 weeks. Three adult mROCK-II−/− mice were sacrificed and examined for gross and histological abnormalities. Examination of tissue sections stained with hematoxylin and eosin revealed no obvious abnormalities in the brain and spinal cord, the heart and aorta, the lung and trachea, the kidney and bladder, endocrine organs (such as the thyroid, pituitary, and adrenal glands), hematopoietic and lymphoid organs (such as the bone marrow, spleen, thymus, and mesenteric lymph nodes), the gastrointestinal tract from the tongue to the rectum, the reproductive organs (such as testis, epididymis, seminal vesicle, prostate gland, ovary, and uterus), and the epidermis and dermis of the skin (data not shown).
FIG. 3.

Growth retardation of ROCK-II−/− mice. Wild-type (ROCK-II+/+) and ROCK-II−/− dpc 18.5 embryos (A) and neonates (B) are shown. Note that the ROCK-II−/− mouse was a runt. Hematomas in the hind limbs of the ROCK-II−/− mouse are indicated by arrows.
High expression of mROCK-II in the labyrinth layer of the placenta and disruption of its architecture in mROCK-II−/− mice.
Because a lacZ reporter gene was knocked in frame with the initiator methionine of mROCK-II in the targeting construct, we examined the pattern of expression of the mROCK-II gene by whole-mount β-galactosidase staining. As shown in a 13.5 dpc ROCK-II+/− embryo, X-Gal staining was observed in many locations throughout the embryo, including the heart, dorsal root ganglions, and umbilical blood vessels (Fig. 4A). Spotted staining was also observed in the liver of the embryo. In the placenta, strong X-Gal staining was observed in the labyrinth layer (Fig. 4B and C). Detailed inspection revealed that trophoblasts in the labyrinth layer were uniformly stained (Fig. 4D). In addition, a blood vessel in the chorionic plate, apparently the umbilical artery, was positively stained (Fig. 4E). In all tissues examined thus far, the patterns of X-Gal staining of ROCK-II+/− and ROCK-II−/− mice were identical or highly similar (data not shown).
FIG. 4.

ROCK-II expression in 13.5-dpc embryo and placenta as revealed by X-Gal staining of the lacZ reporter gene. (A) Expression in the embryo at 13.5 dpc. X-Gal staining is detected in umbilical blood vessels, heart, and dorsal root ganglion. (B and C) Expression in the placenta. Strong X-Gal staining is found in the labyrinth layer of ROCK-II−/− placenta (B), which is rich in vasculature as shown in ROCK-II+/+ placenta (C). (D) Uniform X-Gal staining in trophoblasts in the labyrinth layer. (E) Expression of the ROCK-II-lacZ reporter gene in a blood vessel in the chorionic plate. Parallel staining for actin (data not shown) indicates that the vessel stained with X-Gal (below) is the umbilical artery, while the vessel devoid of the X-Gal staining (above) is a vein. Bars, 100 μm.
The above findings that most ROCK-II−/− mice die in utero and the homozygotes that survived were born runts but subsequently caught up in growth indicated a defect(s) in the interaction of the embryo and placenta in ROCK-II−/− mice. To clarify this issue, a litter of placentas and embryos were isolated at 10.5 to 14.5 dpc from the uterus of a heterozygous mother mated with a heterozygous male and examined. No significant abnormality was observed in eight ROCK-II−/− placentas prior to 11.5 dpc (data not shown). However, as shown in Fig. 5A and B, a number of large blood clots were typically visible in the placentas of ROCK-II−/− mice from 12.5 dpc on, but not in those of wild-type littermates. Approximately 80% of ROCK-II−/− placentas showed this abnormality to various extents. Histological examination of the placentas of ROCK-II−/− mice at 12.5 and 13.5 dpc revealed that multiple large thrombi were present in the labyrinth layer, increased in size and number with time, and disrupted its normal architecture (Fig. 5C to F). In some cases, approximately 10% of the total volume of the labyrinth layer was occupied by thrombi. Small thrombi occasionally formed in the placentas of wild-type and heterozygous littermates (Fig. 5C and data not shown) and grew. However, the prevalence and size of thrombi in the ROCK-II−/− placenta were unusual. It is evident that these thrombi were formed in the labyrinth layer of the maternal circulation system, because they contained enucleated erythrocytes and were not in the fetal circulation system lined by the endothelium (Fig. 5G).
FIG. 5.

Morphology and histology of wild-type (ROCK-II+/+) and ROCK-II−/− embryos and placentas. (A and B) Embryos isolated together with their placentas. Blood clots found in the ROCK-II−/− placenta (arrows) and hemorrhage in the hind limb of the ROCK-II−/− embryo (arrowhead) are indicated. (C to F) Hematoxylin and eosin staining of a 12.5-dpc wild-type placenta (C), a 12.5-dpc ROCK-II−/− placenta (D), and a 13.5-dpc ROCK-II−/− placenta (E). Panel F shows a higher-magnification view of the labyrinth layer shown in panel E. (G) Immunohistochemical staining of the labyrinth layer of a ROCK-II−/− placenta with an anti-PECAM antibody. The arrow indicates a blood clot found in this section. The absence of nucleated blood cells and PECAM1-positive endothelial cells around the clot indicates that the clot had a maternal origin. Bars, 500 μm (C, D, and E), 300 μm (F), and 200 μm (G).
No overt abnormalities in actin fiber structure in the labyrinth layer and in labyrinth layer-derived cultured trophoblasts of ROCK-II−/− mice.
The above results suggested that loss of ROCK-II induced the thrombogenic tendency locally in the labyrinth layer of ROCK-II−/− mice. Because ROCK mediates a Rho signal to organize the actin cytoskeleton involved in cell-substrate adhesion and ROCK-II is abundantly expressed in trophoblasts in the labyrinth layer (Fig. 4B), we first suspected that loss of ROCK-II might affect the cytoarchitecture of trophoblasts and tear them off under the stress of the shearing force. To examine this issue, we first compared the architecture of actin in the labyrinth layer in wild-type and ROCK-II−/− mice. We also cultured trophoblasts from the labyrinth layers of wild-type and ROCK-II−/− mice and compared their actin cytoskeleton.
For an accurate comparison, we placed tissue sections from mice with the two genotypes on the same glass slide and performed staining and microscopic observation of the two simultaneously. We also stained and examined the cell cultures from the two genotypes side by side. As shown in Fig. 6A, a strong and complex network of actin fibers was found throughout the labyrinth layer. However, no significant difference was observed in both the architecture of this network and the strength of actin staining in tissue from wild-type and ROCK-II−/− placentas. Strong actin bundles reminiscent of stress fibers induced by dominant active ROCK (13) were consistently observed in cultured trophoblasts, and there was no difference between ROCK-II+/+- and ROCK-II−/−-derived trophoblasts cultured on a noncoated cover glass (Fig. 6B and C, left panels) or on a fibronectin-coated cover glass (data not shown). We further found that treatment with 10 μM Y-27632 significantly changed the structure of the actin bundles in the trophoblasts obtained from the labyrinth layers of both wild-type and ROCK-II−/− mice. These results suggest that actin bundles observed in the ROCK-II−/− labyrinth layer and cultured trophoblasts are formed by the action of ROCK-I (Fig. 6B and C, right panels).
FIG. 6.

Architecture of actin fibers in the labyrinth layer and cultured trophoblasts from the placentas of wild-type (ROCK-II+/+) and ROCK-II−/− mice. (A) Cytoarchitecture of actin fibers in the labyrinth layer of wild-type and ROCK-II−/− placentas. (B and C) Actin structures in clusters of cultured trophoblasts from wild-type (B) and ROCK-II−/− (C) placentas. Cells were cultured in the absence or presence of 10 μM Y-27632. Note that the structure of actin bundles is not different in the wild-type and ROCK-II−/− cells, and the structures of the actin bundles in the wild-type and ROCK-II−/− cells treated with Y-27632 were similar. Bars, 20 μm (A) and 50 μm (B and C).
Enhanced expression of PAI-1 in the placenta in mROCK-II−/− mice.
To investigate the molecular mechanism underlying thrombus formation and placental dysfunction in mROCK-II−/− mice, we next performed DNA microarray analysis to analyze the alterations in gene expression in the absence of ROCK-II. Placentas were isolated from ROCK-II+/− and ROCK-II−/− mice in the same litter at 13.5 dpc and subjected to analysis. We tested about 10,000 genetic loci that have been identified as clusters by the UNIGENE database. Exclusion of expressed sequence tags and genes with low expression (∼130 to 180 gene clusters) left only a limited number of genes, whose expression in the placenta of ROCK-II−/− mice was different from that of wild-type mice. Table 2 shows three genes with enhanced expression and two genes with decreased expression in the placenta of ROCK-II−/− mice. The three genes with enhanced expression are the prolactin-like protein G precursor, placental lactogen 1, and PAI-1 genes. One of the two genes showing decreased expression in ROCK-II−/− placentas was mROCK-II itself. Notably, there was no significant difference in mROCK-I expression in ROCK-II−/− and ROCK-II+/− placentas. We further confirmed the elevated expression of PAI-1 by RT-PCR and Northern blotting (Fig. 7). mRNA expression of PAI-1 was significantly higher in all the ROCK-II−/− placentas analyzed (n = 5 for RT-PCR and n = 4 for Northern blotting) than in the heterozygous littermates (Fig. 7). Thus, the loss of ROCK-II resulted in increased expression of PAI-1 in the placenta.
TABLE 2.
Gene expression change in ROCK-II−/− placenta analyzed by DNA microarraya
| Gene category and GenBank accession no. | Description | Absolute difference | Fold change |
|---|---|---|---|
| Genes with increased expression in ROCK-II−/− placenta | |||
| AF020525 | Prolactin-like protein G precursor | +1,098 | +2.3 |
| M35662 | Placental lactogen 1 | +204 | +2.1 |
| M33960 | PAI-1 | +748 | +1.7 |
| Genes with decreased expression in ROCK-II−/− placenta | |||
| AJ007909 | Erythroid differentiation regulator | −1,849 | −2.9 |
| U58513 | mROCK-II | −177 | −2.8 |
| Gene with no difference in expression in ROCK-II−/− placenta | |||
| U58512 | mROCK-I | +22 | 1.0 |
The placenta was excised from 13.5-dpc ROCK-II+/− and ROCK-II−/− embryos of the same litter (two embryos for each group). Gene expression of each ROCK-II−/− placenta was compared with that in two ROCK-II+/− placentas, and genes showing a consistent increase or decrease in four combinations of comparison were chosen. The average differences and changes from four comparisons are shown.
FIG. 7.
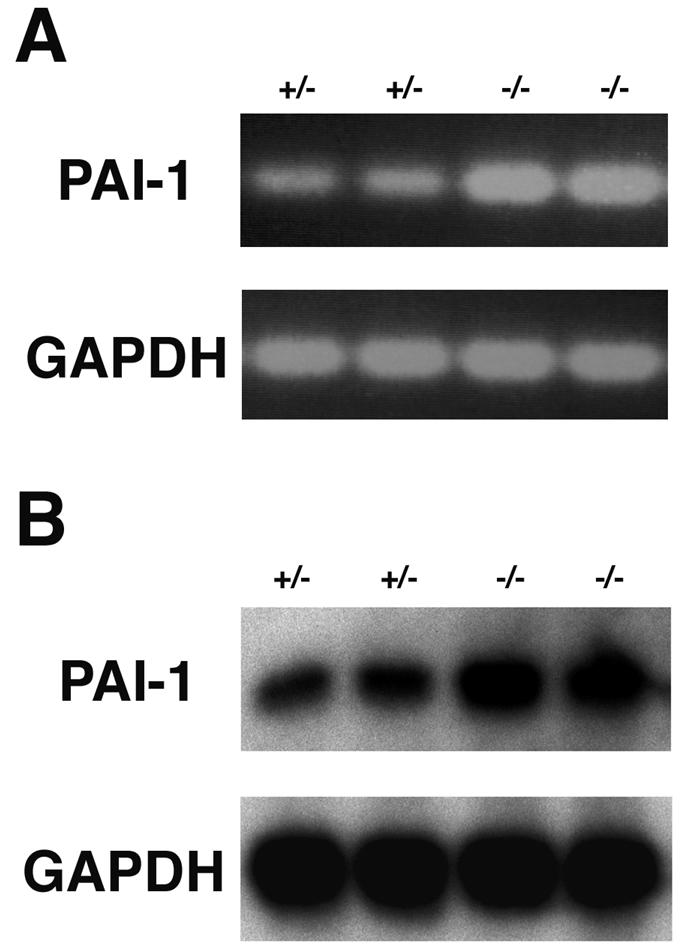
Elevated expression of PAI-1 in ROCK-II−/− placenta. (A) RT-PCR analysis of PAI-1 expression in ROCK-II+/− and ROCK-II−/− 13.5-dpc placentas. (B) Northern blot analysis of PAI-1 mRNA expression in mROCK-II+/− and mROCK-II−/− 13.5-dpc placentas.
Hemorrhage in the hind limbs of ROCK-II−/− mice.
Another feature consistently observed in ROCK-II−/− embryos was hemorrhage in the hind limb bud. Hemorrhage was observed in the tip regions of both hind limb buds in embryos as early as 12.5 dpc, accompanied by swelling of the limb, and persisted until birth (Fig. 3, 4A, 5A and B, and 8A and B). Histological analysis revealed that small capillaries at the tip of the hind limb became significantly dilated at 12.5 dpc and eventually ruptured, causing a hemorrhage at 13.5 dpc (Fig. 8C to E). The bleeding resolved in 3 to 4 days after birth with, in some cases, a deformity left in the feet of adult mice (Fig. 8B). Hemorrhages were also observed in the tip of the tails of ROCK-II−/− embryos but at a lower frequency (2 of 15 dpc 13.5 ROCK-II−/− embryos) (Fig. 8A).
FIG. 8.

Hemorrhage in the hind limb of a ROCK-II−/− mouse. (A) Hemorrhage in a 13.5 dpc ROCK-II−/− embryo. Hemorrhages in both hind limbs and at the tip of the tail are indicated by the arrows. (B) Deformity of the hind limb of a ROCK-II−/− mouse. (C to E) Hematoxylin-and-eosin-stained sections of the hind limb bud from a 12.5-dpc wild-type (ROCK-II+/+) embryo (C) and 12.5-dpc (D) and 13.5-dpc (E) ROCK-II−/− homozygous embryos. Small capillaries in the hind limbs of wild-type and knockout mice are indicated by the arrows. In panels C, D, and E, lower-magnification (left) and higher-magnification (right) views are shown to the right. Bars, 300 μm (left micrographs of panels C, D, and E) and 100 μm (right micrographs of panels C, D, and E).
DISCUSSION
In this study, we disrupted the gene for mROCK-II, one of the two isoforms of ROCK, and found that most ROCK-II-deficient homozygous mice died in utero. A small number of ROCK-II−/− mice that survived were born runts. Although two ROCK-II−/− mice that displayed severe growth retardation died within 4 weeks, most subsequently developed apparently normally. These results indicated that the embryonic lethality found in these mice was caused by a defect(s) not in the embryo per se but in the embryo-placenta interaction. We could place this defect in the labyrinth layer of the placenta. First, using the expression of the mROCK-II-lacZ reporter gene, we found that trophoblasts in this layer highly expressed ROCK-II. We further found that marked thrombosis and the progressive loss of trophoblasts occurred in the labyrinth layer of ROCK-II−/− mice but not in that of wild-type or ROCK-II+/− heterozygous mice.
The question then is how the loss of ROCK-II induces this pathology of the labyrinth layer. Defects in the development of the labyrinth layer and resultant embryonic death have been found in a number of strains of genetically engineered mice (10). However, no histological abnormality was observed in ROCK-II−/− placentas before 12.5 dpc. The phenotype of ROCK-II−/− mice is therefore different from the labyrinth dysplasia found in strains with mutations in the genes encoding fibroblast growth factor receptor 2 (38), hepatocyte growth factor (33), adapter proteins Grb2 (27) and Gab1 (15), Sos1 Ras exchanger (25), Mek1 (8), p38α (1), and Mekk3 (40) and suggests that ROCK-II is not involved in the initial development of the placenta (35). The placenta proper containing the labyrinth layer is fully formed and functional by 12 dpc, and the volume of the labyrinth layer increases to about half of the total placental volume between 12 and 17 dpc (2). During this gestation period, the perfusion pressure of the maternal blood flowing in the labyrinth layer rises and then remains high. Both the volume expansion and the increase in perfusion pressure are necessary to support the drastic growth of the embryo. Because failure of ROCK-II−/− embryos occurs in this period, it is likely that ROCK-II works at this stage of maturation and adaptation of the labyrinth layer. Several adaptive responses could be required. One may be resistance of the labyrinth structure to the shearing force of the blood flow, and the other may be protection from coagulation and thrombosis. The labyrinth sinusoid where maternal blood flows is lined not by endothelium but by special types of trophoblasts (2). ROCK-II is probably involved in such adaptive changes in the trophoblasts in response to blood flow, because they highly express ROCK-II (Fig. 4B and D).
We examined possible defects in the adaptive responses of trophoblasts in the placentas of ROCK-II−/− mice in two ways. First, we examined the possibility that loss of mROCK-II affected the cytoarchitecture of the labyrinth layer by staining F-actin both in tissues and in cultured trophoblasts. No significant differences in the structure of actin cytoskeleton and the strength of phalloidin staining were found in mutant and wild-type tissues and cells. The actin bundling structure in cultured ROCK-II−/− trophoblasts was also sensitive to Y-27632, suggesting that ROCK-I maintains the actin structure in the labyrinth layer in the absence of ROCK-II. However, this does not necessarily exclude the possibility that both ROCK-I and -II are required for proper function of actin structures (e.g., to maintain tissue integrity or to exert contractility in response to stimuli). This issue remains to be investigated. Second, we performed a microarray analysis and examined possible changes of gene expression in the placentas of ROCK-II−/− mice. By this analysis, we detected higher levels of expression of at least three genes, including the prolactin-like protein G and PAI-1 genes (Table 2). We further confirmed the elevated expression of PAI-1 both by RT-PCR and Northern blotting (Fig. 7). Because PAI-1 inhibits fibrinolytic activity of tissue plasminogen activator (4), elevation of PAI-1 expression could very well lead to an enhanced coagulation tendency (29, 39). Indeed, elevated expression of PAI-1 in the placenta has been reported in patients with gestational trophoblastic disease and is proposed to contribute to the hemostatic problems associated with this disorder (6). In addition, an increase in PAI-1 activity of a few-fold could be responsible for the thrombogenic tendency in experimental animals (30). These results therefore suggest that the thrombus formation in the labyrinth layer of the ROCK-II−/− placenta is caused at least partly by enhanced expression of PAI-1. In addition to PAI-1, increased expression of prolactin-like protein G may also lead to the thrombotic tendency. This protein belongs to the family of prolactin-like proteins and is most homologous to prolactin-like proteins E and F (21). While the activity of prolactin-like protein G remains to be tested, prolactin-like proteins E and F are known to stimulate proliferation and differentiation of megakaryocytes via the specific receptor-gp130 pathway to increase the number of platelets in the circulation system (41). It is therefore likely that elevated expression of these genes enhanced the thrombogenic tendency in the ROCK-II−/− placenta. At present, we do not know whether the expression of these genes is a direct consequence of the loss of ROCK-II or is triggered by unknown primary events directly influenced by the loss of this enzyme. In contrast to the findings of this study, previous experiments using Y-27632 in cultured smooth muscle cells and an experimental animal suggest that ROCK mediates the angiotensin-induced PAI-1 expression (16, 32). Whether the PAI-1 expression is a direct or indirect consequence, elevated expression of this gene probably contributes to the induction of the phenotype of ROCK-II−/− mice, given the potent thrombogenic action of this substance.
Elevated expression of PAI-1 in the placentas of ROCK-II−/− mice may also explain the hemorrhages observed in ROCK-II−/− embryos. These hemorrhages, apparently caused by dilation of peripheral capillaries, were found in the hind limbs and the tip of the tails of the embryos, resolved, and did not recur in adult mice. Interestingly, a similar but more severe phenotype was observed in PAI-1 transgenic mice (5), which showed subcutaneous hemorrhage at the tip of the tail and edema of the hind paw. The researchers attributed these symptoms to downstream venous occlusion. Given the elevated expression of PAI-1 in ROCK-II−/− placenta, we examined PAI-1 expression by RT-PCR in the whole embryo at dpc 13.5. However, we could not detect an amplified product (data not shown), suggesting that PAI-1 is not expressed or expressed at a very low level in organs other than the placenta at this embryo stage. It is therefore plausible that PAI-1 generated in the placenta was transported to the fetal circulation system and caused occlusion of particular veins in ROCK-II−/− mice. Such PAI-1 action, if present, may explain the transient nature of the hemorrhage.
Rho and ROCK work downstream of extracellular signaling, and a number of molecules and conditions induce activation of Rho. The question then is what kinds of molecules and conditions induce Rho activation in the labyrinth layer. One of the signaling pathways activating Rho and ROCK is the Wnt-Frizzled pathway (37). It is interesting that mice deficient in Wnt2 showed a placental dysfunction (20) similar to those we found in ROCK-II−/− mice. Wnt2−/− mice were born runts and exhibited an embryonic lethality of 50%. Histological examination showed that about 85% of Wnt2−/− placentas showed defects in the labyrinth layer, including accumulation of abnormally large hematomas and fibrinoid materials. These findings suggest a possibility that ROCK-II works in the Wnt2 pathway in maturation and adaptation of the labyrinth layer in the late stage of gestation.
We thus identified malfunction of the labyrinth layer as the cause of embryonic lethality and growth retardation in ROCK-II−/− mice. While the labyrinth layer malfunction is a major phenotype of ROCK-II deficiency, we do not know whether this phenotype is induced by suppression of actions specific for ROCK-II or suppression to some extent of the combined actions of ROCK-I and -II. Besides this intrauterine phenotype, most ROCK-II−/− mice born alive developed and grew without obvious anatomical and functional abnormalities. This is rather surprising given the critical functions ROCK exerts in organization of the actin cytoskeleton in various types of cells and the different, albeit partially overlapping, patterns of tissue expression of the two isoforms. This is more surprising in the light of the finding that ROCK-I did not increase to compensate for the loss of ROCK-II at least in the embryo, placenta, and brain of adult mice (Fig. 2 and Table 2). It is of course possible that there are abnormalities in ROCK-II-deficient mice, which can be revealed only by detailed functional and histological analyses. This issue will be examined by future studies. The ROCK-II−/− mouse phenotype, including enhanced PAI-1 expression, is strongly reminiscent of human IUGR, for which thrombosis in the placenta is proposed as a major cause (9, 23). IUGR comprises a significant fraction of prenatal morbidity and low-birth-weight babies (29). Understanding molecular mechanisms of the failure of ROCK-II−/− placenta in more detail may provide insight into the physiology and pathophysiology of IUGR and its treatment.
Acknowledgments
This work was supported in part by a grant-in-aid for specially promoted research from the Ministry of Education, Culture, Sports, Science and Technology of Japan, a grant from the Organization for Pharmaceutical Safety and Research, and a grant from Mitsubishi Pharma Corporation. D.T. is a recipient of a Japanese Government Scholarship for Foreign Student.
We thank M. Cole for mouse PAI-1 cDNA; H. Bito and N. Watanabe for critical reading of the manuscript; Y. Arakawa, E. Segi, and T. Tsuji for helpful discussions; Y. Che for technical assistance, and T. Arai and H. Nose for secretarial assistance. D.T. thanks S. Nakanishi for the opportunity to work in the Department of Pharmacology.
REFERENCES
- 1.Adams, R. H., A. Porras, G. Alonso, M. Jones, K. Vintersten, S. Panelli, A. Valladeres, L. Perez, R. Klein, and N. R. Nebreda. 2000. Essential role of p38alpha MAP kinase in placenta but not embryonic cardiovascular development. Mol. Cell 6:109-116. [PubMed] [Google Scholar]
- 2.Adamson, S. L., Y. Lu, K. J. Whitley, D. Holmyard, M. Hemberger, C. Pfarrer, and J. C. Cross. 2002. Interaction between trophoblast cells and the maternal and fetal circulation in the mouse placenta. Dev. Biol. 250:358-373. [DOI] [PubMed] [Google Scholar]
- 3.Bar-Sagi, D., and A. Hall. 2000. Ras and Rho GTPases: a family reunion. Cell 103:227-238. [DOI] [PubMed] [Google Scholar]
- 4.Collen, D. 1999. The plasminogen (fibrinolytic) system. Thromb. Haemostasis 82:259-270. [PubMed] [Google Scholar]
- 5.Erickson, L. A., J. F. Gregory, J. E. Lund, T. P. Boyle, H. G. Polites, and K. R. Marotti. 1990. Development of venous occlusions in mice transgenic for the plasminogen activator inhibitor-1 gene. Nature 346:74-76. [DOI] [PubMed] [Google Scholar]
- 6.Estelles, A., S. Grancha, J. Gilabert, T. Thinnes, M. Chirivella, F. Espana, J. Aznar, and D. J. Loskutoff. 1996. Abnormal expression of plasminogen activator inhibitors in patients with gestational trophoblastic disease. Am. J. Pathol. 149:1229-1239. [PMC free article] [PubMed] [Google Scholar]
- 7.Fukata, Y., M. Amano, and K. Kaibuchi. 2001. Rho-Rho-kinase pathway in smooth muscle contraction and cytoskeletal reorganization of non-muscle cells. Trends Pharmacol. Sci. 22:32-39. [DOI] [PubMed] [Google Scholar]
- 8.Giroux, S., M. Tremblay, D. Bernard, J. F. Cardin-Girard, S. Aubry, L. Larouche, S. Rousseau, J. Hout, J. Laundry, L. Jeannotte, and J. Charron. 1999. Embryonic death of Mek-1 deficient mice reveals a role of this kinase in angiogenesis in the labyrinth region of the placenta. Curr. Biol. 9:369-372. [DOI] [PubMed] [Google Scholar]
- 9.Greer, I. A. 1999. Thrombosis in pregnancy: maternal and fetal issues. Lancet 353:1258-1265. [DOI] [PubMed] [Google Scholar]
- 10.Hemberger, M., and J. C. Cross. 2001. Genes governing placental development. Trends Endocrinol. Metab. 12:162-168. [DOI] [PubMed] [Google Scholar]
- 11.Ishikawa, T., Y. Tamai, A. M. Zorn, H. Yoshida, M. F. Seldin, S. Nishikawa, and M. M. Taketo. 2001. Mouse Wnt receptor gene Fzd5 is essential for yolk sac and placental angiogenesis. Development 128:25-33. [DOI] [PubMed] [Google Scholar]
- 12.Ishizaki, T., M. Maekawa, K. Fujisawa, K. Okawa, A. Iwamatsu, A. Fujita, N. Watanabe, Y. Saito, A. Kakizuka, N. Morii, and S. Narumiya. 1996. The small GTP-binding protein Rho binds to and activates a 160 kDa Ser/Thr protein kinase homologue to myotonic dystrophy kinase. EMBO J. 15:1885-1893. [PMC free article] [PubMed] [Google Scholar]
- 13.Ishizaki, T., M. Naito, K. Fujisawa, M. Maekawa, N. Watanabe, Y. Saito, and S. Narumiya. 1997. p160ROCK, Rho-associated coiled-coil forming protein kinase, works downstream of Rho and induces focal adhesions. FEBS Lett. 404:118-124. [DOI] [PubMed] [Google Scholar]
- 14.Itoh, K., K. Yoshioka, H. Akedo, M. Uehata, T. Ishizaki, and S. Narumiya. 1999. An essential part for Rho-associated kinase in the transcellular invasion of tumor cells. Nat. Med. 5:221-225. [DOI] [PubMed] [Google Scholar]
- 15.Itoh, M., Y. Yoshida, K. Nishida, M. Narimatsu, M. Hibi, and T. Hirano. 2000. Role of Gab1 in heart, placenta, and skin development and growth factor- and cytokine-induced extracellular signal-regulated kinase mitogen-activated protein kinase activation. Mol. Cell. Biol. 20:3695-3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kobayashi, N., S. Nakano, S. Mita, T. Kobayashi, T. Honda, Y. Tsubokou, and H. Matsuoka. 2002. Involvement of Rho-kinase pathway for angiotensin II-induced plasminogen activator inhibitor-1 gene expression and cardiovascular remodeling in hypertensive rat. J. Pharmacol. Exp. Ther. 301:459-466. [DOI] [PubMed] [Google Scholar]
- 17.Leung, T., E. Manser, L. Tan, and L. Lim. 1995. A novel serine/threonine kinase binding the Ras-related RhoA GTPase which translocates the kinase to peripheral membranes. J. Biol. Chem. 270:29051-29054. [DOI] [PubMed] [Google Scholar]
- 18.Maekawa, M., T. Ishizaki, S. Boku, N. Watanabe, A. Fujita, A. Iwamatsu, T. Obinata, K. Ohashi, K. Mizuno, and S. Narumiya. 1999. Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinases. Science 285:895-898. [DOI] [PubMed] [Google Scholar]
- 19.Matsui, T., M. Amano, T. Yamamoto, K. Chihara, M. Nakafuku, M. Ito, T. Nakano, K. Okawa, A. Iwamatsu, and K. Kaibuchi. 1996. Rho-associated kinase, a novel serine/threonine kinase, as a putative target for small GTP binding protein Rho. EMBO J. 15:2208-2216. [PMC free article] [PubMed] [Google Scholar]
- 20.Monkley, S. J., S. J. Delaney, D. J. Pennisi, J. H. Christiansen, and B. J. Wainwright. 1996. Targeted disruption of the Wnt2 gene results in placentation defects. Development 122:3343-3353. [DOI] [PubMed] [Google Scholar]
- 21.Müller, H., K. E. Orwig, and M. J. Soares. 1998. Identification of two new members of the mouse prolactin gene family. Biochim. Biophys. Acta 1396:251-258. [DOI] [PubMed] [Google Scholar]
- 22.Nakagawa, O., K. Fujisawa, T. Ishizaki, Y. Saito, K. Nakao, and S. Narumiya. 1996. ROCK-I and ROCK-II, two isoforms of Rho-associated coiled-coil forming protein serine/threonine kinase in mice. FEBS Lett. 392:189-193. [DOI] [PubMed] [Google Scholar]
- 23.Peeters, L. L. H. 2001. Thrombophilia and fetal growth restriction. Eur. J. Obstet. Gynecol. 95:202-205. [DOI] [PubMed] [Google Scholar]
- 24.Prendergast, G. C., L. E. Diamond, D. Dahl, and M. D. Cole. 1990. The c-myc-regulated gene mr1 encodes plasminogen activator inhibitor 1. Mol. Cell. Biol. 10:1265-1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qian, X., L. Esteban, W. C. Vass, C. Upadhyaya, A. G. Papageorge, K. Yienger, J. M. Ward, D. R. Lowy, and E. Santos. 2000. The Sos1 and Sos2 ras-specific exchange factors: differences in placental expression and signaling properties. EMBO J. 19:642-654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reynolds, L. P., and D. A. Redmer. 2001. Angiogenesis in the placenta. Biol. Reprod. 64:1033-1040. [DOI] [PubMed] [Google Scholar]
- 27.Saxton, T. M., A. M. Cheng, S. H. Ong, Y. Lu, R. Sakai, J. C. Cross, and T. Pawson. 2001. Gene dosage dependent functions for mouse phosphotyrosine-Grb2 signaling during mammalian tissue morphogenesis. Curr. Biol. 11:662-670. [DOI] [PubMed] [Google Scholar]
- 28.Schmitz, A. A. P., E. Govek, B. Bottner, and L. Van Aelst. 2000. Rho GTPases: signaling, migration and invasion. Exp. Cell Res. 261:1-12. [DOI] [PubMed] [Google Scholar]
- 29.Schofield, D., and R. S. Cotran. 1999. Diseases of infancy and childhood, p. 459-491. In R. S. Cotran, V. Kumar, and T. Collins (ed.), Robbins pathologic basis of disease, 6th ed. W. B. Saunders Company, Philadelphia, Pa.
- 30.Shimomura, I., T. Funahashi, M. Takahashi, K. Maeda, K. Kotani, T. Nakamura, S. Yamashita, M. Miura, Y. Fukuda, K. Takemura, K. Tokunaga, and Y. Matsuzawa. 1996. Enhanced expression of PAI-1 in visceral fat: possible contributor to vascular disease in obesity. Nat. Med. 2:800-803. [DOI] [PubMed] [Google Scholar]
- 31.Soares, M. J., K. D. Schaberg, C. S. Pinal, S. K. De, P. Bhatia, and G. K. Andrews. 1987. Establishment of a rat placental cell line expressing characteristics of extraembryonic membranes. Dev. Biol. 124:134-144. [DOI] [PubMed] [Google Scholar]
- 32.Takeda, K., T. Ichiki, T. Tokunou, N. Iino, S. Fujii, A. Kitabatake, H. Shimokawa, and A. Takeshita. 2001. Critical role of Rho-kinase and MEK/ERK pathways for angiotensin II-induced plasminogen activator inhibitor type-1 gene expression. Arterioscler. Thromb. Vasc. Biol. 21:868-873. [DOI] [PubMed] [Google Scholar]
- 33.Uehara, Y., O. Minowa, C. Mori, K. Shiota, J. Kuno, T. Noda, and N. Kitamura. 1995. Placenta defect and embryonic lethality in mice lacking hepatocyte growth factor/scatter factor. Nature 373:702-705. [DOI] [PubMed] [Google Scholar]
- 34.Uehata, M., T. Ishizaki, H. Satoh, T. Ono, T. Kawahara, T. Morishita, H. Tamakawa, K. Yamagami, J. Inui, M. Maekawa, and S. Narumiya. 1997. Calcium sensitization of smooth muscle mediated by Rho-associated protein kinase in hypertension. Nature 389:990-994. [DOI] [PubMed] [Google Scholar]
- 35.Ward, J. M., and D. E. Devor-Henneman. 2000. Gestational mortality in genetically engineered mice: evaluating the extraembryonic placenta and membranes, p. 103-122. In J. M. Ward, J. F. Mahler, R. A. Maronpot, and J. P. Sundberg (ed.), Pathology of genetically engineered mice. Iowa State University Press, Ames, Iowa.
- 36.Wei, L., W. Roberts, L. Wang, M. Yamada, S. Zhang, Z. Zhao, S. A. Rivkees, R. J. Schwartz, and K. Imanaka-Yoshida. 2001. Rho kinases play an obligatory role in vertebrate embryonic organogenesis. Development 128:2953-2962. [DOI] [PubMed] [Google Scholar]
- 37.Wodaz, A., and P. Nusse. 1998. Mechanisms of WNT signaling in development. Annu. Rev. Cell. Dev. Biol. 14:59-88. [DOI] [PubMed] [Google Scholar]
- 38.Xu, X., M. Weinstein, C. Li, M. Naski, R. I. Cohen, D. M. Ornitz, P. Leder, and C. Deng. 1998. Fibroblast growth factor receptor 2 (FGFR2)-mediated reciprocal regulation loop between FGF8 and FGF10 is essential for limb induction. Development 125:753-765. [DOI] [PubMed] [Google Scholar]
- 39.Yamamoto, K., and H. Saito. 1998. A pathological role of increased expression of plasminogen activator inhibitor-1 in human or animal disorders. Int. J. Hematol. 68:371-385. [DOI] [PubMed] [Google Scholar]
- 40.Yang, J., M. Boerm, M. McCarty, C. Bucana, I. J. Fidler, Y. Zhuang, and B. Su. 2000. Mekk3 is essential for early embryonic cardiovascular development. Nat. Genet. 24:309-313. [DOI] [PubMed] [Google Scholar]
- 41.Zhou, B., H. E. Lum, J. Lin, and D. I. H. Linzer. 2002. Two placental hormones are agonists in stimulating megakaryocyte growth and differentiation. Endocrinology 143:4281-4286. [DOI] [PubMed] [Google Scholar]